

Abstract
We report a strategy to create photodegradable poly(ethylene glycol)-based (PEG) hydrogels through rapid polymerization of cytocompatible macromers for remote manipulation of gel properties in situ. Post-gelation control of the gel properties is demonstrated to introduce temporal changes, creation of arbitrarily shaped features, and on-demand pendant functionality release. Channels photodegraded within a hydrogel containing encapsulated cells allow cell migration. Temporal variation of the biochemical gel composition is utilized to influence chondrogenic differentiation of encapsulated stem cells. Photodegradable gels that allow real-time manipulation of material properties or chemistry provide dynamic environments with the scope to answer fundamental questions about material regulation of live cell function and may impact an array of applications from design of drug delivery vehicles to tissue engineering systems.
Hydrogels are hydrophilic polymers swollen by water that are insoluble owing to physical or chemical crosslinks. These water-swollen gels are used extensively as biomaterials for complex device fabrication (1), cell culture for tissue regeneration (2), and targeted drug release (3). Often, sophisticated control of the gel structure in space and time is required to elucidate the dynamic relationship between biomaterial properties and their influence on biological function (4, 5). For example, progenitor cells are often expanded and differentiated in hydrogel microenvironments, and researchers have demonstrated how the initial gel properties, including mechanics (6, 7) and chemical functionality (8), influence cellular fate. In regenerative medicine, the structure and composition of gels are also regulated temporally, through hydrolytic (9) and enzymatic (10-12) degradation mechanisms, to promote cell secretory properties and encourage the development of tissue-like structures in vitro and in vivo. A major challenge is determining which biochemical and biophysical features must be presented in a gel culture environment.
Hydrogel structure and functionality have evolved from the direct encapsulation of cells in simple homogeneous materials to those with highly regulated structures spanning multiple size scales (e.g., through self-assembly (13) or microengineering (14)). These hydrogel structures are further modified locally by cells with the synthetic incorporation of bioresponsive functionalities (15) or externally by advanced patterning to create spatially varying functionalities. For example, the chemical patterning of a gel by the addition of a second, interpenetrating network or peptide tether has been demonstrated by diffusing chemical moieties into a gel and covalently linking these functionalities to the network by photocoupling (16) or reaction with a photolytically uncaged reactive group (17). While these are important advances, such processes do not allow modulation of the gel chemistry in real-time or photodegradation of the gel structure. Few synthetic materials provide a cellular microenvironment in which physical or chemical cues are initially present and subsequently regulated on-demand. We have synthesized monomers capable of polymerizing in the presence of cells to produce photolytically degradable hydrogels whose physical or chemical properties are tunable temporally and spatially with light. The desired gel property for altering cell function or fabricating a device is thus externally-triggered and directed with irradiation by photolytic cleavage and removal of the macromolecules that comprise the gel.
The photodegradable functionality, a nitrobenzyl ether-derived moiety, was selected based on its photolytic efficiency, susceptibility to two-photon photolysis, and previous use in live cell culture and imaging (18, 19). Similar photolabile functionalities have been used in multi-step gel modification processes (20). A base photodegradable monomer was synthesized by acrylation of the photolabile moiety via a pendant hydroxyl group (photodegradable acrylate, PDA, Fig. 1A) and was subsequently attached via a pendant carboxylic acid to PEG-bis-amine to create a photocleavable crosslinking diacrylate macromer (Compound 1, Fig. 1B) from which PEG-based photodegradable hydrogels (Fig. 1C) were synthesized. This approach, where the photolabile moiety is incorporated within the network backbone, is fundamentally different from strategies where reactive groups or biomolecules are asymmetrically caged or capped by a photolabile group (20). Here, the hydrogel itself is cleaved upon light exposure, decreasing the local network crosslink density and resulting in macroscopic property changes such as stiffness, water content, diffusivity, or complete erosion, all in the presence of cells.
Figure 1. Photodegradable hydrogel synthesis and degradation for tuning gel properties.
