

Abstract
Infection with cagA-positive Helicobacter pylori is the strongest risk factor for the development of gastric carcinoma. The cagA gene product CagA, which is delivered into gastric epithelial cells, specifically binds to and aberrantly activates SHP-2 oncoprotein. CagA also interacts with and inhibits partitioning-defective 1 (PAR1)/MARK kinase, which phosphorylates microtubule-associated proteins to destabilize microtubules and thereby causes epithelial polarity defects. In light of the notion that microtubules are not only required for polarity regulation but also essential for the formation of mitotic spindles, we hypothesized that CagA-mediated PAR1 inhibition also influences mitosis. Here, we investigated the effect of CagA on the progression of mitosis. In the presence of CagA, cells displayed a delay in the transition from prophase to metaphase. Furthermore, a fraction of the CagA-expressing cells showed spindle misorientation at the onset of anaphase, followed by chromosomal segregation with abnormal division axis. The effect of CagA on mitosis was abolished by elevated PAR1 expression. Conversely, inhibition of PAR1 kinase elicited mitotic delay similar to that induced by CagA. Thus, CagA-mediated inhibition of PAR1, which perturbs microtubule stability and thereby causes microtubule-based spindle dysfunction, is involved in the prophase/metaphase delay and subsequent spindle misorientation. Consequently, chronic exposure of cells to CagA induces chromosomal instability. Our findings reveal a bifunctional role of CagA as an oncoprotein: CagA elicits uncontrolled cell proliferation by aberrantly activating SHP-2 and at the same time induces chromosomal instability by perturbing the microtubule-based mitotic spindle. The dual function of CagA may cooperatively contribute to the progression of multistep gastric carcinogenesis.
Helicobacter pylori is a spiral-shaped bacterium first described in 1984 by Marshall and Warren (1). H. pylori inhabits at least half of the world's human population. Clinically isolated H. pylori strains can be divided into two major subtypes based on their ability to produce a 120- to 145-kDa protein called cytotoxin-associated gene A antigen (CagA)2 (2–5). More than 90–95% of H. pylori strains isolated in East Asian countries such as Japan, Korea, and China are cagA-positive, whereas 40–50% of those isolated in Western countries are cagA-negative. Infection with a cagA-positive H. pylori strain is associated with severe atrophic gastritis, peptic ulcerations, and gastric adenocarcinoma (6–12).
H. pylori cagA-positive strains deliver the CagA protein into host cells via the cag pathogenicity island-encoded type IV secretion system (4, 5, 13, 14). Translocated CagA then localizes to the inner surface of the plasma membrane, where it undergoes tyrosine phosphorylation by Src family kinases or Abl kinase at the Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs present in the C-terminal region of CagA (15–17). Tyrosine-phosphorylated CagA then binds specifically to SHP-2 tyrosine phosphatase and deregulates its phosphatase activity (18–21). Recent studies have revealed that gain-of-function mutations of SHP-2 are associated with a variety of human malignancies, indicating that SHP-2 is a bona fide human oncoprotein. Furthermore, transgenic expression of CagA in mice induces gastrointestinal and hematological malignancies in a manner that is dependent on CagA tyrosine phosphorylation (22). These findings suggest a critical role of CagA-SHP-2 interaction in the oncogenic potential of CagA.
A polarized epithelial monolayer is characterized by the presence of well developed cell-cell interaction apparatuses such as tight junctions and adherens junctions. The tight junctions act as a paracellular barrier in polarized epithelial cells and play an essential role in the establishment and maintenance of epithelial cell polarity by delimiting the apical and basolateral membrane domains. CagA disrupts the tight junctions and causes loss of epithelial apical-basal polarity (23, 24). The disruption of tight junctions by CagA is mediated by the specific interaction of CagA with partitioning-defective 1 (PAR1) (25, 26). PAR1 is a serine/threonine kinase originally isolated in Caenorhabditis elegans and highly conserved from yeast to humans (27, 28). In mammals, there are four PAR1 isoforms, which may have redundant roles in polarity regulation. PAR1 acts as a master regulator for the regulation of cell polarity in various cell systems. During epithelial polarization, PAR1 specifically localizes to the basolateral membrane, whereas atypical PKC complexed with PAR3 and PAR6 (aPKC complex) specifically localizes to the apical membrane as well as the tight junctions (29–31). This asymmetric distribution of the two kinases, PAR1 and aPKC complex, ensures formation and maintenance of epithelial apical-basal polarity. Notably, mammalian PAR1 kinases were originally identified as microtubule affinity-regulating kinases (MARKs), which phosphorylate microtubule-associated proteins (MAPs) such as Tau, MAP2, and MAP4 on their tubulin-binding repeats. The PAR1/MARK-dependent phosphorylation causes MAPs to detach from and thereby destabilize microtubules (32, 33). Importantly, microtubules form a mitotic spindle, which plays an indispensable role in chromosomal alignment and separation during mitosis, raising the possibility that PAR1 regulates mitosis through controlling stability of the mitotic spindle. Indeed, during mitosis, MAPs undergo a severalfold higher level of phosphorylation (34, 35), and microtubule dynamics increase ∼20-fold (36). This in turn raises the intriguing possibility that CagA influences chromosomal stability by subverting MAP phosphorylation through systemic inhibition of PAR1.
