

Abstract
Using an in situ crosslinkable hydrogel that mimics the extracellular matrix (ECM), cancer cells were encapsulated and injected in vivo following a “tumor engineering” strategy for orthotopic xenografts. Specifically, we created several three-dimensional (3-D) human tumor xenografts and evaluated the tumor response to BrP-LPA, a novel dual function LPA antagonist/ATX inhibitor (LPAa/ATXi). First, we describe the model system and the optimization of semi-synthetic ECM (sECM) compositions and injection parameters for engineered xenografts. Second, we summarize a study to compare angiogenesis inhibition in vivo, comparing BrP-LPA to the kinase inhibitor sunitinib maleate (Sutent). Third, we compare treatment of engineered breast tumors with LPAa/ATXi alone with treatment with Taxol. Fourth, using a re-optimized sECM for non-small cell lung cancer cells, we created reproducibly sized subcutaneous lung tumors and evaluated their response to treatment with LPAa/ATXi. Fifth, we summarize the data on the use of LPAa/ATXi to treat a model for colon cancer metastasis to the liver. Taken together, these improved, more realistic xenografts show considerable utility for evaluating the potential of novel anti-metastatic, anti-proliferative, and anti-angiogenic compounds that modify signal transduction through the LPA signaling pathway.
Keywords: LPA antagonist, ATX, engineered tumor xenograft, lung cancer, synthetic extracellular matrix, angiogenesis assay, phosphonate analog
1. Introduction
To evaluate the anti-cancer potential of signal transduction modifiers in the LPA signaling pathway, clinically relevant human cancer animal models are needed. These models should be predictive for translation of preclinical results to efficacy in human patients, yet most animal models fail to mimic normal human disease, and poorly predict clinical outcomes (1). An injectable, in situ cross-linkable sECM has been developed to deliver and grow cancer cells in vivo (2). This HA-derived sECM was seeded with breast, colon, and ovarian cancer cells prior to gelation, and then injected subcutaneously into mammary fat pads, subserosally in colons, and intracapsularly in ovaries, respectively. When compared with orthotopic injection of cells in serum-free medium, clear advantages emerged for the engineered tumors, including: (i) reduced variability in tumor formation and tumor size, (ii) improved integration of tumor with the surrounding stromal tissue, (iii) increased vascularization and reduced necrosis within the tumor, and (iv) better general health of animals. In another study, encapsulation of human pancreatic cancer cells within the injectable sECM improved tumor growth rate and metastasis in an orthotopic mouse model (3). Most recently, this model has been used for the in vivo evaluation of novel dual-action LPA receptor antagonist/autotaxin inhibitors (4).
This review has two main themes. First, we describe the development of the orthotopic treatment model for producing tumors suitable for evaluating novel anticancer drugs. This optimization of sECM composition and cell density is tested with Taxol, a known anticancer drug. Second, we focus on our most recent results using the sECM engineered tumor xenograft models for the evaluation of metabolically-stabilized analogs of LPA for prevention of angiogenesis and for treatment of cancer.
LPA contributes to tumorigenesis and metastasis, and modulation of LPA signaling is a potential target for developing new anti-cancer therapies (5, 6). The principal biosynthetic source of LPA is the lysophospholipase D activity of autotaxin (ATX) on lysophosphatidylcholine. ATX, which has been reviewed elsewhere (7, 8), is one of the forty most upregulated genes in invasive cancer cells, and is widely implicated in tumor progression, invasion, and metastasis (9, 10).
Expression of LPA GPCRs occurs at different levels in cancer cells from different tissues. LPA1 is the most widely expressed with high mRNA levels in almost all cancer cells (11), while LPA4 is expressed at very low levels in most human cancers. Both LPA2 and LPA3 are aberrantly expressed in cancer cells, particular in ovarian cancer cells, indicating a potential role in the pathophysiology of cancer (12). The recently identified LPA6/GRP87 was significantly overexpressed in squamous cell carcinoma, suggesting it as a possible therapeutic target (13).
An ideal anticancer drug targeting the LPA signaling pathway would simultaneously inhibit signaling through LPA receptors and block LPA production by ATX (4, 7). To address this need, we have synthesized numerous metabolically-stabilized analogs of LPA to identify compounds with (i) longer biological half-lives, (ii) agonist or antagonist activity towards specific LPA GPCRs, and (iii) inhibition of ATX (14). Among these analogs, the α-substituted methylene phosphonate analogs emerged as some of the most interesting (15). In particular, the palmitoyl α-bromomethylene phosphonate BrP-LPA (Figure 1) was selected for further study because of its pan-antagonist activity towards LPA1–4 GPCRs (15). In addition, BrP-LPA showed over 98% inhibition against ATX at 10 µM. With pan-antagonist activity as well as ATX inhibition, the dual function LPA antagonist/ATX inhibitor (LPAa/ATXi) BrP-LPA was examined in a variety of anti-cancer assays in vitro and in vivo.
Figure 1.
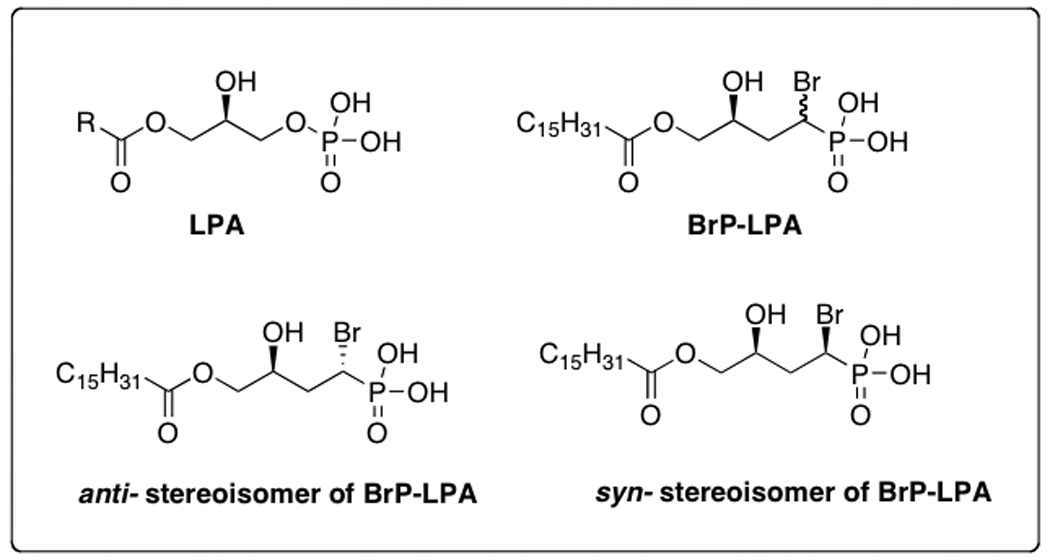
Structures of LPA and bromophosphonate analogs
2. Engineered Tumors in Semi-synthetic ECMs
The most commonly used approach to human tumor xenografts in nude mice employ the subcutaneous or intraperitoneal injection of cancer cells in buffer solution, in serum-free medium, or in Matrigel (16), a tumor-derived basement membrane extract. Matrigel is a murine sarcoma extract that contains proteins, glycoproteins, and growth factors (17). Its high cost, highly variable composition, and need to manipulate at 4 °C make it suboptimal for large-scale use. A room-temperature-injectable 3-D hydrogel vehicle for cell delivery, with a user-controlled composition, that would support the formation of robust, vascularized, orthotopic human cancer tissue in vivo would offer a considerable improvement (2, 18). We turned to the field of tissue engineering to develop an improved method for engineering tumors for drug evaluation.