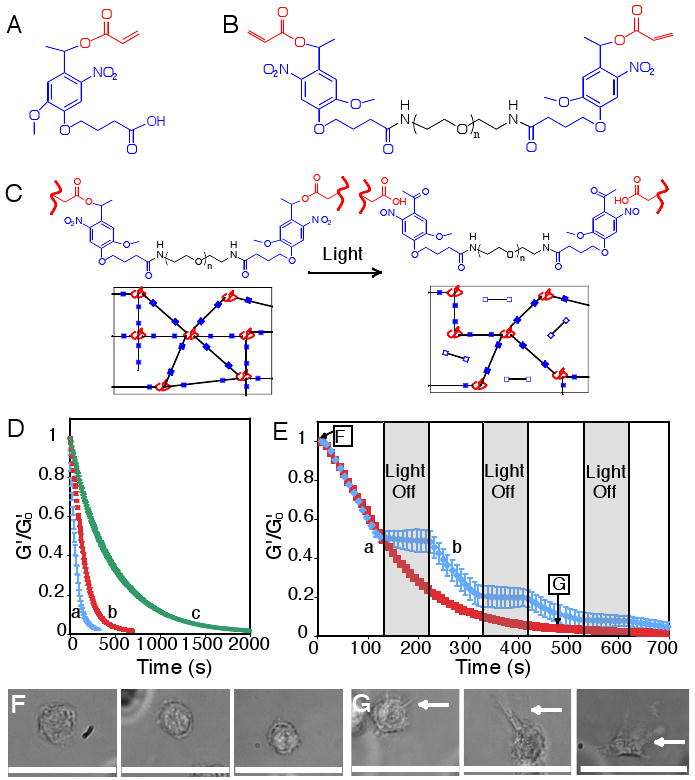
(A) The base photodegradable acrylic monomer was used to synthesize (B) the photodegradable crosslinking macromer (Compound 1, Mn ∼ 4070 g/mol), comprised of PEG (black), photolabile moieties (blue), and acrylic end groups (red). (C) Compound 1 was copolymerized with PEGA (Mn ∼ 375 g/mol), creating gels composed of poly(acrylate) chains (red coils) connected by PEG (black lines) with photolabile groups (solid blue boxes) (left). Upon irradiation, the photolabile moiety cleaves (open blue boxes), decreasing ρx and releasing modified PEG (right). (D) The physical structure of the hydrogel is degraded via photolysis, decreasing ρx and G′. The influence of irradiation on G′, normalized to G′o, was monitored with rheometry. The degradation rate was precisely controlled with irradiation intensity and wavelength: (a) 365 nm at 20 mW/cm2, (b) 365 nm at 10 mW/cm2, and (c) 405 nm at 25 mW/cm2. (E) The gel degradation was modulated by either (a) continuous or (b) periodic irradiation with 365 nm at 10 mW/cm2. The extents of degradation corresponding to the materials used in Fig. 1F and 1G are indicated. (F) hMSCs encapsulated within dense hydrogels exhibit a rounded morphology. (G) Irradiation (480 s, 365 nm at 10 mW/cm2) significantly degrades the gel (ρx/ρxo ∼ 0.04), promoting hMSC spreading after 3 days in culture. Scale bar, 50 μm.
The photodegradable crosslinker, Compound 1, was copolymerized with PEG monoacrylate (PEGA) in phosphate buffered saline (PBS) via redox-initiated free radical polymerization to create photodegradable hydrogels. Upon irradiation, these hydrogels release modified PEG, and after complete degradation they release modified poly(acrylate) chains, as shown in Fig. 1C. The bulk photodegradation of these hydrogels, ensured by flood irradiation through a small thickness of material, was characterized within a parallel-plate rheometer (21). The storage modulus (G′) is proportional to the hydrogel crosslinking density (ρx) (22, 23), allowing calculation of the characteristic photolabile group degradation time (τ) when normalized to its initial value (G′o) via G′/G′o = ρx/ρxo = exp(-2t/τ). At an irradiation intensity of 10 mW/cm2 at 365 nm, typical irradiation conditions for hydrogel photopolymerizations in the presence of cells (24), the hydrogel completely degrades in 10 minutes (τ ∼ 280 s, Fig. 1D). As expected from photolysis kinetics (supporting online text), doubling the intensity halves the time to complete degradation (τ ∼ 140 s, Fig. 1D), while irradiation at 405 nm increases the degradation time inversely with the relative molar absorptivity (τ ∼ 930 s, Fig. 1D). When the irradiation is ceased, the degradation is arrested; degradation is recommenced upon further irradiation (Fig. 1E). The degradation rate and extent and the resulting material properties such as stiffness thus can be predictably manipulated with light intensity and wavelength.