In this study, the effects of CagA on microtubule-dependent cellular events, especially dynamics of the mitotic spindle and chromosomal segregation during mitosis, were examined. The results of this work provide evidence that CagA perturbs mitotic spindle checkpoint and thereby causes chromosomal instability. Given the role of chromosomal instability in cell transformation, the newly identified CagA activity may play a crucial role in the development of gastric carcinoma.
EXPERIMENTAL PROCEDURES
Antibodies
Anti-hemagglutinin (HA, 3F10, Roche Applied Science), anti-phospho histone H3 (Ser-10, Sigma), anti-omni (M-21, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Actin (C-11, Santa Cruz Biotechnology), anti-PAR1b (25) antibodies were used as primary antibodies for immunoblotting and immunostaining.
Cell Lines and Transfection
MKN28 human gastric epithelial cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. WT-A10 cell line was an MKN28-derived stable transfectant clone, in which CagA was inducibly expressed by using the tetracycline-regulatable tet-off system (37). For establishment of stable cell lines that constitutively express a fusion protein of green fluorescence protein (GFP) and human histone H2B, WT-A10 cells were transfected with H2B-GFP expression vector and cultured with 5 μg/ml blasticidin (Funakoshi) (38) for drug selection. Drug-resistant colonies were examined under fluorescence microscopy, and GFP-positive cells were isolated and then single cell cloned by limiting dilution method.
Expression Vectors
The H2B-GFP expression vector was kindly provided by T. Kanda (Aichi Cancer Center, Japan). A recombinant adenovirus that expresses wild-type (WT) PAR1b, dominant-negative PAR1b (PAR1b MC) (25) or β-galactosidase was generated according to the instructions of manufacture (ViraPower Adenoviral Expression System, Invitrogen).
Immunoblotting
Cells were lysed in lysis buffer (50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 5 mm EDTA, 1% Brij-35, 2 mm Na3VO4, 2 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml trypsin inhibitor, 10 μg/ml aprotinin). Immunoblotting was performed as described previously (19).
Immunostaining
Cells were seeded on chamber slide in the presence or absence of 0.2 μg/ml doxycycline (Dox). After 12-h incubation, cells were fixed with 10% formaldehyde for 15 min and permeabilized with 0.25% Triton X-100 for 10 min. The cells were then treated overnight with primary antibody and were visualized with Alexa Fluor-conjugated secondary antibodies (Invitrogen). Images were captured by using a confocal microscope system (Olympus).
Time-lapse Video Microscopy Recording
Cells were cultured on collagen-coated 35-mm culture dishes in the presence or absence of Dox. After 12-h incubation, culture medium was replaced by phenol-red free RPMI 1640 medium (GIBCO) supplemented with 50 mm HEPES (pH 7.3) and 10% fetal bovine serum. Time-lapse fluorescence microscopy was performed on the stage maintained at 37 °C and images were acquired every 3 min with a charge-coupled device camera (Princeton) controlled by Molecular Devices MetaMorph imaging software (Universal Imaging Corp.) using a 20× objective (Olympus).
Cell Cycle Analysis
Cells were collected by trypsinization, washed with phosphate-buffered saline, and fixed in 70% cold ethanol at −20 °C overnight. After centrifugation, cells were washed with phosphate-buffered saline and treated for 20 min at 37 °C with 0.5 mg/ml RNase A (Sigma). Cells were stained at room temperature with 50 μg/ml propidium iodide and subjected to flow cytometric analysis by using FACScan and Cell Quest software (BD Biosciences).
RESULTS
Establishment of CagA-inducible Gastric Epithelial Cells That Stably Express H2B-GFP
It was previously reported that a fusion protein of GFP and human histone H2B (H2B-GFP) is incorporated into nucleosome core particles without perturbing cell cycle progression (39). Accordingly, we employed H2B-GFP to visualize mitotic chromosomal segregation in living cells by time-lapse analysis. To do so, an expression vector for H2B-GFP was transfected into MKN28-derived WT-A10 cells, in which CagA was inducibly expressed by depleting the tetracycline-analog doxycycline (Dox) from the culture supernatant (37). After drug selection and limiting dilution, several transfectant clones with a high level of GFP expression and strict regulation of CagA induction by Dox were chosen for further analysis. One of the established clones, A10/H2B-GFP, was arbitrarily chosen and subsequently used in this work (Fig. 1).
FIGURE 1.

Establishment of CagA-inducible gastric epithelial cells that constitutively express H2B-GFP. A, schematic representation of CagA and H2B-GFP. B, fluorescence of H2B-GFP in the nucleus of A10/H2B-GFP cells. A10/H2B-GFP cells were generated by stably transfecting an H2B-GFP-expression vector into WT-A10 cells, MKN28-derived human gastric epithelial cells in which CagA is inducibly expressed by depleting Doxycycline from the culture. Fluorescence image (upper). Fluorescence image plus phase contrast image (lower). Scale bars, 20 μm. C, inducible expression of HA-tagged CagA in WT-A10 and A10/H2B-GFP cells. Cells were cultured in the presence or absence of Dox and cell lysates were subjected to immunoblotting with the indicated antibodies.
