

Abstract
The matrilins are a family of multidomain extracellular matrix proteins with adapter functions. The oligomeric proteins have a bouquet-like structure and bind to a variety of different ligands whereby the avidity of their interactions is dependent on the number of subunits and domains present. Here we show the contribution of post-translational proteolytic processing to the heterogeneity of matrilins seen in tissue extracts and cell culture supernatants. A cleavage site after two glutamate residues in the hinge region close to the C-terminal coiled-coil oligomerization domain is conserved among the matrilins. Cleavage at this site yields molecules that lack almost complete subunits. The processing is least pronounced in matrilin-1 and particularly complex in matrilin-2, which contains additional cleavage sites. Replacement of the hinge region in matrilin-4 by the matrilin-1 hinge region had no marked effect on the processing. A detailed study revealed that matrilin-4 is processed already in the secretory pathway and that the activation of the responsible enzymes is dependent on proprotein convertase activity. Matrilin-3 and -4, but not matrilin-1 subunits present in matrilin-1/-3 hetero-oligomers, were identified as substrates for ADAMTS4 and ADAMTS5, whereas ADAMTS1 did not cleave any matrilin. A neo-epitope antibody raised against the N terminus of the C-terminal cleavage product of matrilin-4 detected processed matrilin-4 in cultures of primary chondrocytes as well as on cartilage sections showing that the conserved cleavage site is used in vivo.
The matrilins form a four-member family of modular, multisubunit matrix proteins, which are expressed in cartilage and many other forms of extracellular matrix (for review, see Ref. 1). They participate in the formation of fibrillar or filamentous structures (2–7) and mediate interactions between collagen-containing fibrils (8, 9) and other matrix constituents like aggrecan (10), small leucine-rich proteoglycans (9), or COMP (11). Matrilins form homo- and hetero-oligomers by their C-terminal coiled-coil domain. In addition, the subunits contain epidermal growth factor-like and von Willebrand factor A (VWA)2-like domains, where the latter are presumably the major ligand binding domains (11). Mutations in matrilin-3 in humans cause different forms of chondrodysplasia (12–14) and are also linked to the development of hand osteoarthritis (15) and intervertebral disc degeneration (16).
Proteolytic processing of extracellular matrix proteins plays both physiological and pathophysiological roles. Proteolysis is a major post-translational modification used to modify the function of proteins. Tissue homeostasis requires a well balanced synthesis and degradation of extracellular matrix proteins, specifically mediated by protease families like matrix metalloproteinases (17), ADAMs (18), or ADAMTSs (19). The development of degenerative diseases is often accompanied by an activation of such proteases. In addition, the cleavage sometimes releases protein fragments that have completely new functions (20, 21).
Determination of which extracellular proteases cleave which substrates is crucial to understand the physiological function of both (22). Physiological cleavage has been described for most members of the matrilin family (4–6), but was not yet extensively studied. The adapter function of the matrilins may be modulated by physiological proteolysis that causes the loss of single subunits and thereby decreases the binding avidity (5). Interestingly, an earlier identified cleavage site in the hinge region of matrilin-4, N-terminal of the coiled-coil, is conserved throughout the matrilin family (5) and it was recently shown that matrilin-3 is cleaved by ADAMTS4 in vitro at this site (23). Here we studied matrilin processing in some detail and identified another member of the ADAMTS family, ADAMTS5, as being able to cleave matrilin-3 and -4. Such cleavage is likely to alter the cohesion of the extracellular matrix.
MATERIALS AND METHODS
Expression and Purification of Recombinant Wild Type and Mutated Matrilin Proteins
All recombinant matrilin proteins were expressed in the human embryonic kidney cell line 293EBNA (Invitrogen). Expression and affinity purification of wild type full-length matrilin-3 and -4 with an N-terminal BM40 signal peptide and a C-terminal StrepII tag was described earlier (5, 11). The cDNAs encoding murine wild type full-length matrilin-1 and matrilin-2 were amplified by PCR using primers that inserted a SpeI restriction site at the 5′-end and a NotI site at the 3′-end, respectively (5′-matn1spe GCC CAC TAG TCC CCC AGC CCA GAG and 3′-matn1not CAA TGC GGC CGC GAT GAT TCT GTT CTC CAG G; 5′-matn2spe GCC CAC TAG TTA GAG AGC GTC CCC AAG CC and 3′-matn2not CAA TGC GGC CGC TCT GTA TTT TAG GCG ATT TTC). After digestion with SpeI and NotI, the amplified cDNA fragments were inserted into the expression vector pCEP-Pu-StrepII tag (C-terminal) in-frame with the sequence of the signal peptide of BM40 (24). The expression construct for the short matrilin-4 splice variant lacking the VWA1 domain (wt M4 ΔA1) was cloned in the same manner into a modified version of pCEP-Pu carrying a C-terminal His8 tag (pCEP-PuV27, kindly provided by Manuel Koch, Cologne) using 5′-SpeI and 3′-BamHI sites in the primers: m4dA1fw, GCC CAC TAG TAA AGG ACC TGT GTG CTG AGT TGG, and m4dA1rev, CAA TGG ATC CCT TTC GGC TAG CCA GCT GG. The matrilin-4 571EE → AA (M4AA) and 571EE → QQ (M4QQ) mutations were introduced into a matrilin-4 full-length cDNA clone in pBluescript KS using the TransformerTM Site-directed Mutagenesis Kit (Clontech) according to the manufacturer's protocol. Mutagenesis primers were CAG CAT TTG CCC AGC GGC GGG CAT TGG C for M4AA, GCA TTT GCC CAC AGC AGG GCA TTG GC for M4QQ, and GTG ACT GGT GAG GCC TCA ACC AAG TC to switch a ScaI to a StuI restriction site in the vector sequence for easier selection of mutant clones. Matrilin-2 910EE → AA and matrilin-3 429EE → QQ mutant constructs were generated by PCR amplification of two fragments of each cDNA, which overlapped in the region in which the mutations and a new unique restriction site (NheI for M2AA and EcoRV for M3QQ) were introduced. As for the wild type constructs, SpeI and NotI sites were introduced via the primers at the 5′- or 3′-ends, respectively. Primer pairs were 5′-matn2spe (see above) and matn2AAas (GCA TTG GTC CTG GCT AGC TGC CAA AGG GTT TCC TG); 5′-matn3spe (GCC CAC TAG TCC GTT TGG CCC GCG CGA GC) and matn3QQas (GAG GCT TCG GGC TTG TTG GAT ATC TGA ACA TGT C) for the 5′-fragments and matn2AAs (CAG GAA ACC CTT TGG CAG CTA GCC AGG ACC AAT GC) and 3′-matn2not (see above); matn3QQs (CAT GTT CAG ATA TCC AAC AAG CCC GAA GCC TC) and 3′-matn3not (CAA TGC GGC CGC ACG ATG TAC TTG TCC ATA TTC) for the 3′-fragments. The amplified 5′- and 3′-fragments were digested with the appropriate restriction enzymes, ligated, cloned into pBluescript KS for easy screening, and finally cloned into the expression vector pCEP-Pu-Strepll tag. The chimeric matrilin-4 constructs “M1 hinge” and “mut M1 hinge” (Fig. 3) were generated by the same strategy introducing unique NdeI restriction sites in the mutated regions and 5′-SpeI and 3′-BamHI sites at the respective ends. pCEP-PuV27 was used as expression vector. Primer pairs were m4dA1fw (see above) and m4dA1m1as (GGC TTT CGC ATT CGC AGG GGT CCT CCT CTG GGC ATA TGC TGC CTT TGA GA); m4dA1fw (see above) and m4dA1m1QQas (GGC TTT CGC ATT CGC AGG GGT CCT GCT GTG GGC ATA TGC TGC CTT TGA GA) for the 5′-fragments; m4dA1m1s (TCT CAA AGG CAG CAT ATG CCC AGA GGA GGA CCC CTG CGA ATG CGA AAG CC), m4dA1rev (see above), and m4dA1m1QQs (TCT CAA AGG CAG CAT ATG CCC ACA GCA GGA CCC CTG CGA ATG CGA AAG CC) for the 3′-fragments. Each of the expression constructs was transfected into 293EBNA cells with FuGENE 6 (Roche), the cell lines were selected with puromycin (1 μg/ml) and cultured under serum-free conditions prior to harvest of conditioned cell culture supernatants. C-terminal StrepII-tagged M2, M2AA, M3, M3QQ, M4, M4AA, and M4QQ were affinity purified from conditioned supernatants using Streptactin-Sepharose Affinity Resin (IBA, Göttingen) as described (5). For the detection of intracellular matrilin-4, transfected 293EBNA cells were cultured for 2 days under serum-free conditions. Cells were then washed twice with PBS and digested with 0.1% trypsin for 15 min at 37 °C to harvest cells and degrade extracellular and cell surface proteins. Digestion was stopped by addition of 10 volumes of prechilled PBS followed by five washing steps in PBS at 4 °C. The cell pellet was ground under liquid nitrogen, homogenized at 4 °C in 100 mm Tris, pH 8.0, 1 tablet/50 ml of “Complete protease inhibitor mixture” (Roche), and cell debris was removed by centrifugation. The supernatant was subjected to SDS-PAGE.