Precise control of gel crosslinking density was utilized to examine the influence of gel structure on cell spreading. Specifically, human mesenchymal stem cells (hMSCs) were encapsulated within a densely crosslinked gel where they exhibit a rounded morphology (Fig. 1F). When the gel crosslinking density was reduced through photodegradation, cell spreading was observed (Fig. 1G), while the cell viability was maintained (Fig. S4). Thus, cell morphology can be manipulated by irradiation and degradation of these hydrogels at any time in culture. While this control can be useful for allowing cell spreading or extracellular matrix (ECM) elaboration, this molecular scale degradation does not allow for bulk cell migration or long-range cell-cell interactions, which require fabrication of micron-scale channels by gel erosion.
To study gel erosion, degradation was first confined to the irradiation surface by selecting an irradiation wavelength that corresponds to a high photolabile group molar absorptivity (Fig. S1). Since the light intensity is highest within the uppermost layers of a thick gel, these top layers erode away rapidly during light exposure prior to degradation of underlying layers (see schematic, Fig. 2A). Irradiation of a hydrogel through a photomask degrades the material in the exposed regions and creates progressively recessed areas, whereas material in the unexposed areas remains intact (see photographic series, Fig. 2A). Profilometry reveals well-defined ridges, demonstrating reproduction of the irradiation pattern, and feature height increases linearly with exposure time (Fig. 2A). Such photolithographic techniques at highly absorbed wavelengths can be used to create surface features of varying height and width post-gelation, whilst avoiding concomitant dimension changes in the hydrogel bulk, useful for the fabrication and subsequent manipulation of cell culture devices.
Figure 2. Two and three-dimensional patterning of photodegradable hydrogels.
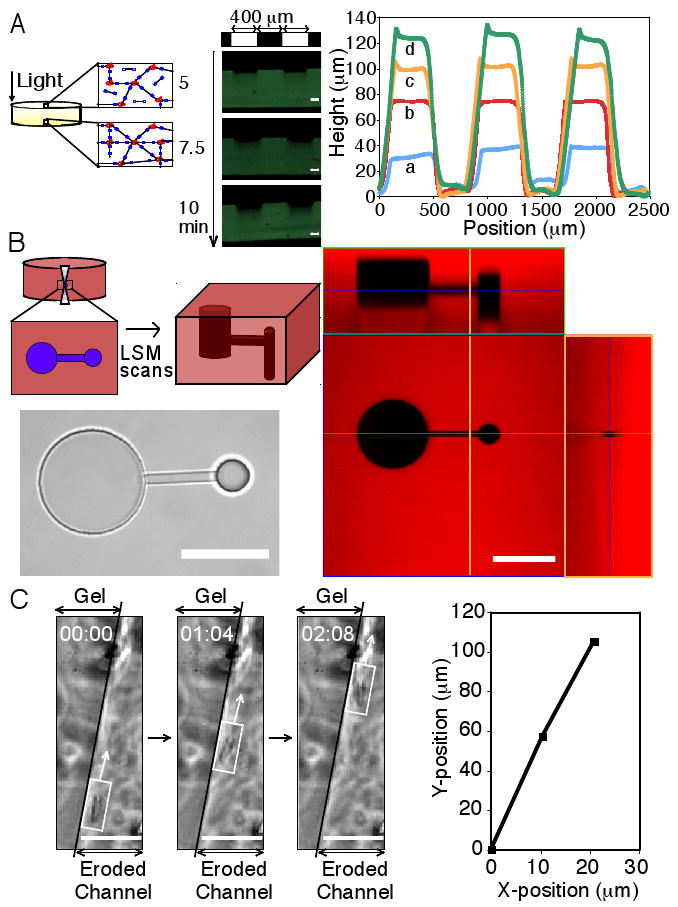
(A) Thick gels demonstrate surface erosion upon irradiation (left). A gel covalently labeled with fluorescein was eroded spatially via masked flood irradiation (320-500 nm at 40 mW/cm2, 400 μm line periodic photomask). Channel depth increased linearly with irradiation, and no changes in hydrogel dimensions due to increased swelling were observed (middle). Feature dimensions were quantified with profilometry: (a) 2.5, (b) 5, (c) 7.5, and (d) 10 minutes irradiation (right). Scale bar, 100 μm. (B) Interconnected three-dimensional channels were fabricated within a photodegradable gel, covalently labeled with rhodamine B, using a two-photon LSM. A thin horizontal channel connected two offset vertical channels of different diameter, which was visualized in brightfield (bottom left) and with confocal LSM (right, intact gel is red and the created feature is black, corresponding cross-sections are noted by blue, green, and orange lines). Scale bar, 100 μm. (C) Channels were eroded within a hydrogel encapsulating fibrosarcoma cells, releasing cells into the degraded channel and enabling migration. Migration of a cell along the edge of a channel is shown in time-lapsed brightfield images (left) and its corresponding position trace (right). Scale bar, 50 μm.