Hyaluronan (HA) is an immunoneutral, non-sulfated glycosaminoglycan (GAG) that is ubiquitous in all tissues as a major constituent of the ECM and has been modified in many ways for tissue engineering (19). To reconstruct an equivalent to the ECM from its simplest components, we developed a covalently cross-linked synthetic ECM (sECM) consisting of a thiol-modified form of HA plus a thiol-modified gelatin (20, 21). Cells can be added prior to crosslinking to produce biocompatible, three-dimensionally encapsulated cells in an injectable format. The selection of the thiol-modified carboxymethylated HA provided further stabilization of the hydrogel towards hyaluronidases, an increase in cross-linking sites, and improved biocompatibility in vivo.
This sECM, now commercially available as Extracel, was developed for engineering of healthy new tissues and for exploring the biology of cells in 3-D. In order to optimize this hydrogel for cancer xenografts, we explored several parameters that would be expected to influence tumor growth, integration with the stromal tissue, and vascularization – all key attributes of making more “patient-like” tumors. Specifically, we explored (a) the composition of the hydrogel, (b) the concentration of cells injected, (c) the dilution of the hydrogel with cell-culture medium, (d) and the use of different cell lines.
The compliance, or stiffness, of the sECM plays an important role for cell morphology and biochemical function (22). Using the standard Extracel sECM as the injectable in situ crosslinkable hydrogels, dilution with cell culture medium reduced stiffness of the hydrogel. Other methods of reducing the sECM compliance include increasing gelatin content or decreasing crosslinking (23). Consistent with our expectation based on the increased stiffness of tumor tissues relative to surrounding stroma, we found that dilution with cell culture medium decreased the growth rate of breast cancer tumors in vivo from sECMs at a constant cell density of MDA-MB-231 cells (Figure 2).
Figure 2.
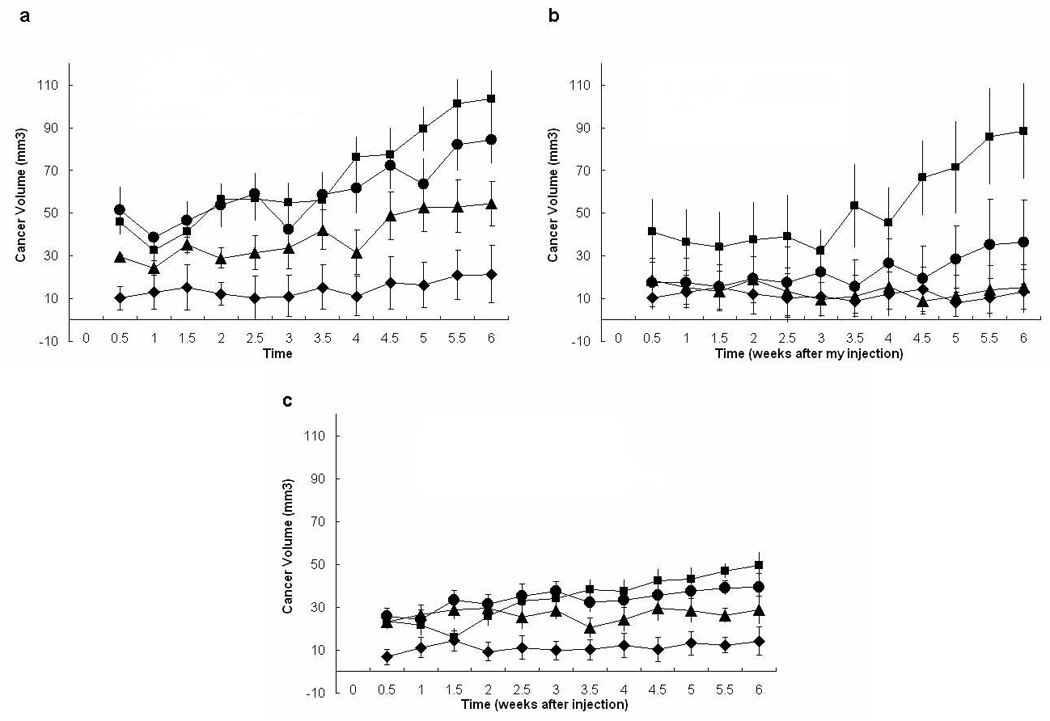
Volumes of sECM-encapsulated cells measured for different starting dilutions and cell densities. Volumes were measured twice per week for 6 weeks after injection of 100 µL of MB-231 cells at different concentrations of 50 × 106 cells/mL (panel a), 10 × 106 cells/mL (panel b), and 2 × 106 cells/mL (panel c). Key: cells were suspended in Extracel (square), Extracel:medium = 3:1 (circle), Extracel:medium = 1:1 (triangle), or medium alone (diamond).
Cell concentration may also affect cell viability in tumor xenograft models. When different cell concentrations were tested, injection of MDA-MB-231 cells at 2 × 106 cells/mL had poorer tumor growth incidence than at 10 × 106 cells/mL and 50 × 106 cells/mL. The apparent tumor volumes began to significantly increase after 2 weeks post-injection; during the first two weeks, cells expand within and degrade the injected bolus of sECM (Figure 2).
We next validated the use of the sECM tumor xenograft model for evaluation of Taxol® (Paclitaxel), a known therapeutic agent for breast cancer for which treatment protocols in xenografts were known (24). Breast cancer MDA-MB-231 cells at a concentration of 50 × 106 cells/mL suspended in the Extracel were injected into the mammary fat pad, and treatment with Taxol® (10 mg/kg) started when tumor volumes reached 200 mm3 (at 2 weeks) (4). After completion of the 2-week treatment, tumor volumes in the treatment group were significantly decreased and a high percentage were no longer visible for measurement. H&E staining of the tumor tissue revealed an irregular arrangement of tumor cells with much larger nuclei and widely varied shapes in the control group (Figure 3a). In addition, cancer cells had invaded the surrounding muscle tissue (Figure 3b). SEM imaging revealed a densely packed layer loaded with cells and extracellular matrix, and this network may facilitate tumor differentiation, proliferation, and neovascularization in vivo (Figure 3c).
