

Abstract
Linoleic acid is required for normal mammalian health and development, but is also prone to oxidation, yielding metabolites with biological effects. We screened linoleic acid, other fatty acids, and some of their derivatives and found that an epoxy-keto derivative of linoleic acid (but neither linoleic acid itself nor others of its oxidation products) strongly activates the antioxidant response element (ARE) in IMR-32 neuroblastoma cells and cerebro-cortical neurons. The active compound, 12,13-epoxy-9-keto-10(trans)-octadecenoic acid (EKODE), induces the expression of ARE-regulated cytoprotective genes such as NQO1 at the transcript and protein levels. EKODE requires transcription factor NRF2 and PI3-kinase for ARE activity. The results suggest that specific oxidation products of linoleic acid may initiate responses that lessen damage caused by oxidative stress.
Keywords: Polyunsaturated Fatty Acids, Fatty Acid Oxidation, Fatty Acid Epoxides, EKODE, Antioxidants, Antioxidant Response Element
1. Introduction
Linoleic acid (C18:2 n-6), the most abundant polyunsaturated fatty acid (PUFA) in human low-density lipoprotein (LDL), is an essential nutrient, but has also been implicated in the pathogenesis of atherosclerosis, hypertension, diabetes, and other diseases [1–4]. The paradox of an essential fatty acid participating in both beneficial and pathological processes may be explained by its conversion to biologically active metabolites [5,6]. As an example of a pathological effect, we (T.L.G. and D.L.B.) have shown that one oxidation product of linoleic acid, 12,13-epoxy-9-keto-10(trans)-octadecenoic acid (EKODE), stimulates aldosterone secretion by rat adrenal cells, providing a potential link between linoleic acid oxidation and hypertension [7].
Effects of polyunsaturated fatty acids differ from cell to cell. For example, in cultured endothelial cells, linoleic acid elicits production of superoxide and nitric oxide, and increases intracellular calcium levels [8]. By contrast, in an insulin-secreting cell line and in primary normal human fibroblasts, linoleic acid protects against oxidative stress [9,10]. The protective effect of linoleic acid described by Beeharry et al. [10] is dependent on PI3-kinase (PI3K) activity. PI3K activity is also necessary for another protective effect of a variety of compounds: activation of the antioxidant response element (ARE) [11]. Because of this common involvement of PI3K, we postulated that the protective effect of linoleic acid derives from its activation of the ARE — an effect of linoleic acid itself or of one of its oxidation products. The experiments described here tested the effects of linoleic acid, other fatty acids, and several oxidation products on ARE activation in two cell types.
2. Materials and methods
2.1 Chemicals, reagents and plasmids
EKODE was synthesized by an adaptation of the process described by Gardner and Crawford [12] and purified by high-pressure liquid chromatography [7]. EKODE employed in this study was an isomeric mixture consisting of primarily one of the isomers, termed trans-EKODE-(E)-1b, according to the nomenclature proposed by Lin et al. [13]. Additional fatty acid derivatives were purchased from Cayman Biochemicals (Ann Arbor, MI) and INDOFINE Chemical Company, Inc. (Hillsborough, NJ). Tert-butylhydroquinone was purchased from Acros Organics (St. Louis, MO). LY294002 and PD98059 were purchased from Calbiochem (San Diego, CA). Media reagents were purchased from Invitrogen (Carlsbad, CA) unless specified otherwise. Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Norcross, GA) and from Hyclone (Logan, UT). FuGENE6 and FuGENE HD transfection reagents were from Roche Diagnostics (Indianapolis, IN) and siLentFect from Bio-Rad Laboratories (Hercules, CA). Detection reagent, BriteLite, for ARE reporter assays was purchased from PerkinElmer Life And Analytical Sciences, Inc. (Waltham, MA). Compounds were dissolved or obtained from the supplier in the solvents indicated in Table 1. All other chemicals were purchased from Fisher Scientific, Inc. (Hampton, NH). The luciferase reporter construct for human NQO1-ARE (ARE-luc) contains the sequence 5′-CTCAGCCTTCCAAATCGCAGTCACAGTGACTCAGCAGAATC-3′) [14]. Small interfering RNAs (siRNAs) against human NRF2 were obtained from Dharmacon, Inc. (Lafayette, CO). The mammalian expression vector for dominant-negative Nrf2 (DN Nrf2) was used as described by Lee et al. [15].
Table 1.