FIGURE 3.

Proteolytic processing of chimeric matrilin-4 proteins. Recombinant matrilin-4 proteins identical to the short splice variant lacking the VWA1 domain were expressed in 293EBNA cells containing either the endogenous matrilin-4 hinge region (wt M4 ΔA1) or the short matrilin-1 hinge region with wild type (M1 hinge) or mutated (mut M1 hinge) cleavage motif. A, amino acid sequence alignment of the hinge region of the different recombinant proteins. The cysteine residues involved in intermolecular disulfide bond formation are underlined. B, SDS-PAGE and subsequent immunoblot of conditioned cell culture media containing the recombinant proteins. The processing patterns were detected with matrilin-4 specific antibodies. For nomenclature see Footnote 3.
Tissue Extraction
Tissues from C57/Bl6 mice were dissected and proteins sequentially extracted at 4 °C with 10 volumes (ml/g wet tissue) of 150 mm NaCl, 50 mm Tris, pH 7.4 (TBS), for 4 h and TBS containing 10 mm EDTA for 16 h. All extraction buffers contained 2 mm phenylmethylsulfonyl fluoride and 2 mm N-ethylmaleimide.
Generation and Purification of Matrilin-4 Neo-epitope Antibodies
To generate a specific cleavage neo-epitope antibody against processed matrilin-4, the peptide matn4pC (H2N-572GIGAGTELRSPC-CONH2) was synthesized and used to immunize two rabbits and two guinea pigs (Pineda Antibody Service, Berlin). The antisera were tested by immunoblot and one rabbit serum, which showed the desired reactivity toward processed matrilin-4, was affinity purified on the matn4pC peptide coupled to CNBr-Sepharose. Antibodies were eluted in 0.1 m glycine, pH 2.5, and neutralized with 1 m Tris, pH 8.8. To assess the specificity, the purified antibody was in some cases incubated with the peptide (1 μg/ml diluted antibody) for 16 h at 4 °C prior to detection by immunoblot or immunofluorescence microscopy.
SDS-Polyacrylamide Gel Electrophoresis, Immunoblotting, Determination of N-terminal Sequences, and Mass Spectrometry
SDS-PAGE was performed as described by Laemmli (25). For immunoblots the proteins were transferred to nitrocellulose and incubated with the appropriate affinity-purified rabbit antibody diluted in TBS containing 5% low fat milk powder. Bound antibodies were detected by luminescence using peroxidase-conjugated swine anti-rabbit IgG (Dako), 3-aminophthalhydrazide (1.25 mm), p-coumaric acid (225 mm), and 0.01% H2O2. Primary antibodies against the following antigens were used: matrilin-1 (26), matrilin-2 (6), matrilin-3 (4), matrilin-4 (5), StrepII-tag (IBA, Göttingen), protein-disulfide isomerase (Stressgen), Golgi matrix protein GM130 (BD Biosciences), protein phosphatase 2A (Santa Cruz), and matn4pC (see above). For N-terminal sequencing, proteins were reduced with 5 mm dithiothreitol and alkylated with ⅓ volume of 6 m acrylamide, subjected to SDS-PAGE, and electroblotted to a polyvinylidene difluoride membrane (Immobilon PSQ, Millipore). Protein bands were cut out and their N-terminal amino acid sequences determined in a Procise Protein Sequencer (Applied Biosystems). Peptide mass fingerprinting (27) and MALDI-TOF mass spectrometry (5, 27) were performed as described. For mass determination of the processed matrilin-3 coiled-coil domains, the affinity purified proteins were reduced in 8 m urea, 10 mm dithiothreitol at 80 °C for 20 min, alkylated with 20 mm iodoacetamide for 1 h in the dark, and desalted on C18 reversed-phase columns (Millipore) with a stepwise elution with 20, 40, 60, and 80% acetonitrile in 0.1% trifluoroacetic acid. Subsequent MALDI-TOF analysis was conducted on a dihydroxybenzoic acid matrix.
Inhibitor Treatments and Pulse-Chase Analysis
Transfected 293EBNA cells were grown to confluence, washed three times with PBS, and cultured for 24 h under serum-free conditions to remove residual serum proteins. Subsequently the cells were cultured in serum-free media supplemented with 0.1, 1, 10, and 30 μm GM 6001, 0.1, 1, and 10 μg/ml aprotinin, E- 64, E-64d, and pepstatin A, 1, 10, and 100 μm amastatin, leupeptin, and decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone, 10, 100, and 1000 μm 1,10-o-phenanthroline, and 20, 200, and 2000 μm 4-(2-aminoethyl)benzenesulfonyl fluoride or only the solvents. The media were changed after 12 and 28 h and the conditioned supernatants harvested at each medium change and after 52 h. For pulse-chase analysis of matrilin-4 processing, transfected 293EBNA cells were grown to confluence, washed in PBS, and cultured in Dulbecco's modified Eagle's medium without l-methionine and l-cysteine. Radiolabeling was performed for 90 min by addition of 50 μCi/ml NEG722 Easytag Express Protein Labeling Mix (PerkinElmer Life Sciences) to the medium (pulse). After two washes with PBS, cells were cultured in serum-free medium and cells and conditioned supernatants were harvested after 30, 60, 90, and 120 min, 6 and 24 h. Cells were lysed in TBS containing 1% Nonidet P-40 and 35S-labeled matrilin-4 was affinity precipitated from cell lysates and conditioned supernatants with 20 μl of Streptactin-Sepharose beads (IBA, Göttingen) per sample for 3 h on a rocking platform. The beads were recovered by centrifugation (1000 × g, 5 min), washed three times in 100 mm Tris, pH 8.0, and eluted in 30 μl of elution buffer (0.1 m Tris, 2.5 mm desthiobiotin). Eluted proteins were separated by SDS-PAGE, the gel dried and exposed to a storage phosphorscreen (GE Healthcare) and detected with a PhosphorImager (GE Healthcare). The autoradiograph was analyzed with ImageQuant 5.1 software (GE Healthcare).
Subcellular Fractionation
Transfected 293EBNA cells expressing recombinant matrilin-4 were grown to confluence, incubated for 24 h in serum-free medium, washed three times in PBS, and recovered from the cell culture dishes with a cell scraper. After three additional washing steps cells were recovered in homogenization buffer (0.25 m sucrose, 10 mm triethanolamine acetate, pH 7.4, 2 mm phenylmethylsulfonyl fluoride, 2 mm N-ethylmaleimide, Complete protease inhibitor mixture (Roche)) and homogenized with 15 strokes in a Dounce homogenizer. Samples were centrifuged at 1000 × g and 4 °C for 10 min. Postnuclear supernatants were separated for 20 h at 38,000 × g in a L7-55 ultracentrifuge (Beckman) equipped with the SW41TI rotor using a 10–40% sucrose gradient, which was poured on a 1-ml cushion of 40% sucrose. Fractions (1 ml) were recovered, proteins were precipitated with ethanol and analyzed by immunoblot.
Isolation of Primary Murine Chondrocytes
Primary murine chondrocytes were isolated from ribcages of newborn C57/Bl6 mice and cultured as described earlier (8).
Immunofluorescence Microscopy
Cultured cells or frozen tissue sections (7 μm) were fixed for 5 min in 95% ethanol, 5% acetic acid at −20 °C, washed three times in TBS, and blocked for 1 h in blocking solution (2% fetal bovine serum in TBS, 0.1% Triton X-100). Frozen sections were in addition digested for 30 min at 37 °C with hyaluronidase (500 units/ml in 100 mm NaH2PO4, 100 mm sodium acetate, pH 5.0) and washed three times in TBS, 0.1% Triton X-100 prior to blocking. Cells or sections were incubated for 3 h with primary antibodies (anti-matrilin-4 (5), anti-matn4pC, or peptide inhibited anti-matn4pC) and 1 h with Alexa Fluor 488 donkey anti-rabbit IgG (Invitrogen) in blocking solution.