CagA Delays Prophase-metaphase Transition during Mitosis
A10/H2B-GFP cells were cultured on collagen-coated dishes with or without Dox, and the progression of mitosis in the presence or absence of CagA was monitored using a time-lapse fluorescence microscope. In the absence of CagA, cells initiated prophase and reached the anaphase without overt abnormality (Fig. 2 and supplemental video 1). In the presence of CagA, however, cells exhibited delayed chromosomal segregation.
FIGURE 2.

Effect of CagA on the progression of mitosis. A10/H2B-GFP cells were cultured in the presence (CagA (−)) or absence (CagA (+)) of Dox. At 12 h after the onset of Dox treatment, cells were subjected to time-lapse fluorescence microscopic analysis. Representative images for the progression of mitosis starting from the onset of prophase (0 min) in A10/H2B-GFP cells with (lower) or without (upper) CagA expression. QuickTime videos of these experiments are included as supplemental videos 1 and 2.
To quantitatively evaluate the effect of CagA on the progression of mitosis from prophase to anaphase, the time required from the onset of chromosome condensation to the onset of chromosomal separation was measured (Fig. 3). In the absence of CagA, cells required ∼30–40 min for the prophase-to-anaphase transition. In contrast, in the presence of CagA, cells took ∼60–80 min on average from prophase to the onset of anaphase. In fact, some of the CagA-expressing cells required >100 min for the transition. To quantitatively evaluate cells entering prophase or metaphase, cells were stained with an antibody specific to phosphorylated Serine 10 of histone H3 (H3-pSer10), which serves as a prophase/metaphase marker, and the number of H3-pSer10-positive cells was counted. As expected, the number of H3-pSer10-positive cells in the presence of CagA was two times larger than that in the absence of CagA (Fig. 4A). To exclude the possibility that the increase in prophase/metaphase cells was due to accelerated cell cycle progression by CagA, cells with CagA induction and those without CagA induction were treated with nocodazole to arrest them in prophase/metaphase. Following nocodazole treatment, there was no difference in the numbers of cells undergoing mitosis in the presence and absence of CagA at each time point after CagA induction (12, 15, and 18 h) (Fig. 4B), indicating that CagA does not accelerate cell cycle progression. Furthermore, CagA expression did not influence the number of cells in anaphase (Fig. 4C). The results indicate that CagA specifically delays the progression from prophase to anaphase during mitosis.
FIGURE 3.
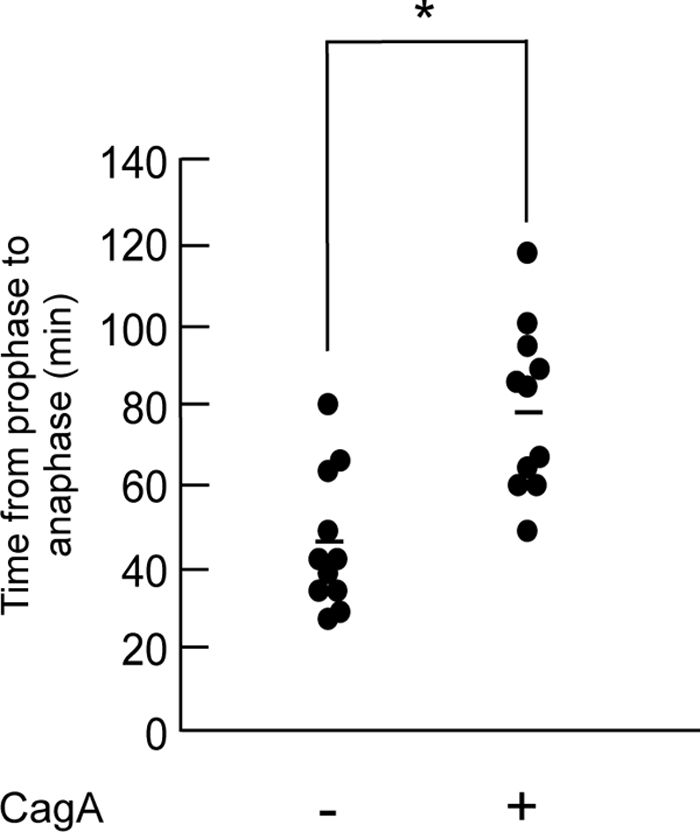
Prolonged mitotic chromosomal segregation in cells expressing CagA. The time length from the onset of chromatin condensation to the initiation of chromosomal separation was measured in A10/H2B-GFP cells with or without CagA expression. *, p < 0.05, statistically significant (Student's t test) (n = 11). Bars indicate mean.
FIGURE 4.