Figure 3.
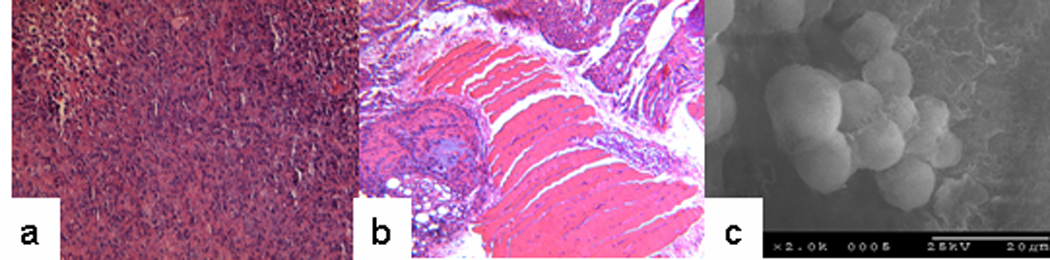
H&E staining of the tissue excised from the breast cancer tumor. Cell shapes vary widely within the tumor (panel a), and cancer cells invaded the surrounding muscle tissue (panel b). SEM microphotograph of MB-231 spheroids showing an ECM network that may facilitate tumor neovascularization (panel c).
3. Angiogenesis Plug Assay for a Dual Function LPAa/ATXi
New blood vessel formation, or neovascularization, promotes the healing of injured tissues but also stimulates tumor growth and inflammatory disease (25, 26). Most tumors cannot grow beyond 2 to 3 mm diameter without the new blood vessels ensuring an adequate supply of oxygen and nutrients (27). Tumor angiogenesis is regulated by both endothelial and non-endothelial cells present within the tumor compartment (28–30). Indeed, with cancer as with many other diseases, angiogenesis constitutes a key organizing principle for drug discovery (31).
The growth and metastasis of most tumors is angiogenesis dependent (32, 33), with a balance between pro-angiogenic molecules and anti-angiogenic molecules released by both tumor cells and the surrounding normal cells (26). Angiogenesis is an essential process for tumor survival and metastasis (34), and a growing number of anti-angiogenic therapies are in development or in the clinic for cancer therapy (35–38). There are currently four main pathways to target angiogenesis: (i) the VEGF/VEGFR signaling pathway (39); (ii) integrin receptors or extracellular matrix (40); (iii) using endogenous angiogenesis inhibitors (36); and (iv) other growth-factor-mediated pathways, such as cytokines (41).
Currently, the Matrigel plug assay is used for in vivo investigation of angiogenesis mechanism and evaluation of anti-angiogenesis compounds (42). However, the drawbacks of Matrigel mentioned above, compounded by the presence of a full spectrum of ECM proteins and growth factors, makes this an assay that lacks adequate control points. Thus, we sought to use the sECM approach to create an angiogenesis plug assay using fully a chemically-defined hydrogel to evaluate angiogenesis inhibitors in vivo. To establish the sECM angiogenesis model we selected a formulation that included with an immobilized thiol-modified heparin to provide controlled release of growth factors (43). In a comparative study (X. Xu, Ph.D. Dissertation, 2008, University of Utah), we injected Extracel-HP subcutaneously in nude mice with or without bFGF and VEGF with growth factor reduced (GFR) Matrigel using the same animal model. The surrounding cells including endothelial cells, which could divide to form a primitive vascular plexus, invaded into the center of the plugs from the margins of the hydrogels.
In the presence of growth factors, cell growth was observed not only in the outer area of the plugs, but also in the inner area of the plugs. Anti-CD31 immunohistochemical staining showed the new blood vessel growth and the blood vessels were calculated within the plugs. With added GFs, Extracel-HP allowed development of as many newly formed blood vessels as Matrigel. Moreover, the dual delivery of VEGF and bFGF led to enhanced neovascularization in vivo relative to GFR-Matrigel alone. Previously, dual delivery of VEGF with either bFGF, angiopoetin-1, or KGF from an sECM had been found to enhance microvascular maturity in a murine ear pinnae model (44). In our revised angiogenesis plug assay, host-derived cells invaded the compliant hydrogel, remodeled it, and secreted a new native ECM specific to the tumor angiogenesis environment.
The LPA signaling pathway plays an important role in tumor growth and angiogenesis (4, 45–48). The signaling mechanism of LPA-regulated VEGF expression in endothelial cells has been reported (49), which suggested that LPA could be a suitable target for therapeutics against tumor angiogenesis. Previously, inhibition of LPA receptors has been shown to cause tumor regression and inhibit angiogenesis (4, 50).
The angiogenesis plug assay using Extracel-HP allows the potential angiogenesis inhibitors to be examined by mixing the sECM with the drug candidates. The details of these experiments will be presented in due course, but we summarize the outcomes briefly here. The pro-angiogenic factors (VEGF and bFGF) and an anticoagulant (heparin) were mixed with the sECM hydrogel and implanted s.c. in nude mice the presence of saline (control), BrP-LPA, or the orally-available small-molecule tyrosine kinase inhibitor sunitinib maleate (Sutent)(51). At day 10, plugs were excised, and microvessel density within the plugs was assessed. Implants containing growth factors showed stronger neovascularization. A greater number of endothelial cells were found in the control saline treatment relative to either the BrP-LPA and Sutent treated groups (Figure 4). In addition, the hemoglobin content of the BrP-LPA and Sutent-containing plugs was decreased over 3-fold relative to the control group (data not shown). Taken together, these studies validate the use of a cell-free sECM to evaluate the anti-angiogenic properties of inhibitors of the LPA signaling pathway, and demonstrate that a dual function LPAa/ATXi has significant potential as an anti-angiogenic agent.
Figure 4.