Digestion of Matrilins with ADAMTSs
Truncated human recombinant ADAMTS1, ADAMTS4, and ADAMTS5 (28) were from Invitek GmbH, Berlin. ADAMTS1 consisted of amino acids Phe253–Asn617, ADAMTS4 of Phe213–Ala579, and ADAMTS5 of Ser262–Gly625. All contained a C-terminal His6 tag. The aggrecanase activity of the recombinant ADAMTSs was determined by cleavage of recombinant aggrecan interglobular domain. Recombinant murine matrilin-3 and -4 were expressed in 293EBNA cells and purified as described above. Matrilin-1/-3 hetero-oligomers were extracted from fetal bovine rib cartilage and purified as described before (29). The matrilins were reconstituted in 150 mm NaCl, 5 mm CaCl2, 50 mm Tris, pH 7.5, at concentrations of 40 μg/ml. Samples (2 μg) were incubated with ADAMTS4 (0.2 μg), ADAMTS1 (0.4 μg), or ADAMTS5 (0.4 μg) in a total volume of 50 μl at 37 °C. The incubation was stopped by addition of 1 ml of ice-cold ethanol, the proteins were precipitated overnight and after centrifugation the pellet was resolved in 10 μl of water. After addition of 10 μl of sample buffer the samples were submitted to SDS-PAGE.
Overexpression and siRNA Knockdown of ADAMTS4 and -5
cDNAs encoding human full-length ADAMTS4 and ADAMTS5 (Invitek GmbH) were cloned into the expression vector pCEP4 (Invitrogen). Each of the expression constructs was transfected into 293EBNA cells expressing recombinant matrilin-4 with LipofectamineTM 2000 (Invitrogen), the cell lines were selected with hygromycin (200 μg/ml) and cultured under serum-free conditions prior to harvest of conditioned cell culture supernatants. Cells transfected with the empty pCEP4 vector were used as control. The cells were washed with PBS and digested with 0.05% trypsin for 20 min at 37 °C to degrade extracellular and cell surface proteins. The harvested cells were counted and identical numbers were lysed in 150 mm NaCl, 5 mm EDTA, 1 mm sodium orthovanadate, 1% SDS, 50 mm HEPES, 1% Triton X-100, 0.5% Nonidet-P40, 10 mm sodium fluoride, 10% glycerol, pH 6.8, and Complete protease inhibitor mixture (Roche).
siRNAs were obtained from Qiagen (Hs_ADAMTS4_1, ACA GAT GTG GTT GCA TCC TAA; Hs_ADAMTS5_2, CCC GTT AAC TTC ATA GCA AAT). Transfections of siRNA were carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfection mixtures were prepared by adding 1 μl (50 μm) of siRNA to 250 μl of Opti-MEM (Invitrogen) and 5 μl of Lipofectamine 2000 solution to 250 μl of Opti-MEM. After a 20-min incubation, the transfection solution was poured over cells previously covered with 500 μl of supplement-free culture medium. Mock transfection without siRNA served as a control. The transfection solution was removed after 4 h to reduce cytotoxicity and the medium added. The cells were cultured for 96 h and transferred to serum-free conditions prior to harvest of conditioned cell culture supernatants and the analysis of these by SDS-PAGE.
RESULTS
Post-translational Processing Contributes to Matrilin Heterogeneity
To investigate the proteolytic processing of matrilins we established stably transfected 293EBNA human embryonic kidney cell lines that recombinantly express full-length murine matrilin-1, -2, -3, or -4 fused to an N-terminal BM40 signal peptide and a C-terminal StrepII tag. Conditioned media were analyzed by SDS-PAGE and immunoblot using antibodies directed either against the different matrilins or against the StrepII tag (Fig. 1A). Most recombinant matrilins showed heterogeneous band patterns. As previously demonstrated by MALDI-TOF mass spectrometry and Edman degradation (5), matrilin-4 consisted of unprocessed trimers (t, 219 kDa),3 processed dimers (d+cc, 153 kDa) and monomers (m+2cc, 87 kDa) containing an intact coiled-coil as well as cleaved-off monomers (m-cc, 66 kDa; Fig. 1, A and B). The cleavage site lies in the hinge region between the VWA2 domain and the coiled-coil oligomerization domain (Fig. 1, B and C). On the basis of the well characterized band pattern of recombinant matrilin-4 we assigned the bands resulting from cleavage of the other matrilins accordingly. Recombinant matrilin-1 gave bands corresponding to intact trimers (t, 156 kDa), processed dimers (d+cc, 108 kDa), and monomers (m+2cc, 61 kDa; m-cc, 47 kDa; Fig. 1A). In the case of matrilin-1, processed fragments were seen only after overexposure and the major part of the sample consisted of uncleaved trimers.
FIGURE 1.

Matrilin processing. A, proteins in conditioned cell culture media of 293EBNA cells transfected with matrilin-1, -2, -3, or -4 (rec) and extracts of murine articular cartilage (cart) were separated by non-reducing SDS-PAGE on 4–12% SDS-PAGE gels and transferred to nitrocellulose. Matrilins were subsequently detected with affinity purified antibodies specific for each matrilin or for the StrepII tag (matn2 rec). Note that the lane showing the recombinant matrilin-1 was overexposed to better visualize also the weak processed bands. For nomenclature, see Footnote 3 (5). B, schematic representation of uncleaved matrilin-4 trimers (t) and fragments resulting from proteolytic processing. Cleavage in the hinge region (arrow) connecting the second von Willebrand factor A-like (VWA) and the C-terminal coiled-coil oligomerization domain generates a mixture of processed fragments comprised of dimers (d+cc) and monomers (m+2cc) with fully assembled trimeric coiled-coils, completely processed trimeric coiled-coils (3cc), as well as the cleaved-off monomer (m-cc) lacking the coiled-coil region. C, amino acid sequence alignment of the murine matrilin hinge regions. The matrilin-4 cleavage motif (bold) is conserved in the protein family. Black vertical arrows mark cleavage sites identified by N-terminal Edman sequencing and MALDI-TOF mass spectrometry. The cleavage site marked by the gray vertical arrow was found only by MALDI-TOF. Horizontal arrows mark the adjacent N-terminal domains and the C-terminal coiled-coil domains, respectively. The cysteine residues involved in intermolecular disulfide bond formation are underlined.
Matrilin-2 and -3 were much more extensively cleaved than matrilin-1. At least half of the recombinant matrilin-3 (q, 220 kDa) was detected in bands corresponding to processed trimers (t+cc, 166 kDa), dimers (d+2cc, 120 kDa), and monomers (m+3cc, 74 kDa; m-cc, 46 kDa; Fig. 1A). Recombinant matrilin-2 also consisted mainly of trimers, dimers, and monomers, presumably with fully assembled coiled-coils, as shown by detection with an antibody directed against the C-terminal StrepII tag (Fig. 1A). Analysis with an affinity-purified antibody against matrilin-2, however, revealed that matrilin-2 is the most extensively processed matrilin (Fig. 2A, left lane). With this antibody, matrilin-2 oligomers were only weakly detected, whereas most of the protein was present in a 97-kDa band, which did not react with the antibody against the StrepII tag (Fig. 1A and not shown) and therefore represents the cleaved-off monomer lacking the coiled-coil region. In addition, matrilin-2 yielded a complex set of fragments with apparent molecular masses ranging from 40 to 90 kDa. None of these protein fragments were immunoreactive with the StrepII antibody (Fig. 1A), nor did they bind to a Streptactin-Sepharose affinity column (not shown). Nevertheless, peptide mass fingerprint analysis showed that all these fragments originated from matrilin-2 (not shown), confirming the immunoblot data.
FIGURE 2.

Immunoblot analysis of recombinant matrilins with mutated cleavage sites. Matrilin-2 (A), matrilin-3 (B), and matrilin-4 (C) with either wild type hinge region (M2, M3, and M4) or a mutated hinge region, in which the conserved motif EE was replaced by two alanine (M2AA and M4AA) or glutamine residues (M3QQ and M4QQ), were expressed in 293EBNA cells and conditioned media subjected to non-reducing SDS-PAGE on 4–12% gels. After transfer to nitrocellulose, matrilins were detected with antibodies specific for matrilin-2, -3, or -4. For nomenclature, see Footnote 3.