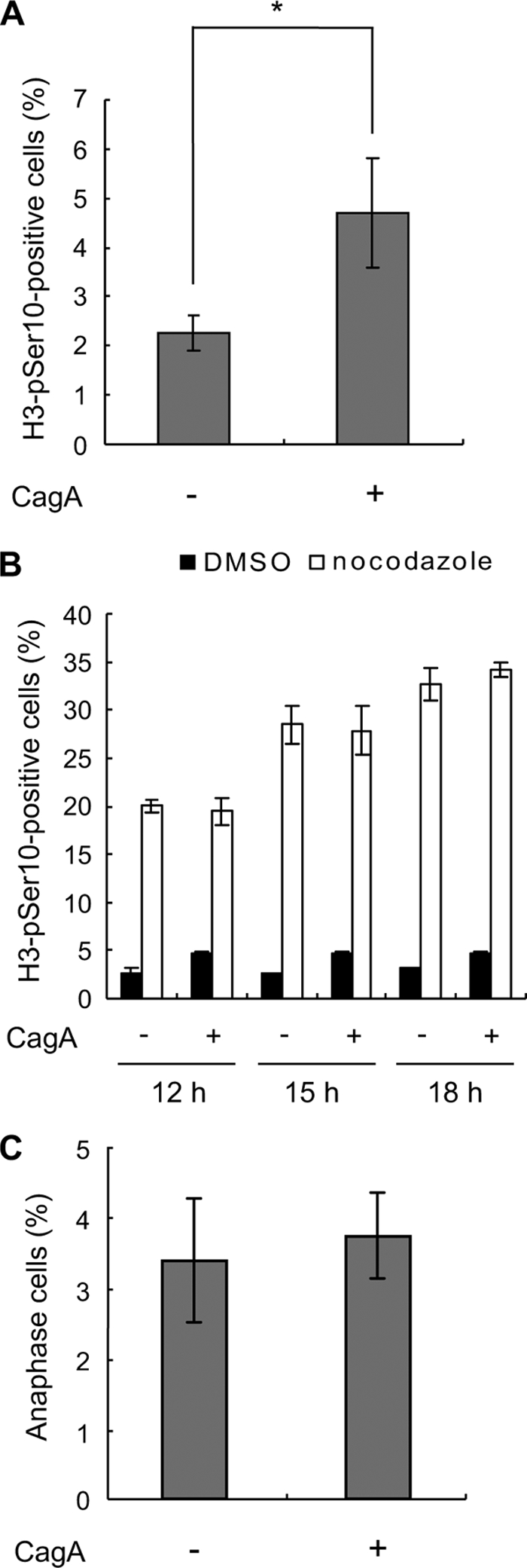
Delay in prophase and metaphase by CagA. A, WT-A10 cells were cultured in the presence or absence of Dox. After 12 h, cells were stained with an antibody against phosphorylated serine 10 on histone H3 (H3-pSer10) to count H3-pSer10-positive cells. Error bars indicate the mean ± S.D. *, p < 0.05, statistically significant (Student's t test) (n = 3). B, WT-A10 cells were induced to express CagA by depleting Dox in the absence or presence of the mitotic inhibitor, nocodazole. After 12, 15, and 18 h of CagA induction, cells were stained with an antibody against H3-pSer10. DMSO was used as a control of nocodazole treatment. Error bars indicate the mean ± S.D. Three hundred cells were subjected to the staining analysis in each of three independent experiments. C, WT-A10 cells were cultured in the presence or absence of Dox. After 12 h, cells were stained with 4′,6-diamidino-2-phenylindole to count anaphase cells. Error bars indicate the mean ± S.D. Two hundred cells were subjected to the staining analysis in each of three independent experiments.
CagA Delays Progression of Mitosis through PAR1b Inhibition
CagA binds PAR1 kinase and inhibits its activity, thereby perturbing microtubule stability (25). To investigate whether inhibition of PAR1 by CagA is involved in the CagA-mediated delay in prophase-metaphase transition, we produced a recombinant adenovirus that expresses omni-tagged PAR1b, one of four PAR1 isoforms in mammals (30). Infection with PAR1b-transducing adenovirus in WT-A10 abolished the ability of CagA to induce the prophase-metaphase delay (Fig. 5). On the other hand, infection with adenovirus transducing a dominant-negative PAR1b (PAR1b MC) increased the number of cells in prophase/metaphase cells in the absence of CagA (Fig. 5). Induction of CagA in cells expressing PAR1b MC did not have an additional effect on prophase-metaphase delay caused by PAR1b MC. These results indicate that CagA delays progression from prophase to anaphase through inhibition of PAR1.
FIGURE 5.

Delay in prophase and metaphase by CagA-mediated PAR1 inhibition. WT-A10 cells were induced to express CagA by depleting Dox in the presence of the indicated adenoviruses (multiplicity of infection = 200 for each virus). After 12 h, cells were stained with an antibody against phosphorylated serine 10 on histone H3 (H3-pSer10) to count H3-pSer10-positive cells. Error bars indicate the mean ± S.D. *, p < 0.05; **, p < 0.01, statistically significant (Student's t test) (n = 3) (left). Expressed protein levels are shown (right). β-Gal, β-galactosidase.
Spindle Misorientation Induced by CagA during Mitosis
Through the time-lapse analysis of mitotic cells, we noticed that a fraction of CagA-expressing cells exhibited aberrant cell division, the direction of which is not parallel to culture dish (Fig. 2 and supplemental video 2). To investigate the mechanism, we sought to examine chromosomal positioning during mitosis in more detail. To this end, we determined the chromosomal orientation of A10/H2B-GFP cells during anaphase by using a confocal fluorescence microscope. In the absence of CagA, most daughter chromosomes separated parallel to the culture dish. In contrast, in the presence of CagA, a significant fraction of cells displayed mispositioning of daughter chromosomes (Fig. 6A). The angles between the inclination of two daughter chromosomes and the culture dish surface (Fig. 6B, left, α) were then measured in anaphase cells. In the presence of CagA, the percentage of cells with spindle misorientation, in which the angle α inclines more than 10°, was significantly increased compared with the percentage of CagA-nonexpressing cells (Fig. 6B). These results indicate that CagA expression causes spindle misorientation and thereby perturbs the cell division axis. As a consequence, two daughter cells failed to attach to the culture dish evenly (supplemental Fig. 1).