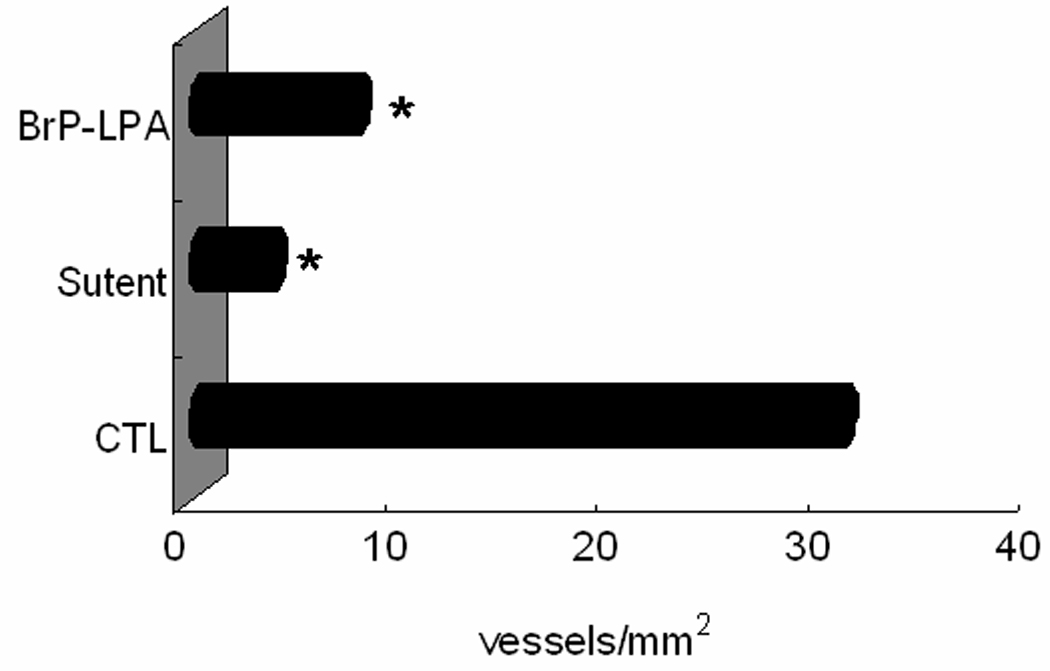
In vivo angiogenesis plug assay in Extracel-HP. Blood vessel density is significantly decreased when 15 mg/kg of either BrP-LPA or sunitinib maleate (Sutent) was added to the plug prior to s.c. injection (p < 0.05).
4. Engineered Breast Tumors Treated with a Dual Function LPAa/ATXi
The first in vivo test of the dual activity agent BrP-LPA was conducted using orthotopic breast tumors in nude mice (4). Subcutaneous injection of MDA-MB-231 cells suspended in the sECM Extracel into the mammary fat pads of nu/nu mice resulted in tumor growth at each site of injection. After 2 weeks of tumor growth, the control group was treated with four intraperitoneal (ip) injections of physiological saline over the course of 2 weeks. Two groups received ip BrP-LPA (as the mixed diastereoisomers, 10 mg/kg) or Taxol alone (10 mg/kg). A third group received a simulated combination drug therapy by giving Taxol for the first week followed by BrP-LPA for the second week. In each treatment group, a reduction of tumor size was observed beginning after the first injection, when compared to the control group. After completion of the 2-week treatment course, tumors in the treatment group were significantly decreased or undetectable (4).
Figure 5a shows the reduction of tumor size with BrP-LPA alone as the therapeutic agent. At necropsy, the largest tumor sample from the BrP-LPA treatment was significantly smaller than the smallest tumor tissue in the control group. Moreover, quantification of the newly formed blood vessels in the tumor samples showed a highly significant reduction of angiogenesis in the mice treated with BrP-LPA relative to either controls or Taxol treatments (p < 0.01) (4).
Figure 5.
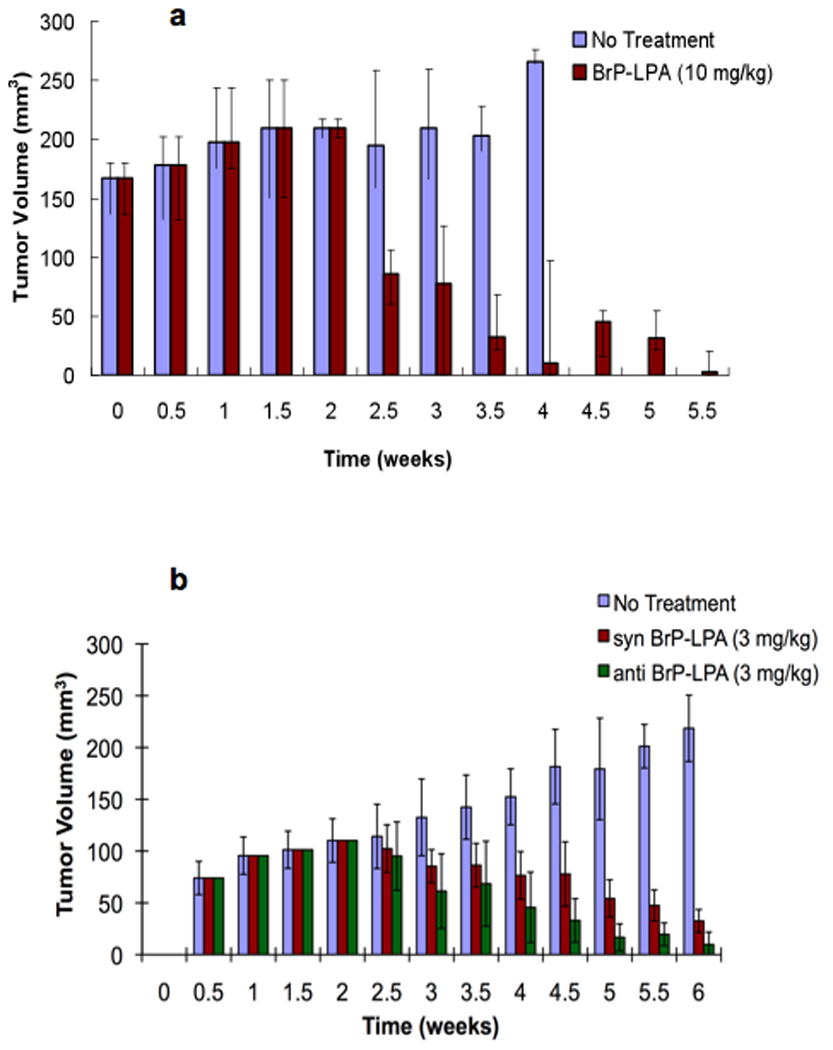
Engineered breast tumor xenograft treatment model. MB-231 cells (50 × 106 cells/mL) were suspended in Extracel and injected subcutaneously into mammary fat pad of nude mice (100 or 200 µL). BrP-LPA (10 mg/kg) was administered in four semi-weekly intraperitoneal injections beginning at week 3 (panel a) to mice with the higher cell loading (200 µL). At 5.5 weeks, tumor volumes in treated animals were significantly different from untreated controls (p < 0.05). The separate syn and anti-diastereoisomers of BrP-LPA (3 mg/kg) were administered similarly to mice with the lower tumor cell loading (200 µL) (panel b). At 6 weeks, tumor volumes in both syn- and anti-diastereomer treated animals were significantly different from untreated controls (p < 0.05) but not from each other (p < 0.1).