The matrilin fragment patterns detected in extracts of murine articular cartilage were in most cases similar to those of the corresponding recombinant proteins, although most of the bands showed slight shifts in electrophoretic mobility that could be attributed to the tag and, possibly, to a different glycosylation (Fig. 1A). In addition to unprocessed molecules, most of the possible fragments with lower subunit numbers were present. As in the recombinant protein, most of matrilin-1 was in the uncleaved homotrimeric form, confirming that matrilin-1 cleavage is very limited in cartilage (Fig. 1A). For the other matrilins, proteolysis in the C-terminal hinge region is frequent also in articular cartilage, as seen by the presence of bands corresponding to processed oligomers (Fig. 1A). The heterogeneity is even greater than among recombinant matrilins. Further degradation of matrilin-2, as observed in 293EBNA cells and several other cell lines and in skin and uterus extracts (Fig. 2A) (6), was not detected in cartilage (Fig. 1B). Alternative splicing of matrilin-2 mRNA also contributes to the heterogeneity. About 50% of the matrilin-2 transcripts lack a region of 57 nucleotides coding for part of the unique domain (6, 30, 31). Matrilin-3, for which alternative splicing has not been detected in mammals (32, 33), showed two additional forms in cartilage extracts with apparent molecular masses of 170 and 120 kDa. These could possibly result from a processing different from that in the C-terminal hinge region (Fig. 1A and not shown). These variants are not produced in 293EBNA cells or COS-7 cells (not shown). As the yield was too low to allow determination of cleavage sites by N-terminal sequencing, they were not further investigated.
In addition to oligomers generated by proteolytic processing, two matrilin-4 variants with apparent molecular masses lower than the trimer and lower than the processed dimer, respectively, were detected in extracts of articular cartilage, calvaria, rib cartilage, spinal chord, and brain (Fig. 1A, not shown, and Ref. 5). From their masses, these two protein bands could represent matrilin-4 trimers with one or two subunits lacking the N-terminal VWA1 domain, corresponding to matrilin-4 in which the sequence for the VWA1 domain has been alternatively spliced out (34).
The Cleavage Site Found in the Hinge Region of Matrilin-4 Is Conserved within the Protein Family
SDS-PAGE analysis of recombinant and tissue matrilins revealed that, with the exception of matrilin-1, most matrilins are extensively processed in the hinge region to yield oligomers of different subunit number. Because the cleavage site found in matrilin-4 is conserved in the sequence of all matrilins (30, 32, 34, 35), we were interested whether this motif is cleaved also in matrilin-2 and -3. Moreover, it was unclear if the presence of the two conserved glutamate residues in the hinge region were essential for cleavage. To address these issues we expressed mutated matrilin-2, -3, and -4 in which the EE cleavage motif was replaced by either two alanine or two glutamine residues and analyzed the corresponding proteins expressed in 293EBNA cells by SDS-PAGE and immunoblots using antibodies raised against the full-length proteins (Fig. 2).
The introduction of the 570EE → AA (M4AA) and 570EE → QQ (M4QQ) mutations into matrilin-4 resulted in a significant reduction of proteolytic processing (Fig. 2C). Although processed dimers and monomers could still be detected for both mutants, the band intensities were reduced by at least 90% when compared with the wild type protein. We then affinity purified the wild type and mutated matrilin-4 proteins using the StrepII tag and analyzed the cleavage products by MALDI-TOF mass spectrometry. After reduction and alkylation the processed C-terminal coiled-coil domains of the wild type, M4AA, and M4QQ proteins gave molecule ion peaks of 7032, 7034, and 7029 Da, respectively, showing that the residual proteolytic processing of the matrilin-4 mutants takes place at the same position as in the wild type protein (not shown).
As shown by immunoblot analysis, the 910EE → AA mutation in the matrilin-2 hinge region leads to a decreased matrilin-2 heterogeneity (Fig. 2A). Although the bands for processed trimers, dimers, and monomers were of comparable intensity in wild type and mutated matrilin-2 (M2AA), the pattern of smaller fragments was less complex in the M2AA mutant, confirming that in wild type matrilin-2 some of the smaller fragments are generated by cleavage at the EE motif. This indicates that the cleavage site identified in matrilin-4 is used in matrilin-2, but that processed oligomers can also be generated by cleavage at additional sites.
Surprisingly, proteolytic processing of matrilin-3 was not affected by the 429EE → QQ mutation in any obvious way (Fig. 2B). N-terminal sequencing of the cleaved-off coiled-coil domains of wild type and mutated matrilin-3 (M3QQ) gave multiple sequences for the two protein fragments. Clear differences in the signal intensities allowed determination of the sequences 429ARSLISIEDA and 431SLIXIED for wild type matrilin-3 and 429ARSLISIXDA and 431SLIXIEXA for M3QQ. Thus, as matrilin-2 and -4, matrilin-3 is cleaved at the conserved motif, but mutation of glutamate residues do not affect cleavage. Furthermore, both wild type and mutated matrilin-3 proteins can be cleaved two residues downstream of the first cleavage site. MALDI-TOF analysis of the reduced and alkylated wild type matrilin-3 coiled-coil (not shown) yielded molecule ion peaks of 6896, 6669, and 6468 Da, showing a cleavage C-terminal of 429EE, 431AR, and 433SL (Fig. 1C). A third sequence obtained for M3QQ showed no similarity to known sequences and could originate from contamination with an artificial peptide out of the protease inhibitor mixture used during purification. Unfortunately the manufacturer was not willing to provide the information needed to confirm this notion.
Taken together, the results show that the conserved glutamate residues in the hinge regions serve as protease cleavage sites in all matrilins that are significantly processed. In contrast to matrilin-4, mutation of the cleavage sites in matrilin-2 and -3 did not affect overall processing of these proteins, a feature due to the presence of alternative cleavage sites. In matrilin-2 these may reside within the unique domain, a 75-amino acid residue N-terminal extension of the hinge region, and in matrilin-3 cleavage takes place in close vicinity of the conserved motif.
Exchanging the Hinge Region Does Not Abolish Cleavage in Matrilin-4
Matrilin-1 is much less processed than the other matrilins. To test whether the hinge region is responsible for the poor cleavage of matrilin-1, we exchanged the hinge region of matrilin-4 with the one from matrilin-1. Hence, we recombinantly expressed the short matrilin-4 splice variant lacking the VWA1 domain in three versions. The wild type control (wt M4 ΔA1) carries the endogenous matrilin-4 hinge region 672GIGAGTELRSP, whereas the other two versions represent chimeric proteins, in which the matrilin-4 hinge region was replaced by the short matrilin-1 linker with either the wild type sequence 458EEDP (M1 hinge) or a mutated version of the cleavage motif QQDP (mut M1 hinge; Fig. 3A).
All three proteins were similarly cleaved, showing unprocessed trimers of 140 kDa, as well as partially cleaved dimers and monomers of 100 and 66 kDa, respectively (Fig. 3B). In the chimeric matrilin-4 carrying the matrilin-1 hinge region the extent of processing was only moderately decreased, an effect that became more pronounced when the EE cleavage motif was mutated. We conclude that the hinge region itself does not decisively influence proteolysis. Lack of the VWA1 domain had no impact on processing, as the recombinant matrilin-4 proteins representing the longer and shorter splice variants were processed to similar extents (compare Figs. 2C, M4, and 3B, wt M4 ΔA1).
Matrilin-4 Is Processed Intracellularly
Next, we wanted to address the question whether matrilin cleavage occurs extra- or intracellularly. It seems conceivable that proteases could cleave the matrilins either extracellularly, after they have been integrated into the extracellular matrix, or that the cleavage could take place already in the secretory pathway. Extended incubation of conditioned media up to 8 days at 37 °C did not yield any further degradation (not shown) indicating that the cleavage is either intracellular or takes place at the cell surface. The topology of the processing was studied in depth for matrilin-4. In a pulse-chase experiment the newly synthesized proteins were labeled with [35S]methionine and [5S]cysteine. At 30 min after the end of the 90-min pulse, matrilin-4 could be detected as trimers, dimers, and monomers in the conditioned medium (Fig. 4A). The intensity of the bands in the medium increased with time (Fig. 4A), whereas the band intensity in the cellular fraction decreased and was undetectable after 24 h (not shown). A quantitative analysis of the secreted forms of matrilin-4 showed that the ratio between the different oligomeric forms did not significantly change within 24 h and a dimer band could be detected in the cellular fraction already after 2 h (Fig. 4A, right panel). To exclude an influence by cell surface-bound matrilins, matrilin-4 expressing cells were extensively washed and incubated with trypsin for 15 min, followed by a renewed wash and lysis. Also after this treatment dimers and monomers were detected in the cell pellet (Fig. 4B), indicating intracellular proteolysis. To further examine the cleavage, a site-specific neo-epitope antibody against the newly formed N terminus was generated using the peptide H2N-572GIGAGTELRSPC-CONH2 (matn4pC) as antigen for immunization. The matn4pC antibody was specific for processed forms that contain at least one neo-epitope (Fig. 4B). In contrast to the antibody against full-length matrilin-4, the neo-epitope antibody detected mainly the monomer carrying coiled-coil peptides from cleaved subunits (m+2cc) in the cell pellet, a result clearly indicating an intracellular cleavage.