FIGURE 6.

Impaired cell division axis by CagA. A, confocal x-z plane views of spindle orientation in anaphase. A10/H2B-GFP cells were cultured in the presence or absence of Dox. After 12 h, cells were fixed, and segregation of chromosomes was visualized by confocal microscopy. B, percentages of cells with spindle misorientation in the presence or absence of CagA. In this experiment, cells in which the angles of the two daughter chromosomes (α) incline more than 10° were judged to have destabilized cell division axis (left). Fifteen anaphase cells were investigated in each of three independent experiments. Error bars indicate the mean ± S.D. *, p < 0.05, statistically significant (Student's t test) (right).
CagA Induces Chromosomal Instability
Perturbation of mitotic progression by CagA suggests that chronic exposure of cells to CagA causes chromosomal instability in cells. To test this possibility, WT-A10 cells were cultured for 6 days with or without intermittent induction of CagA by depleting Dox from the culture on days 1, 3, and 5. Consistent with the above-described data showing mitotic delay by CagA, flow cytometric analysis of DNA contents revealed that CagA exposure increases cells in mitosis (M-phase). The results also revealed the appearance of cells with an 8N peak only in cells chronically exposed to CagA (Fig. 7). In these CagA-expressing cells, the percentage of cells with DNA content of more than 4N was about three-times greater than that of control cells. On the other hand, the number of cells in G1 phase was significantly reduced in the presence of CagA, most probably due to an increase in the number of cells in G2/M as well as dramatic accumulation of cells with >4N. Effect of CagA on S-phase was marginal. These results indicate that chronic exposure to CagA induces aneuploidy and polyploidy in cells. The results are also in agreement with the results of a previous study showing that checkpoint activation (mitotic delay) is often transient, with some cells slipping past the arrest and producing a tetraploid population due to defective cell division (40). From the observations, we conclude that repeated exposure to CagA, which represents chronic infection with cagA-positive H. pylori in vivo, induces chromosomal instability in host cells.
FIGURE 7.
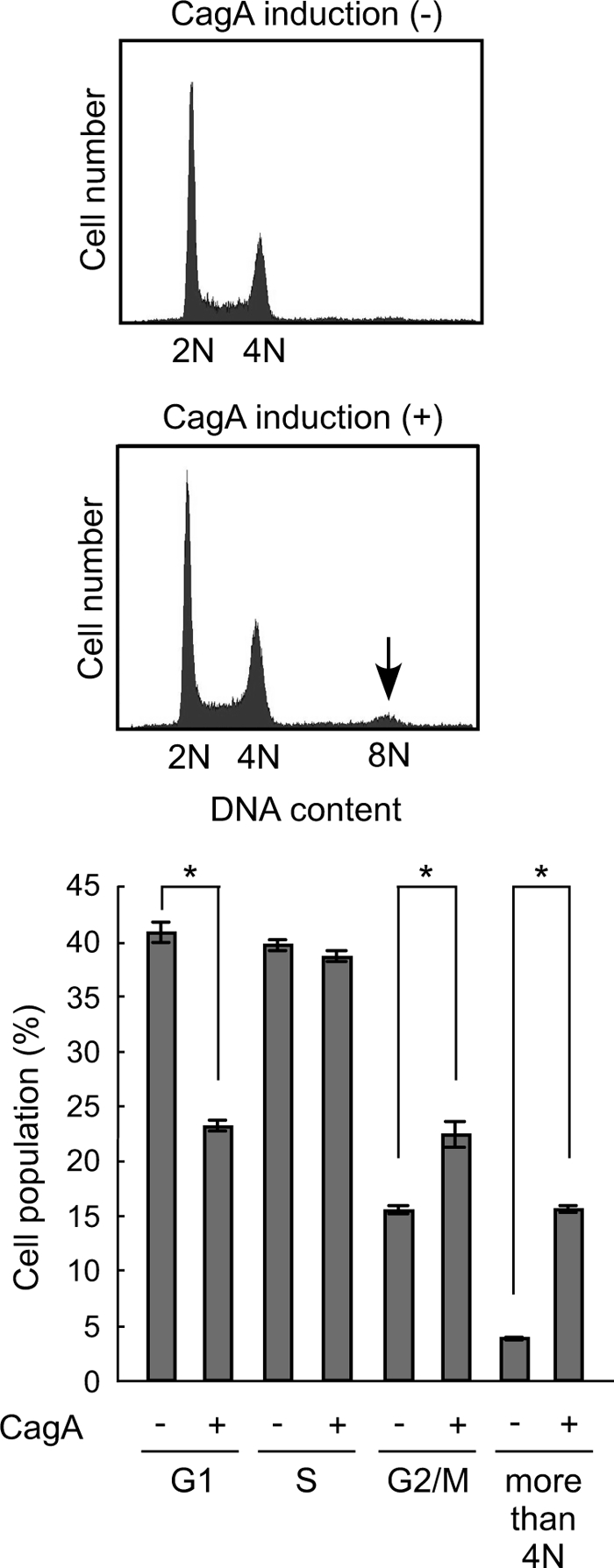
Chromosomal instability caused by repeated exposure to CagA. WT-A10 cells were cultured for 6 days with intermittent induction of CagA on days 1, 3, and 5 by depleting Dox. WT-A10 cells without CagA induction during the culture were used as a control. After the culture, cells were harvested, stained with propidium iodide, and subjected to DNA histogram analysis using flow cytometry. The arrow shows tetraploid (8N) (upper). Percentages of cells in each cell cycle phases were determined by use of CellQuest and ModFit cell cycle analysis software. Error bars indicate the mean ± S.D. *, p < 0.001, statistically significant (Student's t test) (lower).