Fatty acids and their acronyms, controls and respective solvents used in this study
| Acronym | IUPAC name | Solvent |
|---|---|---|
| EKODE | 12,13-Epoxy-9-keto-(10)trans-octadecenoic acid | DMSO |
| (+)-Vernolic acid | (12S,13R)-Epoxy-9(Z)-octadecenoic acid | Ethanol |
| (−)-Vernolic acid | (12R,13S)-Epoxy-9(Z)-octadecenoic acid | Ethanol |
| (+)-Coronaric acid | (9R,10S)-Epoxy-12Z-octadecenoic acid | Ethanol |
| 15-Oxo-ETE | 15-Oxo-5Z,8Z,11Z,13E-eicosatetraenoic acid | Ethanol |
| 13-Oxo-ODE | 13-Oxo-9Z,11E-octadecadienoic acid | Ethanol |
| 9-Oxo-ODE | 9-Oxo-10E,12Z-octadecadienoic acid | Ethanol |
| (±)-9-HETE | (±)-9-Hydroxy-5Z,7E,11Z,14Z-eicosatetraenoic acid | Ethanol |
| (±)-12-HETE | (±)-12-Hydroxy-5E,8Z,10Z,14Z-eicosatetraenoic acid | Ethanol |
| (±)-15-HETE | (±)-15-Hydroxy-5E,8Z,11Z,13Z-eicosatetraenoic acid | Ethanol |
| (9S)-HODE | (9S)-Hydroxy-10E,12Z- octadecadienoic acid | Ethanol |
| (13S)-HODE | (13S)-Hydroxy-9Z,11E-octadecadienoic acid | Ethanol |
| Linoleic acid | 9Z,12Z-Octadecadienoic acid | Ethanol* |
| Arachidonic acid | 5Z,8Z,11Z,14Z-Eicosatetraenoic acid | Ethanol* |
| α-Linolenic acid | 9Z,12Z,15Z-Octadecatrienoic acid | Ethanol |
| γ-Linolenic acid | 6Z,9Z,12Z-Octadecatrienoic acid | Ethanol |
| Oleic acid | 9Z-Octadecenoic acid | Ethanol |
| Prostaglandin E2 (PGE2) | 9-Oxo-11 α-(15S)-dihydroxy-prosta-5Z,13E- dien-1-oic acid | DMSO |
| Prostaglandin J2 (PGJ2) | 11-Oxo-(15S)-hydroxy-prosta-5Z,9,13E-trien-1-oic acid | Methyl acetate |
| tBHQ | Tert-butylhydroquinone | DMSO |
| DEM | Diethyl maleate | DMSO |
in 0.1% 2,6-di-tert-butyl-4-hydroxytoluene (BHT) to prevent autoxidation to peroxides.
2.2 ARE activity profiling of fatty acid derivatives in IMR-32 cells
ARE-luciferase reporter plasmid (100 ng/well), CMV-GFP (10 ng/well, for monitoring transfection efficiency) and actin-lacZ (20 ng/well, for normalization) were cotransfected into IMR-32 human neuroblastoma cells (3 × 104 cells/well) using FuGENE HD transfection reagent following the manufacturer’s instructions. The transfected cells were dispensed in 96-well plates and cultured in Eagle’s Minimum Essential Medium (ATCC, Manassas, VA) supplemented with 10% FBS at 37 °C in a humidified 5% CO2 atmosphere. After 24 h, cells were treated with individual compounds and incubated at the same condition for another 24 h. ARE activities were detected by using BriteLite detection reagent for luminescence. Each compound was tested in quadruplicate. Normalized values are given.
2.3 ARE activity in IMR-32 cells in the presence of antioxidant (catalase) or dominant-negative (DN) Nrf2
IMR-32 human neuroblastoma cells were plated at a density of 1 × 104 cells/well in a 96-well plate and cultured for 24 h in Dulbecco’s modified eagle media supplemented with 10% FBS at 37 °C in a humidified 10% CO2 atmosphere. Following the incubation, cells were cotransfected with the ARE-luciferase reporter plasmid (80 ng/well) and CMV-lacZ (20 ng/well) using FuGENE6 transfection reagent following the manufacturer’s instructions. For DN Nrf2 cotransfection experiments, the DN Nrf2 expression vector was added to the transfection experiment (5 ng/well). Total DNA amount in the transfection mixture was normalized using an empty vector. After 24 h, cells were treated with catalase (100 U) immediately followed by compound addition. An additional 24 h later, luciferase and β-galactosidase activities were measured [14] and compared with compound treatment alone. The data are presented as the ratio of luciferase activity in relative light units to β-galactosidase activity to account for any differences in transfection efficiencies between experiments.