Controlled creation of three-dimensional cell culture microenvironments facilitates the examination of cell-matrix and cell-cell interactions. The post-gelation addition of chemical moieties, such as peptides, to hydrogels in three dimensions has been demonstrated in a multi-step technique using a two-photon laser scanning microscope (LSM) (16, 17). Using a photodegradable hydrogel in conjunction with a single (405nm) (Fig. S2) or two-photon (740nm) LSM (Fig. 2B), we generate three-dimensional features within a hydrogel by local photodegradation with scan parameters similar to those used for cell imaging and without the addition of chemical moieties. This rapid patterning (seconds to minutes) is achieved by controlled rastering of the laser focus within the hydrogel, locally degrading the polymer network and creating arbitrary three-dimensional features on the micron scale (schematic, Fig. 2B). Interconnected channels, potentially useful for directing cell connectivity and/or migration, were created within a rhodamine-labeled photodegradable hydrogel with a two-photon LSM and visualized using brightfield and fluorescence microscopy (Fig. 2B). Other features of arbitrary shape and size for desired applications such as device fabrication can be precisely and rapidly patterned within these photodegradable hydrogels in situ using either a single or two-photon LSM with standard imaging parameters (Fig. S2).
To demonstrate how erosion of these photodegradable hydrogels can be used to direct cell migration, fibrosarcoma cells were encapsulated within a hydrogel. A portion of these entrapped cells was subsequently released by degrading channels within the gel using photolithography. Cells near the periphery of the channel or released by erosion of the channel were then allowed to migrate along the channel length (Fig. 2C). With the ability to pattern micro to macroscopic structures in the presence of encapsulated cells, these photodegradable hydrogels provide a cell culture platform to allow unique studies of directed cell migration (25, 26) and cell connectivity (27) by enabling real-time manipulation of the cell microenvironment. It is important to note, however, that cell migration exclusively occurs in regions of complete or near-complete gel erosion. In many cases, the culturing of cells in an environment that allows bulk migration is desirable with subsequent direction of cell-cell interactions at a certain point in time or space. To achieve this, complementary chemistries, such as enzymatically degradable moieties, can be incorporated within these gels by, for example, Michael addition (9) of the photodegradable crosslinker with an enzymatically cleavable, thiol-containing peptide (2).
The aforementioned techniques have focused on the patterning of hydrogels for degradation of the physical network structure; however, photodegradable linkages can also be exploited to locally modify the chemical environment within a hydrogel via incorporation of tethered, but photolabile, biologically active functionalities. To demonstrate, the PDA monomer was coupled through its pendant carboxylic acid to the ubiquitous adhesion peptide sequence RGDS (28), producing an asymmetric, photodegradable, biofunctional acrylic monomer (Fig. 3A). Copolymerization of this photo-releasable macromer with a non-degradable PEG diacrylate (PEGDA) yields a hydrogel with photolytic control over its chemical interaction with encapsulated cells (Fig. 3B). Upon irradiation, the modified peptide is cleaved from the polymer network and released into solution, where it quickly diffuses out of the gel (Fig. S3). Peptide presentation within the microenvironment can thus be controlled with irradiation at a rate based on the photolabile group characteristic degradation time (Fig. 1D). More generally, this technique can be used for controlling the chemical structure of hydrogels in three dimensions and in time, where the peptide RGDS can be replaced with any biomolecule of interest, creating an additional tool for the modulation of ligand presentation (4).