DISCUSSION
In the present study, we demonstrated that expression of H. pylori CagA results in delay of the progression from prophase to metaphase in mitosis and causes spindle misorientation at the onset of anaphase due to a destabilized cell division axis, resulting in induction of chromosomal instability. We previously demonstrated that CagA specifically binds to PAR1, comprising four isoforms (PAR1a, b, c, and d), and inhibits the kinase activity (25). In mammalian cells, PAR1 acts as an MARK and thereby plays an important role in the regulation of microtubule stability upon MAP phosphorylation (32, 33). PAR1-mediated MAP phosphorylation causes dissociation of MAPs from the microtubules and destabilizes microtubules. Given that microtubules are essentially involved in mitosis as components of the mitotic spindle, our results indicate that CagA perturbs mitosis by binding to PAR1 kinase and inhibiting its activity.
In metazoans, the spindle-assembly checkpoint plays a critical role in mitosis to ensure appropriate alignment of chromosomes prior to the onset of anaphase to enable segregation of genetic materials to each daughter cell (41, 42). At the beginning of mitosis, when kinetochores are unattached, the spindle-assembly checkpoint proteins are activated to coordinate the variable duration of microtubule-chromosome attachment and chromosomal alignment with subsequent loss of sister-chromatid cohesion at metaphase to anaphase transition. Using time-lapse microscopy, we found that CagA delays chromosomal separation. The observations imply that CagA subverts mitotic spindle organization and/or attachment of microtubules to kinetochores, both of which are attributable to the impaired microtubule functions caused by CagA-mediated PAR1 inhibition, thereby keeping the spindle checkpoint machinery in its active state.
Another abnormality that was found to be associated with CagA expression during mitosis is spindle misorientation at the end of metaphase. Equal attachment of two daughter cells to the culture dish is ensured by the positioning of the spindle axis parallel to the long cell axis during mitosis. In turn, spindle misorientation causes uneven attachment of the two daughter cells to the dish following mitosis, as observed in cells expressing CagA. Théry et al. reported that the spindle orientation is controlled by cell adhesion to the extracellular matrix (43). Toyoshima and Nishida showed that the spindle is oriented parallel to the cell-extracellular matrix adhesion plane via β1-integrin-mediated signaling (44). This observation provides a molecular basis for the spindle misorientation in cells expressing CagA: CagA inhibits PAR1 and thereby perturbs microtubule stability, which in turn causes malfunctioning of microtubules, including astral microtubules. Consequently, cells expressing CagA generate an inadequate cell division axis and thereby initiate aberrant cell division, the direction of which is not parallel to culture dish. Interestingly, when ectopically expressed in Chinese hamster ovary cells, PAR1a, PAR1b, and PAR1c are distributed uniformly to the cytoplasm, whereas PAR1d preferentially localizes to the centrosome and microtubules. Notably, acentrosomal cells lack radial arrays of astral microtubules and are therefore defective in spindle positioning, indicating that the centrosome ensures the fidelity of chromosomal segregation (45). Thus, PAR1d might be a primary target of CagA in perturbing the mitotic process. Also, it is noted that mitotic spindles are oriented parallel to the substratum through a mechanism that is dependent on integrin-mediated cell-substrate adhesion (44, 46). In this regard, CagA-deregulated SHP-2 is known to dephosphorylate FAK and thereby inhibit focal adhesions. Accordingly, CagA-mediated SHP-2 activation may cooperate with CagA-mediated inhibition of PAR1 to induce destabilization of spindle orientation and formation of an improper cell division axis.
Inhibition of proteins involved in spindle assembly causes mitotic delay and ultimately promotes segregation errors and aneuploidy, leading to chromosomal instability (47). Our study also showed that chronic exposure to CagA induces chromosomal instability in cells. This CagA activity, caused by malfunctioning of the mitotic process, may significantly contribute to carcinogenesis. Notably, the effect of CagA on mitosis is relatively mild. However, compared with drastic perturbations of chromosomal segregation that would immediately trigger apoptosis, mild perturbation of chromosomal alignment is more likely to increase the chance to generate variant cells that can escape from the mitotic spindle checkpoint (41, 48).
Our present work reveals that CagA exploits the chromosomal segregation system for pathogenesis. The newly identified CagA activity increases the chance for cells to acquire additional genetic alterations that contribute to tumor progression toward more aggressive phenotypes. Our findings therefore reveal a bifunctional role of CagA as an oncoprotein. On the one hand, CagA stimulates unscheduled cell proliferation by targeting signal transducing molecules such as SHP-2, and, on the other hand, it increases genetic instability by disturbing the microtubule-based mitotic spindles. The dual function of CagA may cooperatively contribute to the progression of multistep gastric carcinogenesis.