The pharmacological activities of the individual diastereoisomers of BrP-LPA (see Figure 1) against LPA1–5, inhibition of the lysophospholipase D activity of ATX, inhibition of cell migration, and inhibition of cell invasion were also examined (4). Subtle isoform-selective differences in receptor antagonism were evident for the separate diastereomers. For example, the anti-stereoisomer was 8-fold more potent as an ATX inhibitor, and slightly more potent in inhibiting MB-231 cell migration in a scratch wound assay. The stereoisomers were equipotent in inhibiting invasion. Finally, the orthotopic xenograft model was repeated using a lower treatment dosage of 3 mg/kg and a lower injection volume of cells. As shown in Figure 5b and 5c, both diastereoisomers significantly decreased the tumor volume. With this sample size, we only observed a trend suggesting that the anti isomer might be more potent than syn isomer (p < 0.1).
5. Treatment of Engineered Lung Carcinoma with a Dual Function LPAa/ATXi
Most lung cancer patients have non-small cell lung cancer (NSCLC), and chemotherapy is the primary first-line treatment option for the advanced or metastatic cancer patients (52). New anti-angiogenic and anti-metastatic compounds are under development, and to expedite the evaluation of these treatment strategies, it is necessary to have a suitable and reproducible animal model of lung cancer. However, human lung cancer cells do not grow readily in athymic mice, even when a large number of cells (53, 54) and/or immunosuppressive agents are used to obtain tumors (54).
We recently described an engineered lung cancer xenograft model using human A549 NSCLC cells encapsulated in sECM, and the evaluation of BrP-LPA in this model (55). We explored several customized compositions of sECM for in vivo xenografts of the A-549 cells. Since an intratumoral vascular network is important in pulmonary malignancies (56, 57), we used our angiogenesis plug strategy, selecting Extracel-HP as the sECM for retention of growth factors (18). Extracel-HP, which contains an immobilized form of heparin (43), mimics the heparin sulfate proteoglycans normally present in the ECM to help protect growth factors (GFs) from proteolysis and to slow their release to attached cells. In addition, we explored a higher stiffness 2% (w/v) sECM hydrogel, Extracel-XX, in recognition of the preference of epithelial like cells for stiffer substrata and a more stretched actin cytoskeleton.
Subcutaneous injection of A-549 cells suspended in Extracel-HP, Extracel-HP plus growth factors and a laminin peptide, Extracel-XX or PBS in nu/nu mice resulted in tumor growth at all sites of injection. The rank order of tumor growth rates was Extracel-HP plus growth factors and laminin ≈ Extracel-HP alone > Extracel-XX > buffer alone (55). H&E staining and CD31 immunohistochemistry CD31 showed microvascular patterns typical of NSCLC. That the addition of growth factors and laminin did not significantly improve tumor growth and vascularization in the sECM was unexpected, and we speculated that this was due to the ability of the Extracel-HP to retain growth factors secreted by the A549 cells. In any case, the ability to use a simpler, less expensive model was viewed as highly desirable in going forward with testing the activity of BrP-LPA against lung cancer.
Recently, it has been found that LPA reduced the cellular abundance of the tumor suppressor p53 in A549 lung carcinoma cells, which express endogenous LPA receptors (58). This led us to explore the BrP-LPA as a potential therapy agent in our engineered lung cancer treatment model (55). We compared encapsulation of A-549 cells in Matrigel with encapsulation in the chemically-defined Extracel-HP. As illustrated in Figure 6, tumors in both untreated control groups grew as large as 1000 mm3 after 7 weeks, with a somewhat higher growth rate in Matrigel, a tumor-derived ECM rich in growth factors and other components which provide cancer cells with a naturally favorable mixture environment. After 2 weeks of tumor growth, the treatment groups in Matrigel or in Extracel-HP received intraperitoneal injections of the anti-stereoisomer of BrP-LPA (3 mg/kg), twice per week for 2 weeks. The dosage of BrP-LPA did not reduce the body weight significantly indicating that the dual activity agent was well tolerated (55).
Figure 6.
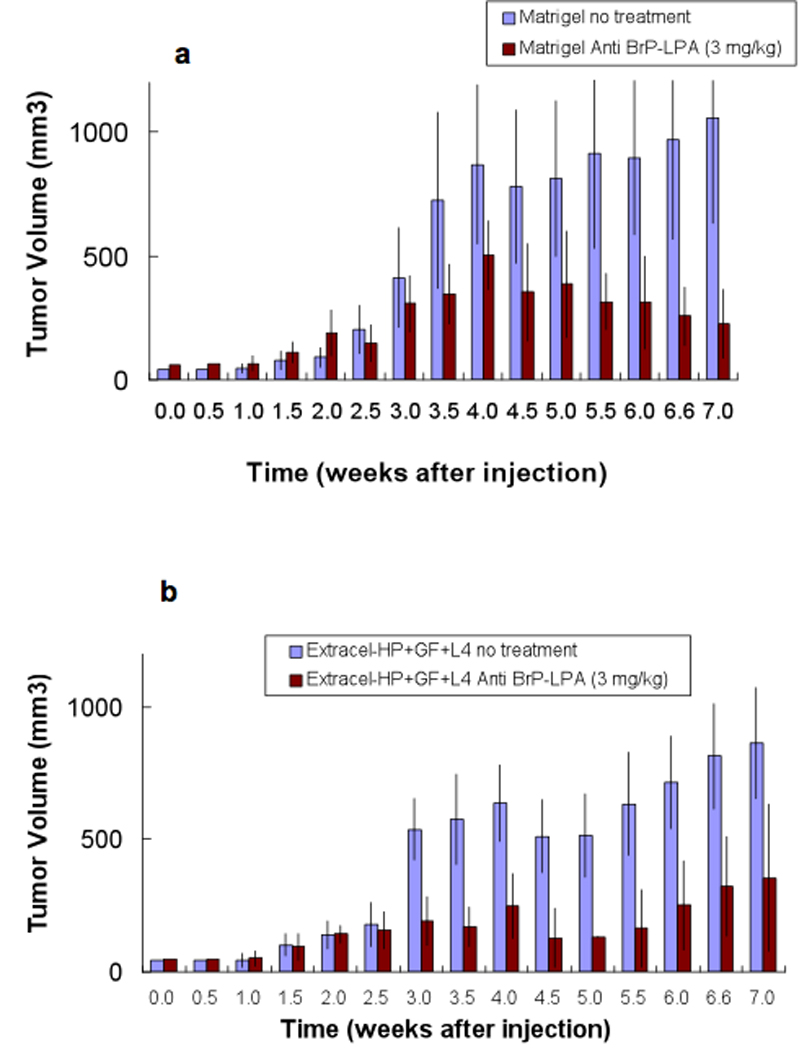
Engineered NSCLC lung xenograft model. A-549 cells (50 × 106 cells/mL) were suspended in Matrigel (panel a) or in Extracel-HP (panel b) modified with a laminin peptide (L-4) and injected subcutaneously into the backs of nude mice (200 µL). The anti-diastereoisomer of BrP-LPA (3 mg/kg) was administered in four semi-weekly intraperitoneal injections beginning at week 3 to both groups of mice. For all time points at 3.5 weeks and continuingly through to 7 weeks, tumor volumes in all treated animals were significantly different from untreated controls (p < 0.05).