Figure 3. Photolabile RGDS tether synthesis and utilization for dynamic changes in microenvironment chemistry.

(A) A photolabile asymmetric biofunctional acrylic monomer (Compound 2) was synthesized containing the adhesion peptide RGDS (black) attached to the PDA (acrylate in red and photolabile moiety in blue). (B) Compound 2 was polymerized (green triangle with closed blue box) into a non-degradable gel (red poly(acrylate) coils connected by black non-degradable PEG crosslinks) whose chemical composition is controlled with light exposure by photolytic release of the tethered biomolecule RGDS (bottom, green triangle with open blue box). hMSCs were encapsulated in non-degradable PEG gels (b) with or (a) without photoreleasable RGDS. The presentation of RGDS was temporally altered by (c) photocleavage of RGDS from the gel on Day 10 in culture. (C) RGDS presentation maintains hMSC viability within PEG-based gels (inset table). RGDS photolytic removal on Day 10 directs hMSC chondrogenesis, (c) increasing GAG production four-fold over (b) persistently-presented RGDS or (a) PEG-only gels by Day 21 (p<0.05), an indicator of hMSC chondrogenesis.
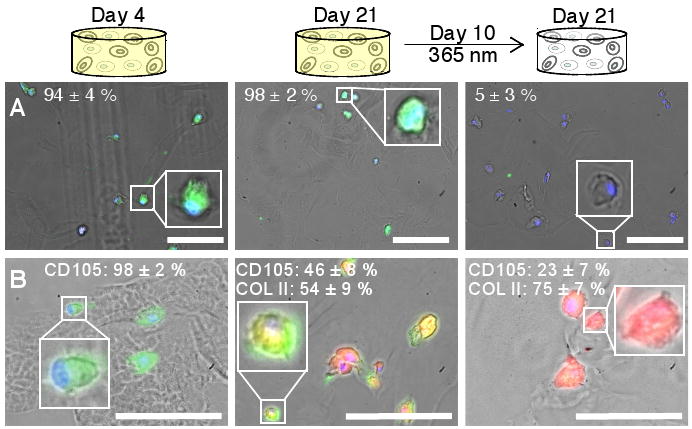
To demonstrate the utility of this photolabile tether platform, hMSCs were encapsulated in non-degradable PEGDA hydrogels, both with and without the photoreleasable RGDS moiety, to examine the effect of temporal RGDS presentation on hMSC viability and chondrogenic differentiation. Control of the local biochemical environment enables promotion of chondrogenesis. While numerous researchers have incorporated the fibronectin epitope RGD into biomaterial microenvironments to promote the survival of hMSCs (5, 29, 30), few studies investigate the importance of the persistence of this signal on the differentiation fate (31-33). Natively, hMSCs differentiating into chondrocytes initially produce the adhesion protein fibronectin, which is subsequently downregulated between Day 7 and 10 while the excreted ECM is remodeled through enzyme production (31, 34). After Day 10, these cells upregulate production of glycosaminoglycans (GAGs) and type II collagen (COLII), two major components of cartilage and specific indicators of chondrogenic differentiation (31, 34). To examine this temporal change in vitro, a portion of the cell-gel constructs containing the photolabile RGDS tether were irradiated on Day 10 in culture to remove the fibronectin epitope RGDS from the encapsulated cell microenvironment (Fig. 3B).