Supplementary Material
Acknowledgment
We thank Dr. Teru Kanda for the H2B-GFP expression vector.
This work was supported by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1 and videos 1 and 2.
- CagA
- cytotoxin-associated gene A
- cag
- cytotoxin-associated gene
- Dox
- doxycycline
- GFP
- green fluorescent protein
- H3-pSer10
- phosphorylated serine-10 of histone H3
- H2B-GFP
- a fusion protein of human histone H2B and GFP
- MAP
- microtubule-associated protein
- MARK
- microtubule affinity-regulating kinase
- PAR1
- partitioning-defective 1
- SHP-2
- Src homology 2 domain-containing protein tyrosine phiosphatase-2
- WT
- wild type
- PKC
- protein kinase C.
REFERENCES
- 1.Marshall B. J., Warren J. R. (1984) Lancet 1, 1311–1315 [DOI] [PubMed] [Google Scholar]
- 2.Covacci A., Censini S., Bugnoli M., Petracca R., Burroni D., Macchia G., Massone A., Papini E., Xiang Z., Figura N., Rappuoli R. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 5791–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tummuru M. K., Cover T. L., Blaser M. J. (1993) Infect. Immun. 61, 1799–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomb J. F., White O., Kerlavage A. R., Clayton R. A., Sutton G. G., Fleischmann R. D., Ketchum K. A., Klenk H. P., Gill S., Dougherty B. A., Nelson K., Quackenbush J., Zhou L., Kirkness E. F., Peterson S., Loftus B., Richardson D., Dodson R., Khalak H. G., Glodek A., McKenney K., Fitzegerald L. M., Lee N., Adams M. D., Hickey E. K., Berg D. E., Gocayne J. D., Utterback T. R., Peterson J. D., Kelley J. M., Cotton M. D., Weidman J. M., Fujii C., Bowman C., Watthey L., Wallin E., Hayes W. S., Borodovsky M., Karp P. D., Smith H. O., Fraser C. M., Venter J. C. (1997) Nature 388, 539–547 [DOI] [PubMed] [Google Scholar]
- 5.Covacci A., Telford J. L., Del Giudice G., Parsonnet J., Ruppuoli R. (1999) Science 284, 1328–1333 [DOI] [PubMed] [Google Scholar]
- 6.Blaser M. J., Perez-Perez G. I., Kleanthous H., Cover T. L., Peek R. M., Chyou P. H., Stemmermann G. N., Nomura A. (1995) Cancer Res. 55, 2111–2115 [PubMed] [Google Scholar]
- 7.Parsonnet J., Friedman G. D., Orentreich N., Vogelman H. (1997) Gut 40, 297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rugge M., Busatto G., Cassaro M., Shiao Y. H., Russo V., Leandro G., Avellini C., Fabiano A., Sidoni A., Covacci A. (1999) Cancer 85, 2506–2511 [PubMed] [Google Scholar]
- 9.Rudi J., Kolb C., Maiwald M., Zuna I., vov Herbay A., Galle P. R., Stremmel W. (1997) Dig. Dis. Sci. 42, 1652–1659 [DOI] [PubMed] [Google Scholar]
- 10.Torres J., Pérez-Pérez G. I., Leal-Herrera Y., Muñoz O. (1998) Int. J. Cancer 78, 298–300 [DOI] [PubMed] [Google Scholar]
- 11.Shimoyama T., Fukuda S., Tanaka M., Mikami T., Munakata A., Crabtree J. E. (1998) J. Clin. Pathol. 51, 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nomura A. M., Lee J., Stemmermann G. N., Nomura R. Y., Pérez-Pérez G. I., Blaser M. J. (2002) J. Infect. Dis 186, 1138–1144 [DOI] [PubMed] [Google Scholar]
- 13.Segal E. D., Cha J., Lo J., Falkow S., Tompkins L. S. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 14559–14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odenbreit S., Püls J., Sedlmaier B., Gerland E., Fischer W., Haas R. (2000) Science 287, 1497–1500 [DOI] [PubMed] [Google Scholar]
- 15.Stein M., Bagnoli F., Halenbeck R., Rappuoli R., Fantl W. J., Covacci A. (2002) Mol. Microbiol. 43, 971–980 [DOI] [PubMed] [Google Scholar]
- 16.Backert S., Moese S., Selbach M., Brinkmann V., Meyer T. F. (2001) Mol. Microbiol. 42, 631–644 [DOI] [PubMed] [Google Scholar]
- 17.Poppe M., Feller S. M., Römer G., Wessler S. (2007) Oncogene 26, 3462–3472 [DOI] [PubMed] [Google Scholar]
- 18.Higashi H., Tsutsumi R., Muto S., Sugiyama T., Azuma T., Asaka M., Hatakeyama M. (2002) Science 295, 683–686 [DOI] [PubMed] [Google Scholar]
- 19.Higashi H., Tsutsumi R., Fujita A., Yamazaki S., Asaka M., Azuma T., Hatakeyama M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 14428–14433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamazaki S., Yamakawa A., Ito Y., Ohtani M., Higashi H., Hatakeyama M., Azuma T. (2003) J. Infect. Dis. 187, 334–337 [DOI] [PubMed] [Google Scholar]