After completion of the 2-week treatment course, tumors in both BrP-LPA treatment groups were significantly smaller than the tumor tissue in the control group (Figure 6)(55). Moreover, the surface of the untreated tumor was significantly rougher, suggesting that BrP-LPA reduced the metastatic potential of the rapidly growing tumors. An endothelial layer covering tumor vasculature was observed using CD31 immunohistochemical staining and the quantification in six different fields of three slides for each treatment group showed highly significant reduction of angiogenesis (p < 0.05). Taken together, the sECM model demonstrated that Extracel-HP provides a useful new material for A549 xenografts, and that a dual activity LPAa/ATXi such as BrP-LPA may contribute a new arrow to the quiver of anti-cancer agents for NSCLC (55).
6. Treatment of Engineered Hepatic Colon Tumor with Dual Function LPAa/ATXi
Colon cancer is the third most common form of cancer and the second leading cause of cancer-related death in the western world (59). Treatment of primary colon cancer with surgical resection, combined with chemotherapy or radiation therapy, can be curative for some patients. However, nearly half of patients will develop liver metastases during the course of their disease. As with many types of cancer, death from colon cancer is often a result of metastatic disease (60, 61). Approximately 60% of patients with colorectal cancer develop hepatic metastases (62, 63), and few are acceptable candidates for surgical resection. For the vast majority of patients with colorectal cancer liver metastases, additional chemotherapy is often the only option (64).
LPA signaling is important in colon cancer: signaling through both LPA2, and LPA3 promotes the proliferation in HCT-116 cells via inactivation of GSK-3β and nuclear translocation of β-catenin (65). LPA2 is the major LPA receptor that mediates mitogenic signals and cytokine induction in colonic epithelial cells. In addition, LPA has been found to be over-expressed in the SW 480 and HCT-116 colon cancer cell lines (66).
In order to evaluate the effects BrP-LPA on tumor growth of human colorectal cancer on the liver, HCT-116 colon cancer cells were encapsulated in Extracel, and the cell suspension was directly injected into the livers of eight nude mice (2, 3). The full details of this study will be presented in due course (G. Yang, H. Zhang, and G. D. Prestwich, unpublished results). Herein we summarize the model and outcomes to illustrate the generality of the use of BrP-LPA for tumor regression in engineered tumor models. Based on the dosage required to achieve reduction of tumor size and vascularity by BrP-LPA (mixed diastereomers) in an orthotopic breast cancer model (4), we selected a two-week treatment course of 10 mg/kg, injected i.p. twice per week starting after one week of tumor growth.
All mice in both groups developed liver tumors. The HCT-116 cells encapsulated in Extracel grew rapidly and formed extensive tumor masses attached to the liver in the untreated group. In contrast, the tumor masses in the BrP-LPA treatment group were significantly smaller than those of the controls (14). The BrP-LPA treated group showed a marked reduction of hepatic tumor burden (p < 0.05) and tumor volume (p < 0.05). The histopathological analysis revealed a distinct interface between hepatic colon cancer and mouse native liver.
7. Concluding remarks
Current cancer animal models have low rates of tumor growth and metastasis (67). We have demonstrated herein engineered tumors in breast cancer, non-small cell lung cancer, and a simulated hepatic colon cancer metastasis models. Well isolated, readily measurable, and uniform-sized tumors are produced with the ability to monitor tumor growth in vivo. We found that the use of Extracel-HP for certain cell types greatly improved tumor growth and metastasis.
We further studied the anticancer and anti-angiogenesis efficacy of a dual LPAa/ATXi compound, BrP-LPA, using these cancer xenograft models. Orthotopic engineered tumors were established in the mammary fat pads of nude mice by injection of MDA-MB-231 breast cancer cells, in the livers using encapsulated HCT-116 colon cancer cells, and subcutaneously using A549 lung cancer cells in customized sECM (Table 1). In each case, mice treated with BrP-LPA all showed significantly reduced tumor burdens, and LPAa/ATXi treatment was superior to Taxol treatment in reducing blood vessel density in tumor tissue. Moreover, the chemically-defined sECM offers a “tabula rasa” hydrogel, lacking the inherent mixture of growth factors present in Matrigel. Similarly, a cell-free sECM was used to validated as an angiogenesis plug assay; a proof-of-concept drug evaluation using this new model showed that Br-LPA has acceptable toxicity profiles (68, 69) and potentially enhanced anticancer activity.
Table 1.
Summary of validated engineered tumor xenograft models
| Xenograft Type or Model |
Cancer Type | Cell lines | sECM Used |
|---|---|---|---|
| Orthotopic | Breast cancer | MCF7, MDA-MB-231, MDA-MB-468 |
Extracel |
| Colorectal cancer | HCT-116, CaCo-2 |
Extracel | |
| Pancreatic cancer | MiaPaCa-2 | Extracel | |
| Ovarian cancer | SK-OV-3, OVCAR-3 |
Extracel | |
| Subcutaneous | NSCLC lung cancer | A-549 | Extracel-HP |
| Angiogenesis Plug | Extracel-HP, Heprasil |
||
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006;66:3351–3354. doi: 10.1158/0008-5472.CAN-05-3627. discussion 3354. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Shu XZ, Prestwich GD. Tumor engineering: orthotopic cancer models in mice using cell-loaded, injectable, cross-linked hyaluronan-derived hydrogels. Tissue Eng. 2007;13:1091–1101. doi: 10.1089/ten.2006.0297. [DOI] [PubMed] [Google Scholar]
- 3.Scaife CL, Shea JE, Dai Q, et al. Synthetic extracellular matrix enhances tumor growth and metastasis in an orthotopic mouse model of pancreatic adenocarcinoma. J. Gastrointest. Surg. 2008;12:1074–1080. doi: 10.1007/s11605-007-0425-3. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Xu X, Tsukahara R, et al. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor suppresses breast cancer cell migration and invasion in vitro and causes tumor regression in vivo. Cancer Res. 2009;69:5441–5449. doi: 10.1158/0008-5472.CAN-09-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 6.Umezu-Goto M, Tanyi J, Lahad J, et al. Lysophosphatidic acid production and action: validated targets in cancer? J. Cellular Biochem. 2004;92:1115–1140. doi: 10.1002/jcb.20113. [DOI] [PubMed] [Google Scholar]