FIGURE 4.

Intracellular processing of recombinant matrilin-4. A, 293EBNA cells stably transfected with StrepII-tagged matrilin-4 were exposed to a pulse of 35S-labeled methionine and cysteine. Cell culture supernatants were harvested at the indicated time points (chase). For the 2-h time point both the supernatant (sn) and the cell lysate (cl) are shown. Radiolabeled matrilin-4 was precipitated using Streptactin-Sepharose beads. The precipitated proteins were separated by non-reducing SDS-PAGE, the gel was dried and radioactivity detected with a phosphorimager. B, immunoblot of recombinant matrilin-4 either from cell culture supernatants (sn) or cell lysates (cl) after removing potential cell surface matrilin-4 by trypsin treatment. The processing patterns were detected using antibodies against all matrilin-4 forms (anti-matn4) or exclusively against the N-terminal neo-epitope of the cleaved C-terminal coiled-coil fragment (anti-matn4pC), which is present in processed dimers and monomers with still trimeric coiled-coils (d+cc, m+2cc) (see Footnote 3). Note that the lane showing the cell lysate probed with the neo-epitope antibody was overexposed to better visualize the bands. C, cells expressing recombinant matrilin-4 were homogenized and subcellular fractions were separated by centrifugation in a 10–40% sucrose gradient, poured on a cushion of 40% sucrose. Fractions 1 to 11 and a pellet fraction (p) were applied to SDS-PAGE and analyzed in subsequent immunoblots with antibodies specific for the cis-Golgi marker GM130, the endoplasmic reticulum resident protein-disulfide isomerase (PDI), the cytoplasmic protein phosphatase 2A (PP2A), and matrilin-4 (matn4).
A subcellular fractionation was also performed and the extent of proteolysis was determined by SDS-PAGE and immunoblot of fractions obtained after sucrose gradient centrifugation (Fig. 4C). Fractions corresponding to smooth and rough endoplasmic reticulum, the cis-Golgi apparatus, and cytosol were identified by use of antibodies against protein-disulfide isomerase, Golgi matrix protein GM130, and protein phosphatase 2A, respectively. Both full-length and processed matrilin-4 were detected in the lower two-thirds of the gradient, however, a clear assignment to a specific compartment was not possible. To exclude that the presence of matrilin-4 in the high density fractions was due to protein aggregation rather than containment in membrane vesicles, the fractionation was repeated in the presence of 1% Triton X-100. Under these conditions, matrilin-4 and all the markers were shifted to lower density fractions indicating that they occur in lipid vesicles (not shown).
To analyze the cleavage in chondrocytes, conditioned medium from primary murine chondrocytes was subjected to immunoblot analysis. Probing with the antibody against full-length matrilin-4 showed a processing pattern similar to that of recombinant matrilin-4, although the cleaved-off monomer (m-cc) was missing (Fig. 5). The matn4pC neo-epitope antibody detected the processed monomer (m+2cc), thereby showing the same cleavage specificity in chondrocytes as in 293EBNA cells. Preincubation of the antibody with the peptide used for immunization resulted in a loss of all signals.
FIGURE 5.
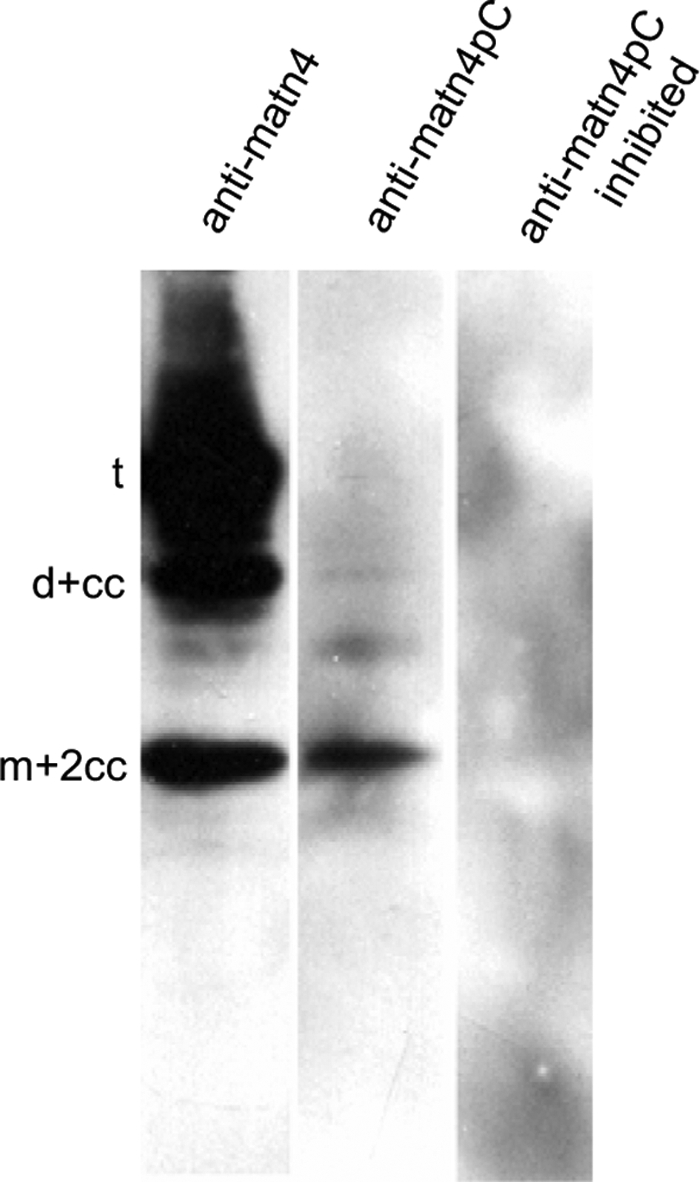
Immunoblot detection of processed matrilin-4 secreted by primary murine chondrocytes. Cell culture supernatants of untransfected primary murine chondrocytes were separated by non-reducing SDS-PAGE and subjected to immunoblot using antibodies detecting all matrilin-4 forms (anti-matn4) or exclusively directed against the N-terminal neo-epitope of the cleaved C-terminal coiled-coil fragment (anti-matn4pC), which is present in processed dimers and monomers with still trimeric coiled-coils (d+cc, m+2cc) (see Footnote 3). To demonstrate the specificity, the anti-matn4pC antibody was preincubated with the peptide against which it was raised (anti-matn4pC inhibited).
We also analyzed 293EBNA cells expressing matrilin-4 by immunohistochemistry. The protein could be detected intracellularly, presumably in compartments representing the secretory pathway, whereas the cells did not deposit extracellular networks containing matrilin-4 (Fig. 6A). The matn4pC neo-epitope, representing processed matrilin-4, could be detected in intracellular compartments (Fig. 6B), whereas peptide inhibition led to a complete loss of the signal (Fig. 6C). This supports the conclusion that matrilin-4 processing takes place in the secretory pathway. In cryosections of newborn mice, the neo-epitope was detected mainly in cartilaginous tissues, e.g. nasal septum and periost of ossifying calvaria, long bones, phalanges of the autopods, ribs, and the vertebrae (Fig. 6E and not shown). In sections of 15.5-day-old mouse embryos the signals were significantly weaker and mainly present in the perichondrium of vertebral bodies and ribs (not shown), indicating that processing of matrilin-4 starts during late gestation and is more pronounced in postnatal development.
FIGURE 6.