The absence of RGDS in three dimensional cell culture yields a statistically significant decrease (p<0.05) in hMSC viability within the first 7 days in PEG-only gels (see inset, Fig. 3C), in agreement with other experimental results (29, 30) (supporting online text). Upon photolytic removal of RGDS from the photolabile tether gels on Day 10, cell viability was unaffected; however, by day 21, a four-fold statistical increase (p<0.05) in the production GAGs occurred relative to the persistently presented RGDS or PEG-only hydrogels, indicating further differentiation of the hMSCs down the chondrogenic pathway in response to the microenvironment modification (Fig. 3C, Fig. S5). To examine cell interaction with this dynamic microenvironment, fixed sections were immunostained for the expression of the αvβ3 integrin, one of the cell surface integrins by which cells interact with RGDS. Cells persistently presented with RGDS express the integrin while most cells with RGDS photoreleased cease integrin expression by Day 21 (Fig. 4A), indicating that the cells have locally sensed and responded to the chemical change in their environment. Additionally, to determine that the elevated GAG production is associated with chondrogenesis, sections of the cell-hydrogel constructs were immunostained for the hMSC marker CD105 (TGFβ receptor) and the chondrocyte marker COLII. As hMSCs differentiate into chondrocytes in three-dimensional culture, a decrease in CD105 expression is observed along with onset of COLII production (35). The photolytic removal of RGDS on Day 10 is accompanied by a decrease in CD105 expression and elevated COLII production as compared to persistently presented RGDS on Day 21 (Fig. 4B), indicating increased chondrogenesis. With this photodegradable tether approach, the dynamic influence of other biomolecules on cell function can be similarly studied, expanding the ability to fabricate material environments that control cell function. While manipulation of a single signal was examined and utilized to direct hMSC differentiation herein, this gel system can be readily functionalized with photolabile moieties of different light absorbances and cleavage efficiencies, enabling independent control over multiple signals via selection of irradiation wavelength and intensity.
Figure 4. Influence of dynamic microenvironment chemistry on integrin expression and differentiation.
(A) Cells (DAPI-labeled nuclei, blue) cultured with persistent RGDS express the cell-surface integrin αvβ3 (FITC-labeled, green) on at Day 4 (left) and Day 21 (middle) in culture. Cells with photocleaved RGDS have decreased expression of the αvβ3 integrin by Day 21 (right), indicating that the cells have responded to the removal of RGDS. (Representative images shown with average cell percentages noted.) (B) Chondrogenic differentiation of hMSCs was verified by immunostaining for the hMSC marker CD105 (FITC, green) and the chondrocyte marker COLII (TRITC, red). Within error, no cells initially produce COLII (Day 4, left). By Day 21, half of the cells presented persistently with RGDS strongly expressed CD105 and the other half produced COLII (middle). With photolytic removal of RGDS on Day 10, one-fourth of the cells strongly expressed CD105 and three-fourths produced COLII (right), supporting that photolytic removal of RGDS increases chondrogenesis. (Representative images shown with average cell percentages strongly expressing the marker noted.) Scale bars, 100 μm.
We have demonstrated the synthesis of photodegradable macromers and their subsequent polymerization to form hydrogels whose physical, chemical, and biological properties can be tuned in situ and in the presence of cells via UV, visible, or two-photon irradiation. These photodegradable hydrogels show promise as in vitro three-dimensional cell culture platforms where cell-material interactions are dynamically and externally directed to elucidate how cells receive and process information from their environment. Ways to promote or suppress desired cell functions may become possible with temporal and spatial regulation of the 3D culture microenvironment, leading to applications in fields ranging from controlled drug delivery to tissue regeneration.
Supplementary Material
Acknowledgments
The authors would like to thank C. Bowman, T. Scott, and C. Kloxin for comments on early versions of this manuscript and valuable discussions; A. Aimetti for peptide synthesis training; J. McCall and S. Anderson for cell culture assistance; M. Schwartz for cell migration discussions; C. Bowman and his laboratory for use of and assistance with the rheometer and profilometer; C. Kloxin for assistance with image analysis; C. DeForest, Carl Zeiss, Inc., and the Howard Hughes Medical Institute Janelia Farms campus for assistance with and use of the Zeiss 710 two-photon confocal LSM; and the Howard Hughes Medical Institute and the NIH (grants DE12998 and DE16523) for research support. A. Kloxin thanks the NASA GSRP fellowship and the DoEd GAANN fellowship for support. A patent related to this work has been submitted (U.S. Patent Application No. 11/374,471).