2.4 Mouse primary cortical cultures
Primary cerebral cortical cultures were prepared from an ARE-driven, heat-stable human alkaline phosphatase (ARE-hPAP) transgenic reporter mouse line [16]. On embryonic day 16 (E16), the cortices from mouse pups were dissected and transferred to Hank’s balanced salt solution (HBSS). The tissue was dissociated with 0.05% trypsin in HBSS for 10 min at 37 °C, and then filtered through a 70-μm cell strainer. Cells were plated on poly-D-lysine-coated plates at approximately 3.2 × 105 cells/cm2 in Complete Essential Minimum Eagle’s Media (CEMEM: Essential Minimum Eagle’s Media with 10% fetal bovine serum, 10% horse serum, 2 mM L-glutamine, and 1% penicillin/streptomycin) and allowed to adhere to the plate for 45 min in a 37 °C humidified incubator (5% CO2 and 5% O2) prior to a media exchange to remove non-adherent cells. The plated cells were incubated in CEMEM for 48 h prior to a media change to Neurobasal media with a B27 supplement containing antioxidants to specifically enhance neuronal growth. On the following day, the experimental treatments were performed. For analysis of human placental alkaline phosphatase (hPAP) activity, the mouse primary cortical cultures were lysed and the whole cell extracts incubated at 65 °C for 25 min to inactivate endogenous phosphatase activity. Analysis of the hPAP activity was performed using the phospholight chemiluminescent substrate per the manufacturer’s directions (Tropix, Bedford, MA).
2.5 RNA extraction, quantitative RT-PCR and validation
IMR-32 cells (6 × 105 cells/well) were plated in a 6-well plate 24 h prior to treatment. Cells were treated with vehicle (0.5% DMSO), tBHQ (10 μM) and EKODE (1 and 10 μM). After 8 h of incubation, total RNA was extracted with RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was synthesized from 2 μg of total RNA by using SuperScript II Reverse Transcriptase (Invitrogen) and Oligo (dT)12–18 Primer (Invitrogen). The cDNA served as a template for quantitative PCR (qPCR) using TaqMan probes (Applied Biosystems, Foster City, CA). qPCR was performed by using 12.5 μL of TaqMan 2× universal master mix, 1.25 μL of 20× TaqMan gene expression assay mix, 2 μL of cDNA and 9.25 μL of RNase-free sterile water, in a total volume of 25 μL per well reaction in a 96-well plate (Applied Biosystems) by using the ABI 7300 sequence detection systems (Applied Biosystems). The thermocycler program consisted of 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Each assay was carried out in triplicate. GAPDH expression was used as internal control for normalization.
2.6 Immunoblot analysis
IMR-32 cells (6 × 105 cells/well) were plated into each well of 6-well plates 24 h prior to treatment. Cells were treated with vehicle (0.5% DMSO) and EKODE at 1 and 10 μM. After 24 h of incubation, whole-cell lysates were prepared by using PhosphoSafe lysis buffer (Novagen, Gibbstown, NJ). The protein concentrations of the cell lysates were measured by using the BCA assay (Pierce, Rockford, IL). Samples containing equal amount of protein were separated by SDS-PAGE, transferred to PVDF membrane, probed with antibodies and detected with Supersignal Femto Western Blotting Kit (Pierce). Anti-NQO1 antibody was purchased from Abcam (Cambridge, MA) and the secondary anti-goat antibody from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-β-actin and the secondary anti-rabbit antibodies were obtained from Cell Signaling (Boston, MA).
2.7 Glutathione assays
IMR-32 cells were seeded at 3 × 106 per plate (10-cm dish) or 1.5 × 106 per well (6-well plate). When the desired confluency was reached, the cells were treated with EKODE (10 μM) or vehicle (DMSO, 0.5%) for 2, 8, 16 or 24 h. Treated cells were then washed twice with PBS and harvested in 1 mL (10-cm dish) or 200 μL (6-well plate) of PBS. The collected cells were centrifuged at 600 g for 10 min at 4 °C. After removal of the supernatant, the volume of the cell pellet was measured and resuspended in 3 volumes of 5% sulfosalicylic acid (SSA) solution. The cell suspensions were frozen (in liquid nitrogen) and thawed (in 37 °C water bath) twice, incubated for 5 min at 4 °C, and centrifuged at 10,000 g for 10 min at 4 °C. The supernatant was transferred to a separated microcentrifuge tube and used immediately as glutathione stock. The concentrations of total (reduced and oxidized) glutathione (GSH + GSSG) were assessed using Glutathione Assay Kit (Sigma, St. Louis, MO), following the manufacturer’s instructions. Briefly, the total glutathione was measured based on a kinetic reaction in which catalytic amounts (nmoles) of GSH cause a continuous reduction of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) to 5-thio-2-nitrobenzoic acid (TNB) and the GSSG formed is recycled by glutathione reductase and NADPH, which also gives a positive value in this reaction. The yellow product, TNB is measured spectrophotometrically at 412 nm. The total glutathione levels were determined from a standard curve of reduced glutathione. Each of the four time points was measured in duplicate. Fresh 10-fold dilutions of GSH stocks were used. Values given are normalized to glutathione content in vehicle-treated cells.