Immunofluorescence microscopy of matrilin-4-transfected 293EBNA cells and mouse nasal cartilage sections. Total matrilin-4 and the cleavage site neo-epitope were detected with affinity purified antibodies raised against full-length matrilin-4 (anti-matn4) or the N-terminal neo-epitope of the cleaved C-terminal coiled-coil fragment of matrilin-4 (anti-matn4pc), followed by an Alexa 488TM-conjugated secondary antibody. To demonstrate the specificity, the anti-matn4pC antibody was preincubated with the same peptide that was used for immunization (anti-matn4pC inhibited). A–C, cells were transfected with StrepII-tagged matrilin-4 cDNA and grown on chamber slides. D–F, immunohistochemistry was performed on frozen tissue of newborn mice. Total matrilin-4 was detected throughout the nasal septum cartilage (ns). The neo-epitope antibody detects matrilin-4 in the perichondrium and weaker in cartilage. Arrows indicate nonspecific mucosa (mu) staining that is not affected by preincubation with the neo-epitope peptide used for antibody generation. The bars are 50 μm.
Cleavage of Matrilin-4 Depends on Proprotein Convertase Activity
In a first attempt to identify the proteases involved in matrilin cleavage, matrilin-4 expressing 293EBNA cells were cultivated in the presence of a panel of inhibitors (see “Materials and Methods”) acting on a great variety of proteases. The metalloproteinase inhibitor 1,10-o-phenanthrolin and the serine protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride were cytotoxic when used at high concentrations for longer than 12 h. Among the non-toxic inhibitors only the proprotein convertase-specific inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone was able to inhibit proteolytic processing, with 1 μm giving a reduction of processed matrilin-4 and 100 μm nearly completely blocking cleavage (Fig. 7). However, matrilins do not contain a proprotein convertase cleavage consensus sequence RX(K/R)R at their C termini and it is therefore unlikely that proprotein convertases act directly on matrilins.
FIGURE 7.

Processing of matrilin-4 depends on proprotein convertase activity. Immunoblot analysis of non-reduced samples of cell culture supernatants harvested from matrilin-4 expressing 293EBNA cells after treatment with different concentrations (1, 10, and 100 μm) of the proprotein convertase inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone, the vehicle (dimethyl sulfoxide, DMSO), or without treatment (C). Treatment lasted for 12, 28, or 52 h. For nomenclature, see Footnote 3.
Matrilin-3 and -4 Are Cleaved in Vitro by ADAMTS4 and ADAMTS5 but Not by ADAMTS1
Human matrilin-3 is a substrate for ADAMTS4 and is cleaved C-terminal to the conserved EE motif (23). Furthermore, the proprotein convertases are essential for ADAMTS4 activation (36). We therefore also tested murine matrilin-4 and matrilin-1/-3 hetero-oligomers purified from bovine fetal cartilage for cleavage with ADAMTS4. Homo-oligomeric murine matrilin-3 was used as control. In contrast to the previous study on matrilin-3 cleavage (23) we used a truncated form of ADAMTS4 consisting of the catalytic domain, the disintegrin domain, and the thrombospondin type 1 domain that has been shown to cleave the aggrecan interglobular domain (28). Already the purified matrilins, recombinantly expressed in 293EBNA cells, showed a complex band pattern under non-reducing conditions in SDS-PAGE, due to proteolytic processing during their secretion from 293EBNA cells. However, both matrilin-3 and -4 were further cleaved by ADAMTS4 and non-reducing SDS-PAGE showed the presence of cleaved-off monomers in the digest (Fig. 8A). In contrast, with matrilin-1/-3 hetero-oligomers a complex band pattern appeared after incubation with ADAMTS4, reminiscent of that seen for partially cleaved recombinant matrilin-3 and -4. Bands migrated at the positions of trimers, dimers, and monomers with a complete coiled-coil. In-gel trypsin digestion and MALDI-TOF analysis revealed that all higher bands contained matrilin-1 peptides and that only matrilin-3 subunits had been cleaved. This indicates that the matrilin-1 subunits are not a substrate for ADAMTS4. We also tested C-terminal truncated forms of ADAMTS1 and ADAMTS5 (28) for cleavage of matrilins. ADAMTS5, but not ADAMTS1, was able to cleave matrilin-3 and -4 (Fig. 8B and not shown). Interestingly the matrilin-1/-3 hetero-oligomers were digested by neither ADAMTS5 nor ADAMTS1 (Fig. 8B and not shown), suggesting that formation of hetero-oligomers between matrilin-1 and -3 could represent a mechanism by which matrilin-3 subunits become protected against degradation by ADAMTS5, possibly through a sterical hindrance.
FIGURE 8.

Processing of matrilins by ADAMTS4 and ADAMTS5 in vitro. Recombinant matrilin-3 and -4 homo-oligomers and matrilin-1/-3 hetero-oligomers were incubated with truncated forms of ADAMTS4 (A) or ADAMTS5 (B) for the indicated periods time at 37 °C. The digests were separated on non-reducing 4–12% SDS-polyacrylamide gels. Bands were visualized with Coomassie Brilliant Blue. For nomenclature see, Footnote 3.
Both Overexpression and siRNA Knockdown of ADAMTS4 and ADAMTS5 Influence Matrilin-4 Processing in Cell Culture
It has been reported that truncated ADAMTS4 has a broader specificity for non-aggrecan substrates than the wild type protease (37). We therefore studied matrilin processing by ADAMTS4 and -5, also by transfection of matrilin-4 expressing 293EBNA cells with cDNAs coding for the full-length proteases. The cell lysates and supernatants were analyzed by SDS-PAGE and immunoblot using the neo-epitope antibody as well as the polyclonal matrilin-4 antibody (Fig. 9, A–D). Both transfections resulted in a significantly increased proportion of processed matrilin-4 fragments (Fig. 9, C and D) and as expected, some of the bands reacted also with the neo-epitope antibody (Fig. 9, A and B). In addition, partial siRNA knockdowns of ADAMTS4 and ADAMTS5 in matrilin-4 expressing 293EBNA cells led to decreased matrilin-4 cleavage. Together with the results from the in vitro digestions with truncated ADAMTS4 and ADAMTS5 these experiments clearly show the participation of ADAMTS4 and ADAMTS5 in the intracellular degradation of matrilin-4.
FIGURE 9.

Overexpression and knockdown of ADAMTS4 and ADAMTS5 in matrilin-4 expressing 293EBNA cells. ADAMTS4 and ADAMTS5 were either overexpressed together with matrilin-4 (A–D) or knocked down by specific siRNAs (E) in matrilin-4 expressing 293EBNA cells. Cell lysates (A and C) or conditioned media (B, D, and E) were subjected to non-reducing SDS-PAGE on 4–12% gels. After transfer to nitrocellulose, matrilins were detected with antibodies specific for the N-terminal neo-epitope of the cleaved C-terminal coiled-coil fragment of matrilin-4 (A and B) (anti-matn4pc) or full-length matrilin-4 (A and B) (anti-matn4). For nomenclature, see Footnote 3.
DISCUSSION
We have characterized the proteolytic processing of matrilins, a family of oligomeric proteins that have been shown to act as adapters in the assembly of the extracellular matrix (1, 8, 9, 11, 38). It has been proposed that limited proteolysis releasing almost complete subunits regulates their highly cooperative ligand interactions (11). Previous studies revealed that recombinant matrilin-2, -3, and -4 show a pronounced structural heterogeneity in SDS-PAGE and electron microscopy (4–6). We now show that the pattern of cleavage products is similar in matrilins extracted from tissues as in recombinantly expressed matrilins, indicating that a related processing occurs also in vivo. Proteolytic processing is the major cause for heterogeneity in the matrilin fragment pattern seen in tissue extracts, even though the alternative splicing described for mammalian matrilin-2 and -4 (30, 34, 39) also contributes.
All matrilins contain a hinge region that connects the short C-terminal oligomerization domain with the arms of the bouquet-shaped molecule and is therefore an ideal substrate for proteases to release nearly complete subunits (Fig. 1B). In matrilin-1 and -4 the hinge region is positioned C-terminal to the VWA2 domain, in matrilin-2 C-terminal to the unique domain and in matrilin-3 C-terminal to the epidermal growth factor domains. For matrilin-4 it has been demonstrated that cleavage occurs after two glutamate residues, occurring N-terminal of the coiled-coil, which are conserved in all matrilins (Fig. 1C) (5). The physiological relevance of this processing was now confirmed by the use of a neo-epitope antibody that detects the N-terminal sequence of the cleaved-off coiled-coil in cartilaginous tissues (Fig. 6E). We further demonstrate that murine matrilin-3 is cleaved at the same position as matrilin-4. However, in contrast to matrilin-4, matrilin-3 is also cleaved two and four amino acid residues N-terminal of the cleavage site after the two glutamate residues (Fig. 1C). The importance of the two glutamate residues for cleavage differs between the four matrilins. Whereas their exchange significantly reduced the processing of matrilin-4, in matrilin-2 it leads to a less complex band pattern and in matrilin-3 to no effect. Replacement of the hinge region of matrilin-4 by that of matrilin-1 indicated that the cleavage site in the hinge region is not the only determinant for cleavage. Although matrilin-1 is only poorly cleaved, the exchange had only a moderate effect on matrilin-4 processing. This suggests that at least in the case of matrilin-4 the responsible enzyme also detects other domains via exosite interactions and can cleave at any convenient site in the hinge.