References and notes
- 1.Weibel DB, DiLuzio WR, Whitesides GM. Nat Rev Microbiol. 2007 Mar;5:209. doi: 10.1038/nrmicro1616. [DOI] [PubMed] [Google Scholar]
- 2.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005 Jan;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 3.Langer R, Peppas NA. Aiche J. 2003 Dec;49:2990. [Google Scholar]
- 4.Stevens MM, George JH. Science. 2005 Nov 18;310:1135. doi: 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- 5.Chan G, Mooney DJ. Trends Biotechnol. 2008 Jul;26:382. doi: 10.1016/j.tibtech.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006 Aug 25;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 7.Engler AJ, et al. J Cell Biol. 2004 Sep 13;166:877. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenny PA, Bissell MJ. Int J Cancer. 2003 Dec 10;107:688. doi: 10.1002/ijc.11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metters A, Hubbell J. Biomacromolecules. 2005 Jan-Feb;6:290. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 10.Rice MA, Sanchez-Adams J, Anseth KS. Biomacromolecules. 2006 Jun;7:1968. doi: 10.1021/bm060086+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. J Biomed Mater Res Part A. 2004 Mar 15;68A:704. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 12.Ehrbar M, et al. Circ Res. 2004 Apr 30;94:1124. doi: 10.1161/01.RES.0000126411.29641.08. [DOI] [PubMed] [Google Scholar]
- 13.Silva GA, et al. Science. 2004 Feb 27;303:1352. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 14.Khademhosseini A, Langer R. Biomaterials. 2007 Dec;28:5087. doi: 10.1016/j.biomaterials.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Sands RW, Mooney DJ. Curr Opin Biotechnol. 2007 Oct;18:448. doi: 10.1016/j.copbio.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hahn MS, Miller JS, West JL. Adv Mater. 2006 Oct 17;18:2679. [Google Scholar]
- 17.Wosnick JH, Shoichet MS. Chem Mat. 2008 Jan 8;20:55. [Google Scholar]
- 18.Zhao YR, et al. Journal of The American Chemical Society. 2004 Apr 14;126:4653. doi: 10.1021/ja036958m. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez M, et al. Adv Mater. 2008 Dec;20:4563. [Google Scholar]
- 20.Luo Y, Shoichet MS. Nat Mater. 2004 Apr;3:249. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 21.Khan SA, Plitz IM, Frantz RA. Rheol Acta. 1992 Mar-Apr;31:151. [Google Scholar]
- 22.Young RJ, Lovell PA. Introduction to polymers. 2nd. Chapman & Hall; London, U. K.: 1991. p. 443. [Google Scholar]
- 23.Bryant SJ, Anseth KS. In: Scaffolding in Tissue Engineering. Ma PX, Elisseeff J, editors. Marcel Dekker, Inc.; 2005. [Google Scholar]
- 24.Bryant SJ, Nuttelman CR, Anseth KS. J Biomater Sci-Polym Ed. 2000;11:439. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 25.Tayalia P, Mendonca CR, Baldacchini T, Mooney DJ, Mazur E. Adv Mater. 2008 Dec;20:4494. [Google Scholar]
- 26.Raeber GP, Lutolf MP, Hubbell JA. Biophys J. 2005 Aug;89:1374. doi: 10.1529/biophysj.104.050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahoney MJ, Anseth KS. Biomaterials. 2006 Apr;27:2265. doi: 10.1016/j.biomaterials.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Ruoslahti E, Pierschbacher MD. Science. 1987 Oct 23;238:491. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 29.Boontheekul T, Mooney DJ. Curr Opin Biotechnol. 2003 Oct;14:559. doi: 10.1016/j.copbio.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Hudalla GA, Eng TS, Murphy WL. Biomacromolecules. 2008 Mar;9:842. doi: 10.1021/bm701179s. [DOI] [PubMed] [Google Scholar]
- 31.Tavella S, et al. J Cell Sci. 1997 Sep;110:2261. doi: 10.1242/jcs.110.18.2261. [DOI] [PubMed] [Google Scholar]
- 32.Nuttelman CR, Tripodi MC, Anseth KS. Matrix Biol. 2005 May;24:208. doi: 10.1016/j.matbio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Salinas CN, Anseth KS. Biomaterials. 2008;29:2370. doi: 10.1016/j.biomaterials.2008.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLise AM, Fischer L, Tuan RS. Osteoarthritis Cartilage. 2000 Sep;8:309. doi: 10.1053/joca.1999.0306. [DOI] [PubMed] [Google Scholar]
- 35.Salinas CN, Cole BB, Kasko AM, Anseth KS. Tissue Eng. 2007 May;13:1025. doi: 10.1089/ten.2006.0126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.