- 7.Federico L, Pamuklar Z, Smyth S, Morris A. Therapeutic potential of autotaxin/lysophospholipase D inhibitors. Curr. Drug Targets. 2008;9:698–708. doi: 10.2174/138945008785132439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Meeteren L, Moolenaar W. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Tokumura A. Physiological and pathophysiological roles of lysophosphatidic acids produced by secretory lysophospholipase D in body fluids. Biochim Biophys Acta. 2002;1582:18–25. doi: 10.1016/s1388-1981(02)00133-6. [DOI] [PubMed] [Google Scholar]
- 10.Umezu-Goto M, Kishi Y, Taira A, et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hecht JH, Weiner JA, Post SR, Chun J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulte KM, Beyer A, Kohrer K, Oberhauser S, Roher HD. Lysophosphatidic acid, a novel lipid growth factor for human thyroid cells: over-expression of the high-affinity receptor edg4 in differentiated thyroid cancer. Int J Cancer. 2001;92:249–256. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1166>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 13.Gugger M, White R, Song S, et al. GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis Markers. 2008;24:41–50. doi: 10.1155/2008/857474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prestwich GD, Gajewiak J, Zhang H, Yang G, Serban MA. Phosphatase-resistant analogues of lysophosphatidic acid: Agonists promote healing, antagonists and autotaxin inhibitors treat cancer. Biochim. Biophys. Acta. 2008;1781:588–594. doi: 10.1016/j.bbalip.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang G, Xu Y, Fujiwara Y, et al. Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. ChemMedChem. 2007;2:679–690. doi: 10.1002/cmdc.200600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyrick BJ, Ozawa T, Lamborn KR, Bollen AW, Deen DF. Effects of Matrigel on the SF-767 malignant glioma athymic mouse tumor model. Anticancer Res. 1997;17:2419–2425. [PubMed] [Google Scholar]
- 17.Kleinman HK, McGarvey ML, Hassell JR, et al. Basement membrane complexes with biological activity. Biochemistry. 1986;25:312–318. doi: 10.1021/bi00350a005. [DOI] [PubMed] [Google Scholar]
- 18.Prestwich GD. Evaluating drug efficacy and toxicology in three dimensions: using synthetic extracellular matrices in drug discovery. Acc Chem Res. 2008;41:139–148. doi: 10.1021/ar7000827. [DOI] [PubMed] [Google Scholar]
- 19.Allison DD, Grande-Allen KJ. Review. Hyaluronan: a powerful tissue engineering tool. Tissue Eng. 2006;12:2131–2140. doi: 10.1089/ten.2006.12.2131. [DOI] [PubMed] [Google Scholar]
- 20.Prestwich GD. Simplifying the extracellular matrix for 3-D cell culture and tissue engineering: a pragmatic approach. J Cell Biochem. 2007;101:1370–1383. doi: 10.1002/jcb.21386. [DOI] [PubMed] [Google Scholar]
- 21.Prestwich GD. Engineering a clinically-useful matrix for cell therapy. Organogenesis. 2008;4:42–47. doi: 10.4161/org.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 23.Vanderhooft JL, Acoutlabi M, Magda JJ, Prestwich GD. Rheological properties of crosslinked hyaluronan-gelatin hydrogels for tissue engineering. Macromol. Biosci. 2008;9:20–28. doi: 10.1002/mabi.200800141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sui M, Chen F, Chen Z, Fan W. Glucocorticoids interfere with therapeutic efficacy of paclitaxel against human breast and ovarian xenograft tumors. Int J Cancer. 2006;119:712–717. doi: 10.1002/ijc.21743. [DOI] [PubMed] [Google Scholar]
- 25.Bikfalvi A. Angiogenesis: health and disease. Ann Oncol. 2006;17 Suppl 10:x65–x70. doi: 10.1093/annonc/mdl239. [DOI] [PubMed] [Google Scholar]
- 26.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 27.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 28.Warren BA, Greenblatt M, Kommineni VR. Tumour angiogenesis: ultrastructure of endothelial cells in mitosis. Br J Exp Pathol. 1972;53:216–224. [PMC free article] [PubMed] [Google Scholar]
- 29.Folkman J, Haudenschild C. Angiogenesis by capillary endothelial cells in culture. Trans Ophthalmol Soc U K. 1980;100:346–353. [PubMed] [Google Scholar]
- 30.Munaron L. Intracellular calcium, endothelial cells and angiogenesis. Recent Patents Anticancer Drug Discov. 2006;1:105–119. doi: 10.2174/157489206775246502. [DOI] [PubMed] [Google Scholar]
- 31.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nature Rev. Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 32.Nicosia RF, Tchao R, Leighton J. Angiogenesis-dependent tumor spread in reinforced fibrin clot culture. Cancer Res. 1983;43:2159–2166. [PubMed] [Google Scholar]
- 33.Kim JS, Chang JH, Yu HK, et al. Inhibition of angiogenesis and angiogenesis-dependent tumor growth by the cryptic kringle fragments of human apolipoprotein(a) J Biol Chem. 2003;278:29000–29008. doi: 10.1074/jbc.M301042200. [DOI] [PubMed] [Google Scholar]
- 34.Blood CH, Zetter BR. Tumor interactions with the vasculature: angiogenesis and tumor metastasis. Biochim Biophys Acta. 1990;1032:89–118. doi: 10.1016/0304-419x(90)90014-r. [DOI] [PubMed] [Google Scholar]
- 35.Takano S, Kamiyama H, Tsuboi K, Matsumura A. Angiogenesis and antiangiogenic therapy for malignant gliomas. Brain Tumor Pathol. 2004;21:69–73. doi: 10.1007/BF02484513. [DOI] [PubMed] [Google Scholar]
- 36.Rege TA, Fears CY, Gladson CL. Endogenous inhibitors of angiogenesis in malignant gliomas: nature's antiangiogenic therapy. Neuro Oncol. 2005;7:106–121. doi: 10.1215/S115285170400119X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou S. Combination therapy with bacteria and angiogenesis inhibitors: strangling cancer without mercy. Cancer Biol Ther. 2005;4:846–847. doi: 10.4161/cbt.4.8.2021. [DOI] [PubMed] [Google Scholar]
- 38.Gulati R, Simari RD. Cell therapy for angiogenesis: embracing diversity. Circulation. 2005;112:1522–1524. doi: 10.1161/CIRCULATIONAHA.105.566497. [DOI] [PubMed] [Google Scholar]
- 39.Kamiyama M, Ichikawa Y, Ishikawa T, et al. VEGF receptor antisense therapy inhibits angiogenesis and peritoneal dissemination of human gastric cancer in nude mice. Cancer Gene Ther. 2002;9:197–201. doi: 10.1038/sj.cgt.7700428. [DOI] [PubMed] [Google Scholar]