The use of inhibitors to identify the enzymes responsible for the cleavage was only partially successful. Only a proprotein convertase inhibitor was able to inhibit matrilin-4 processing in cell culture (Fig. 7). In particular, we could not show a direct effect of metalloproteinase inhibitors. It is, however, possible that the intracellular inhibitor concentrations did not reach the required levels when applied in reduced amounts to avoid cytotoxicity. In a screen of a peptide phage library, human matrilin-3 was recently identified as an in vitro substrate of ADAMTS4 (23). The cleavage site identified is located after the two glutamate residues in the hinge that are conserved in all matrilins and that we have identified as cleavage sites (Fig. 1C) (5). The second glutamate residue (Glu-434 in human matrilin-3) at P1 was shown to be important for binding and catalysis in vitro (23). We could demonstrate that ADAMTS4 is able to also cleave murine matrilin-3 in vitro, indicating a conservation of the processing. Although the cleavage site 430EARSLISI is slightly different in mouse, the ADAMTS4 cleavage motif E-(AFVLMY)-X(0,1)-(RK)-X(2,3)-(ST)-(VYIFWMLA) is fully contained within the murine sequence. As ADAMTS4 is expressed in 293EBNA cells and primary human chondrocytes (not shown), ADAMTS4 could be responsible for the cleavage. Surprisingly, mutating both glutamate residues at positions P1 and P2 to glutamine did not affect the cleavage observed in cell culture. In contrast, it was shown that peptides having the d-isomeric form of glutamate or an aspartic acid residue at P1 are not cleaved in vitro (23). It could well be that in vivo the closely related ADAMTS5, expressed by 293EBNA cells and primary human chondrocytes, is able to use this altered cleavage site. Alternatively, ADAMTS4 may be less strictly dependent on the glutamate residue at P1 in the context of the matrilin-3 cleavage site. This is supported by the finding that in 16% of the ADAMTS4 substrate sites glutamate is not present at P1 (23). In addition, in 293EBNA cells the wild type and the mutated forms of murine matrilin-3 were cleaved further downstream. It is not known whether this cleavage was also catalyzed by ADAMTS4.
We now show that not only matrilin-3 but also matrilin-4 is cleaved by ADAMTS4 in vitro. If ADAMTS4 is responsible for the cleavage in the hinge region at 571E↓GIGAGT, this is not in complete agreement with the proposed consensus site. Only the glutamate residue at P1 and the threonine residue at P6′ are conserved, whereas in particular, the well conserved basic amino acid residue at either P2′ or P3′ is missing. Nevertheless, mutation of the conserved glutamate residues at P1 and P2 leads to a drastically reduced cleavage of matrilin-4 (Fig. 2C), which could point to a more strictly defined cleavage site for ADAMTS4 in matrilin-4. On the other hand it could be that matrilin-4 is cleaved by another enzyme, possibly ADAMTS5.
As ADAMTS5 probably plays a key role in aggrecan degradation, at least in mouse models of arthritis (40, 41), we further studied the role of ADAMTS5 in matrilin processing. We could indeed show that ADAMTS5 is able to cleave matrilin-4 in vitro and in contrast to published results (23), we could also show in vitro cleavage of matrilin-3 by ADAMTS5 (Fig. 8B). Explanations for the conflicting results could be that the matrilins used were from different species or that different forms of ADAMTS5 were used. The published results (23) were obtained with the full-length enzyme, whereas we used a truncated form lacking the C-terminal part consisting of the cysteine-rich domain, the spacer domain, and the second thrombospondin type I domain (28). It has been shown that the truncated form of ADAMTS5 is active and able to cleave aggrecan (42) or aggrecan fragments (28), although recently it was found that the sequential inclusion of C-terminal domains enhanced its activity toward several both aggrecan and non-aggrecan substrates (43). In the case of ADAMTS4 it was shown that the loss of C-terminal domains leads to an enhanced activity for one and a reduced activity for another cleavage site in aggrecan (44) and the loss of the spacer domain enhanced more general activity against non-aggrecan substrates (37). To further support a physiological role of ADAMTS4 and -5 in matrilin processing we transfected matrilin-4 expressing 293EBNA cells with cDNA coding for the full-length proteases. Both transfections significantly increased matrilin-4 cleavage, whereas siRNA knockdown decreased processing. As compared with ADAMTS4, biochemical characterization of ADAMTS5 is incomplete (43) and the addition of matrilin-3 and -4 to the list of ADAMTS5 substrates will help to define the physiological role of this enzyme. In contrast, neither matrilin-3 nor matrilin-4 are cleaved by ADAMTS1 (not shown), further demonstrating the specific nature of the cleavage.
Matrilin-4 cleavage takes place intracellularly (Figs. 4 and 6B). This is consistent with an intracellular role of ADAMTS4 in in vivo processing, particularly as the proprotein convertase furin that is essential for matrilin-4 processing mediates the intracellular ADAMTS4 activation (36). To our knowledge it has not been reported that ADAMTS5 is activated already inside the cell. At least in cartilage it has been proposed that ADAMTS5 is activated in the extracellular milieu (45). It could well be that the potential role of ADAMTS5 in the intracellular cleavage of matrilins in 293EBNA cells differs from its role in the etiology of osteoarthritis in cartilage.
Matrilins are adapter proteins that connect different extracellular macromolecular networks. The avidity of the interactions is dependent on their ability to enter multiple interactions with a ligand (11). The number of protein binding VWA domains present is influenced by alternative splicing and the number of subunits in the oligomeric complex by proteolytic processing. The characterization of this processing and the identification of members of the ADAMTS family responsible for matrilin processing will lead to a better understanding of tissue remodelling in development and disease.
Acknowledgments
We thank Gabi Klinger, Brigitte Paul-Klausch, and Birgit Kobbe for excellent technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft Grant WA 1338/2-6 and the Köln Fortune program of the Medical Faculty of the University of Cologne.
To allow simple identification of protein components, we introduced a nomenclature where q represents tetramer, t represents trimer, d represents dimer, m represents monomer, and cc represents coiled-coil (5). These designations are followed by a −cc or a +cc. The −cc indicates that the coiled-coil region is absent from the subunit, and +cc shows that the fragment carries additional coiled-coil regions bound by disulfide bonds and derived from other cleaved subunits. The digit before cc refers to the number of these coiled-coil fragments when more than one is carried. For example, m+2cc means a monomer connected by disulfide bonds to two additional coiled-coil region fragments thereby forming a triple coiled-coil α-helix. For a schematic depiction of this nomenclature, see Fig. 1B.
- VWA
- von Willebrand factor A
- ADAM
- a disintegrin and metalloproteinase
- ADAMTS
- a disintegrin and metalloproteinase with thrombospondin-1 motif
- PBS
- phosphate-buffered saline
- PP2A
- protein phosphatase 2A
- TBS
- Tris-buffered saline
- siRNA
- small interfering RNA
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight.