- 21.Hatakeyama M. (2004) Nat. Rev. Cancer 4, 688–694 [DOI] [PubMed] [Google Scholar]
- 22.Ohnishi N., Yuasa H., Tanaka S., Sawa H., Miura M., Matsui A., Higashi H., Musashi M., Iwabuchi K., Suzuki M., Yamada G., Azuma T., Hatakeyama M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amieva M. R., Vogelmann R., Covacci A., Tompkins L. S., Nelson W. J., Falkow S. (2003) Science 300, 1430–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bagnoli F., Buti L., Tompkins L., Covacci A., Amieva M. R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16339–16344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saadat I., Higashi H., Obuse C., Umeda M., Murata-Kamiya N., Saito Y., Lu H., Ohnishi N., Azuma T., Suzuki A., Ohno S., Hatakeyama M. (2007) Nature 447, 330–333 [DOI] [PubMed] [Google Scholar]
- 26.Lu H. S., Saito Y., Umeda M., Murata-Kamiya N., Zhang H. M., Higashi H., Hatakeyama M. (2008) Cancer Sci. 99, 2004–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kemphues K. J., Priess J. R., Morton D. G., Cheng N. S. (1988) Cell 52, 311–320 [DOI] [PubMed] [Google Scholar]
- 28.Watts J. L., Etemad-Moghadam B., Guo S., Boyd L., Draper B. W., Mello C. C., Priess J. R., Kemphues K. J. (1996) Development. 122, 3133–3140 [DOI] [PubMed] [Google Scholar]
- 29.Suzuki A., Hirata M., Kamimura K., Maniwa R., Yamanaka T., Mizuno K., Kishikawa M., Hirose H., Amano Y., Izumi N., Miwa Y., Ohno S. (2004) Curr. Biol. 14, 1425–1435 [DOI] [PubMed] [Google Scholar]
- 30.Macara I. G. (2004) Nat. Rev. Mol. Cell Biol. 5, 220–231 [DOI] [PubMed] [Google Scholar]
- 31.Hurov J. B., Watkins J. L., Piwnica-Worms H. (2004) Curr. Biol. 14, 736–741 [DOI] [PubMed] [Google Scholar]
- 32.Drewes G., Ebneth A., Preuss U., Mandelkow E. M., Mandelkow E. (1997) Cell 89, 297–308 [DOI] [PubMed] [Google Scholar]
- 33.Ebneth A., Drewes G., Mandelkow E. M., Mandelkow E. (1999) Cell Motil. Cytoskeleton 44, 209–224 [DOI] [PubMed] [Google Scholar]
- 34.Vandré D. D., Centonze V. E., Peloquin J., Tombes R. M., Borisy G. G. (1991) J. Cell Sci. 98, 577–588 [DOI] [PubMed] [Google Scholar]
- 35.Preuss U., Doring F., Illenberger S., Mandelkow E. M. (1995) Mol. Biol. Cell 6, 1397–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxton W. M., Stemple D. L., Leslie R. J., Salmon E. D., Zavortink M., Mclntosh J. R. (1984) J. Cell Biol. 99, 2175–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murata-Kamiya N., Kurashima Y., Teishikata Y., Yamahashi Y., Saito Y., Higashi H., Aburatani H., Akiyama T., Peek R. M., Jr., Azuma T., Hatakeyama M. (2007) Oncogene 26, 4617–4626 [DOI] [PubMed] [Google Scholar]
- 38.Izumi M., Miyazawa H., Kamakura T., Yamaguchi I., Endo T., Hanaoka F. (1991) Exp. Cell Res. 197, 229–233 [DOI] [PubMed] [Google Scholar]
- 39.Kanda T., Sullivan K. F., Wahl G. M. (1998) Curr. Biol. 8, 377–385 [DOI] [PubMed] [Google Scholar]
- 40.Storchova Z., Pellman D. (2004) Nat. Rev. Mol. Cell Biol. 5, 45–54 [DOI] [PubMed] [Google Scholar]
- 41.Kops G. J., Weaver B. A., Cleveland D. W. (2005) Nat. Rev. Cancer 5, 773–785 [DOI] [PubMed] [Google Scholar]
- 42.Musacchio A., Salmon E. D. (2007) Nat. Rev. Mol. Cell Biol. 8, 379–393 [DOI] [PubMed] [Google Scholar]
- 43.Théry M., Racine V., Pépin A., Piel M., Chen Y., Sibarita J. B., Bornens M. (2005) Nat. Cell Biol. 7, 947–953 [DOI] [PubMed] [Google Scholar]
- 44.Toyoshima F., Nishida E. (2007) EMBO J. 26, 1487–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varmark H. (2004) J. Cell Biochem. 91, 904–914 [DOI] [PubMed] [Google Scholar]
- 46.Toyoshima F., Matsumura S., Morimoto H., Mitsushima M., Nishida E. (2007) Dev. Cell 13, 796–811 [DOI] [PubMed] [Google Scholar]
- 47.Wu G., Lin Y. T., Wei R., Chen Y., Shan Z., Lee W. H. (2008) Mol. Cell Biol. 28, 3652–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weaver B. A., Silk A. D., Montagna C., Verdier-Pinard P., Cleveland D. W. (2007) Cancer Cell 11, 25–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.