- 40.Lim EH, Danthi N, Bednarski M, Li KC. A review: Integrin alphavbeta3-targeted molecular imaging and therapy in angiogenesis. Nanomedicine. 2005;1:110–114. doi: 10.1016/j.nano.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Kuhlmann MT, Klocke R, Nikol S. Therapeutic angiogenesis for peripheral artery disease: cytokine therapy. Vasa. 2007;36:253–260. doi: 10.1024/0301-1526.36.4.253. [DOI] [PubMed] [Google Scholar]
- 42.Passaniti A. Extracellular matrix-cell interactions: Matrigel and complex cellular pattern formation. Lab Invest. 1992;67:804. author reply 804–808. [PubMed] [Google Scholar]
- 43.Cai S, Liu Y, Zheng Shu X, Prestwich GD. Injectable glycosaminoglycan hydrogels for controlled release of human basic fibroblast growth factor. Biomaterials. 2005;26:6054–6067. doi: 10.1016/j.biomaterials.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 44.Hosack L, Firpo MA, Scott JA, Prestwich GD, Peattie RA. Microvascular maturity elicited in tissue treated with cytokine-loaded hyaluronan-based hydrogels. Biomaterials. 2008;29:2336–2347. doi: 10.1016/j.biomaterials.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tigyi G, Dyer DL, Miledi R. Lysophosphatidic acid possesses dual action in cell proliferation. Proc Natl Acad Sci U S A. 1994;91:1908–1912. doi: 10.1073/pnas.91.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panetti TS, Nowlen J, Mosher DF. Sphingosine-1-phosphate and lysophosphatidic acid stimulate endothelial cell migration. Arterioscler Thromb Vasc Biol. 2000;20:1013–1019. doi: 10.1161/01.atv.20.4.1013. [DOI] [PubMed] [Google Scholar]
- 47.Piazza GA, Ritter JL, Baracka CA. Lysophosphatidic acid induction of transforming growth factors alpha and beta: modulation of proliferation and differentiation in cultured human keratinocytes and mouse skin. Exp Cell Res. 1995;216:51–64. doi: 10.1006/excr.1995.1007. [DOI] [PubMed] [Google Scholar]
- 48.Goetzl EJ, Kong Y, Mei B. Lysophosphatidic acid and sphingosine 1-phosphate protection of T cells from apoptosis in association with suppression of Bax. J Immunol. 1999;162:2049–2056. [PubMed] [Google Scholar]
- 49.Lin CI, Chen CN, Huang MT, et al. Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation in human endothelial cells through LPA(1/3), COX-2, and NF-kappaB activation- and EGFR transactivation-dependent mechanisms. Cell Signal. 2008;20:1804–1814. doi: 10.1016/j.cellsig.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Rivera-Lopez CM, Tucker AL, Lynch KR. Lysophosphatidic acid (LPA) and angiogenesis. Angiogenesis. 2008;11:301–310. doi: 10.1007/s10456-008-9113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chow L, Eckhardt S. Sunitinib: from rational design to clinical efficacy. J. Clin. Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 52.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 53.McLemore TL, Liu MC, Blacker PC, et al. Novel intrapulmonary model for orthotopic propagation of human lung cancers in athymic nude mice. Cancer Res. 1987;47:5132–5140. [PubMed] [Google Scholar]
- 54.Gazdar AF, Carney DN, Sims HL, Simmons A. Heterotransplantation of small-cell carcinoma of the lung into nude mice: comparison of intracranial and subcutaneous routes. Int J Cancer. 1981;28:777–783. doi: 10.1002/ijc.2910280617. [DOI] [PubMed] [Google Scholar]
- 55.Xu X, Prestwich GD. Inhibition of tumor growth and angiogenesis by a lysophosphatidic acid antagonist in a engineered three-dimensional lung cancer xenograft model. Cancer: in press. 2009 doi: 10.1002/cncr.24907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goudar RK, Vlahovic G. Hypoxia, angiogenesis, and lung cancer. Curr Oncol Rep. 2008;10:277–282. doi: 10.1007/s11912-008-0043-6. [DOI] [PubMed] [Google Scholar]
- 57.Sasaki T, Tanno S, Shibukawa K, et al. Administration of VEGF receptor tyrosine kinase inhibitor increases VEGF production causing angiogenesis in human small-cell lung cancer xenografts. Int J Oncol. 2008;33:525–532. [PubMed] [Google Scholar]
- 58.Murph MM, Hurst-Kennedy J, Newton V, Brindley DN, Radhakrishna H. Lysophosphatidic acid decreases the nuclear localization and cellular abundance of the p53 tumor suppressor in A549 lung carcinoma cells. Mol Cancer Res. 2007;5:1201–1211. doi: 10.1158/1541-7786.MCR-06-0338. [DOI] [PubMed] [Google Scholar]
- 59.Espey DK, Wu XC, Swan J, et al. Annual report to the nation on the status of cancer, 1975–2004, featuring cancer in American Indians and Alaska Natives. Cancer. 2007;110:2119–2152. doi: 10.1002/cncr.23044. [DOI] [PubMed] [Google Scholar]
- 60.Ballantyne GH, Quin J. Surgical treatment of liver metastases in patients with colorectal cancer. Cancer. 1993;71:4252–4266. doi: 10.1002/1097-0142(19930615)71:12+<4252::aid-cncr2820711815>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 61.Wolpin BM, Meyerhardt JA, Mamon HJ, Mayer RJ. Adjuvant treatment of colorectal cancer. CA Cancer J Clin. 2007;57:168–185. doi: 10.3322/canjclin.57.3.168. [DOI] [PubMed] [Google Scholar]
- 62.Russell AH, Tong D, Dawson LE, Wisbeck W. Adenocarcinoma of the proximal colon. Sites of initial dissemination and patterns of recurrence following surgery alone. Cancer. 1984;53:360–367. doi: 10.1002/1097-0142(19840115)53:2<360::aid-cncr2820530232>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 63.Dizon DS, Kemeny NE. Intrahepatic arterial infusion of chemotherapy: clinical results. Semin Oncol. 2002;29:126–135. doi: 10.1053/sonc.2002.31680. [DOI] [PubMed] [Google Scholar]
- 64.Silen W. Hepatic resection for metastases from colorectal carcinoma is of dubious value. Arch. Surg. 1989;124:1021–1022. doi: 10.1001/archsurg.1989.01410090027004. [DOI] [PubMed] [Google Scholar]
- 65.Yang M, Zhong W, Srivastava N, et al. G-protein coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the beta-catenin pathway. Proc. Natl. Acad. Sci. USA. 2005;102:6027–6032. doi: 10.1073/pnas.0501535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yun CC, Sun H, Wang D, et al. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol Cell Physiol. 2005;289:C2–C11. doi: 10.1152/ajpcell.00610.2004. [DOI] [PubMed] [Google Scholar]
- 67.Jackson T, Chougule MB, Ichite N, Patlolla RR, Singh M. Antitumor activity of noscapine in human non-small cell lung cancer xenograft model. Cancer Chemother Pharmacol. 2008;63:117–126. doi: 10.1007/s00280-008-0720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun L, Liang C, Shirazian S, et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid(2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46:1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- 69.Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]