REFERENCES
- 1.Wagener R., Ehlen H. W., Ko Y. P., Kobbe B., Mann H. H., Sengle G., Paulsson M. (2005) FEBS Lett. 579, 3323–3329 [DOI] [PubMed] [Google Scholar]
- 2.Chen Q., Johnson D. M., Haudenschild D. R., Tondravi M. M., Goetinck P. F. (1995) Mol. Biol. Cell 6, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Q., Zhang Y., Johnson D. M., Goetinck P. F. (1999) Mol. Biol. Cell 10, 2149–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klatt A. R., Nitsche D. P., Kobbe B., Mörgelin M., Paulsson M., Wagener R. (2000) J. Biol. Chem. 275, 3999–4006 [DOI] [PubMed] [Google Scholar]
- 5.Klatt A. R., Nitsche D. P., Kobbe B., Macht M., Paulsson M., Wagener R. (2001) J. Biol. Chem. 276, 17267–17275 [DOI] [PubMed] [Google Scholar]
- 6.Piecha D., Muratoglu S., Mörgelin M., Hauser N., Studer D., Kiss I., Paulsson M., Deák F. (1999) J. Biol. Chem. 274, 13353–13361 [DOI] [PubMed] [Google Scholar]
- 7.Winterbottom N., Tondravi M. M., Harrington T. L., Klier F. G., Vertel B. M., Goetinck P. F. (1992) Dev. Dyn. 193, 266–276 [DOI] [PubMed] [Google Scholar]
- 8.Budde B., Blumbach K., Ylöstalo J., Zaucke F., Ehlen H. W., Wagener R., Ala-Kokko L., Paulsson M., Bruckner P., Grässel S. (2005) Mol. Cell. Biol. 25, 10465–10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiberg C., Klatt A. R., Wagener R., Paulsson M., Bateman J. F., Heinegård D., Mörgelin M. (2003) J. Biol. Chem. 278, 37698–37704 [DOI] [PubMed] [Google Scholar]
- 10.Hauser N., Paulsson M., Heinegârd D., Mörgelin M. (1996) J. Biol. Chem. 271, 32247–32252 [DOI] [PubMed] [Google Scholar]
- 11.Mann H. H., Ozbek S., Engel J., Paulsson M., Wagener R. (2004) J. Biol. Chem. 279, 25294–25298 [DOI] [PubMed] [Google Scholar]
- 12.Chapman K. L., Mortier G. R., Chapman K., Loughlin J., Grant M. E., Briggs M. D. (2001) Nat. Genet. 28, 393–396 [DOI] [PubMed] [Google Scholar]
- 13.Mostert A. K., Dijkstra P. F., Jansen B. R., van Horn J. R., de Graaf B., Heutink P., Lindhout D. (2003) Am. J. Med. Genet. A 120, 490–497 [DOI] [PubMed] [Google Scholar]
- 14.Borochowitz Z. U., Scheffer D., Adir V., Dagoneau N., Munnich A., Cormier-Daire V. (2004) J. Med. Genet. 41, 366–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefánsson S. E., Jónsson H., Ingvarsson T., Manolescu I., Jónsson H. H., Olafsdóttir G., Pálsdóttir E., Stefánsdóttir G., Sveinbjörnsdóttir G., Frigge M. L., Kong A., Gulcher J. R., Stefánsson K. (2003) Am. J. Hum. Genet. 72, 1448–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min J. L., Meulenbelt I., Riyazi N., Kloppenburg M., Houwing-Duistermaat J. J., Seymour A. B., van Duijn C. M., Slagboom P. E. (2006) Ann. Rheum. Dis. 65, 1060–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sternlicht M. D., Werb Z. (2001) Annu. Rev. Cell Dev. Biol. 17, 463–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blobel C. P. (2005) Nat. Rev. Mol. Cell Biol. 6, 32–43 [DOI] [PubMed] [Google Scholar]
- 19.Porter S., Clark I. M., Kevorkian L., Edwards D. R. (2005) Biochem. J. 386, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Reilly M. S., Holmgren L., Chen C., Folkman J. (1996) Nat. Med. 2, 689–692 [DOI] [PubMed] [Google Scholar]
- 21.Bergers G., Javaherian K., Lo K. M., Folkman J., Hanahan D. (1999) Science 284, 808–812 [DOI] [PubMed] [Google Scholar]
- 22.Overall C. M., Blobel C. P. (2007) Nat. Rev. Mol. Cell Biol. 8, 245–257 [DOI] [PubMed] [Google Scholar]
- 23.Hills R., Mazzarella R., Fok K., Liu M., Nemirovskiy O., Leone J., Zack M. D., Arner E. C., Viswanathan M., Abujoub A., Muruganandam A., Sexton D. J., Bassill G. J., Sato A. K., Malfait A. M., Tortorella M. D. (2007) J. Biol. Chem. 282, 11101–11109 [DOI] [PubMed] [Google Scholar]
- 24.Kohfeldt E., Maurer P., Vannahme C., Timpl R. (1997) FEBS Lett. 414, 557–561 [DOI] [PubMed] [Google Scholar]
- 25.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 26.Hauser N., Paulsson M. (1994) J. Biol. Chem. 269, 25747–25753 [PubMed] [Google Scholar]
- 27.Sengle G., Kobbe B., Morgelin M., Paulsson M., Wagener R. (2003) J. Biol. Chem. 278, 50240–50249 [DOI] [PubMed] [Google Scholar]
- 28.Will H., Dettloff M., Bendzkô P., Sveshnikov P. (2005) J. Biomol. Tech. 16, 459–472 [PMC free article] [PubMed] [Google Scholar]
- 29.Mann H. H., Sengle G., Gebauer J. M., Eble J. A., Paulsson M., Wagener R. (2007) Matrix Biol. 26, 167–174 [DOI] [PubMed] [Google Scholar]
- 30.Deák F., Piecha D., Bachrati C., Paulsson M., Kiss I. (1997) J. Biol. Chem. 272, 9268–9274 [DOI] [PubMed] [Google Scholar]
- 31.Muratoglu S., Krysan K., Balázs M., Sheng H., Zákány R., Módis L., Kiss I., Deák F. (2000) Cytogenet. Cell Genet. 90, 323–327 [DOI] [PubMed] [Google Scholar]
- 32.Wagener R., Kobbe B., Paulsson M. (1997) FEBS Lett. 413, 129–134 [DOI] [PubMed] [Google Scholar]
- 33.Wagener R., Kobbe B., Aszódi A., Liu Z., Beier D. R., Paulsson M. (2000) Mamm. Genome 11, 85–90 [DOI] [PubMed] [Google Scholar]
- 34.Wagener R., Kobbe B., Paulsson M. (1998) FEBS Lett. 436, 123–127 [DOI] [PubMed] [Google Scholar]
- 35.Aszódi A., Hauser N., Studer D., Paulsson M., Hiripi L., Bösze Z. (1996) Eur. J. Biochem. 236, 970–977 [DOI] [PubMed] [Google Scholar]
- 36.Wang P., Tortorella M., England K., Malfait A. M., Thomas G., Arner E. C., Pei D. (2004) J. Biol. Chem. 279, 15434–15440 [DOI] [PubMed] [Google Scholar]
- 37.Fushimi K., Troeberg L., Nakamura H., Lim N. H., Nagase H. (2008) J. Biol. Chem. 283, 6706–6716 [DOI] [PubMed] [Google Scholar]
- 38.Paulsson M., Heinegård D. (1979) Biochem. J. 183, 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagener R., Kobbe B., Paulsson M. (1998) FEBS Lett. 438, 165–170 [DOI] [PubMed] [Google Scholar]
- 40.Glasson S. S., Askew R., Sheppard B., Carito B., Blanchet T., Ma H. L., Flannery C. R., Peluso D., Kanki K., Yang Z., Majumdar M. K., Morris E. A. (2005) Nature 434, 644–648 [DOI] [PubMed] [Google Scholar]
- 41.Stanton H., Rogerson F. M., East C. J., Golub S. B., Lawlor K. E., Meeker C. T., Little C. B., Last K., Farmer P. J., Campbell I. K., Fourie A. M., Fosang A. J. (2005) Nature 434, 648–652 [DOI] [PubMed] [Google Scholar]
- 42.Zeng W., Corcoran C., Collins-Racie L. A., Lavallie E. R., Morris E. A., Flannery C. R. (2006) Biochim. Biophys. Acta 1760, 517–524 [DOI] [PubMed] [Google Scholar]
- 43.Gendron C., Kashiwagi M., Lim N. H., Enghild J. J., Thøgersen I. B., Hughes C., Caterson B., Nagase H. (2007) J. Biol. Chem. 282, 18294–18306 [DOI] [PubMed] [Google Scholar]
- 44.Kashiwagi M., Enghild J. J., Gendron C., Hughes C., Caterson B., Itoh Y., Nagase H. (2004) J. Biol. Chem. 279, 10109–10119 [DOI] [PubMed] [Google Scholar]
- 45.Malfait A. M., Arner E. C., Song R. H., Alston J. T., Markosyan S., Staten N., Yang Z., Griggs D. W., Tortorella M. D. (2008) Arch. Biochem. Biophys. 478, 43–51 [DOI] [PubMed] [Google Scholar]