2.8 RNA interference experiments
24 h prior to transfection, IMR-32 cells (1 × 105 cells/well) were plated in a 24-well plate. The nontargeting control siRNA and siGENOME SMARTpool siRNA reagents targeting NRF2 were obtained from Dharmacon. Using siLentFect as a transfection reagent, siNRF2 (50 nM, 330 ng/well) was cotransfected with ARE-luc (250 ng/well), CMV-GFP (50 ng/well, for monitoring transfection efficiency) and actin-lacZ (100 ng/well, for normalization). After 48 h, the cells were treated with vehicle (0.5% DMSO), tBHQ (10 μM) and EKODE (10 μM) and incubated for an additional 24 h before luciferase and β-galactosidase activities were measured [14,17]. Each experiment was carried out in quadruplicate. The data are presented as the ratio of luciferase activity in relative light units to β-galactosidase activity to account for effects on viability and any differences in transfection efficiencies between experiments.
2.9 Inhibitor studies
IMR-32 cells (1 × 105 cells/well) were transfected with ARE-luc (594 ng/well), CMV-GFP (59.4 ng/well, for monitoring transfection efficiency) and actin-lacZ (118 ng/well, for normalization) in 24-well plates. After 24 h, the cells were pre-treated with vehicle (0.5% DMSO), LY294002 (25 μM, PI3K inhibitor) and PD98059 (50 μM, MEK1 inhibitor) for 30 min prior to treatment with vehicle or EKODE (10 μM). Luciferase and β-galactosidase activities were detected another 24 h later. Each experiment was carried out in quadruplicate. The data are presented as the ratio of luciferase activity in relative light units to β-galactosidase activity to account for any differences in transfection efficiencies between experiments and for potential effects of inhibitors on cell viability.
3. Results
3.1 Profiling of fatty acids and related metabolites for ARE activation in IMR-32 cells
We screened linoleic acid and 18 related fatty acids and oxidation products (for structures, see Figure S1) for ARE activation at 10 μM in IMR-32 human neuroblastoma cells — a validated cellular model of oxidative stress [11,14,15] — using an ARE-luciferase based reporter gene assay [14]. These fatty acids included four other nonconjugated unsaturated fatty acids without additional functional groups, five allylic alcohols, three α,β,γ,δ-unsaturated carbonyl systems, four epoxides and two prostaglandins, including prostaglandin J2 (PGJ2), an arachidonic acid metabolite that is known to induce the expression of ARE-regulated genes [18]. Linoleic acid itself did not induce the ARE in IMR-32 cells; however, the epoxy-keto linoleic acid EKODE exhibited activity (23.1-fold) comparable to the model activator tert-butylhydroquinone (tBHQ) at 10 μM (34.9-fold), while only four other compounds (15-oxo-ETE, 13-oxo-ODE, 9-oxo-ODE, PGJ2) activated the ARE more than 2-fold (4.7-, 3.9-, 2.3-, and 4.4-fold, respectively). All of the active compounds are Michael acceptors — conjugated π-systems commonly consisting of α,β-unsaturated carbonyl groups (enones) which can act as electrophiles (Figure 1). The activity of PGJ2 was accompanied by apparent toxicity at 10 μM in this cell type; subsequent dose-response analysis suggested that PGJ2’s ARE activity was the highest at 3.2 μM (30.9-fold) without toxicity. Based on the remarkable activity of EKODE, without apparent toxicity at 10μM, we focused on this compound in subsequent experiments.
Figure 1.

ARE activation by EKODE and other linoleic acid derivatives in IMR-32 cells at 10 μM using an ARE-luciferase reporter gene assay. Like tBHQ, EKODE is a strong activator of the ARE. Four other Michael acceptors (15-oxo-ETE, 13-oxo-ODE, 9-oxo-ODE, and PGJ2) exhibited weak ARE activation. ARE activation for each compound was normalized to the respective solvent (Table 1). Cotransfection of actin-lacZ served to account for differences in cell number due to effects on viability. Structures of active compounds are given. Results are the means ± standard deviation (n = 4).
3.2 ARE activation by EKODE in the presence of antioxidant and validation in primary cortical neurons
Many Michael acceptors are known for their ability to activate the ARE and react with sulfhydryl groups, including glutathione, which could mimic an oxidative insult [19]. Consequently, their ARE activity may be reduced in the presence of scavenging antioxidants. Indeed, EKODE’s ARE activity was significantly diminished when catalase was added to the culture medium (Figure 2A). The same effect was observed with diethyl maleate (DEM, 20 μM), a classical electrophile that reacts via conjugate addition, while the activity of tBHQ was largely unaffected as previously described (Figure 2A) [15]. Dose response analysis indicated that an EKODE concentration of 10 μM was required for potent ARE activity (here: 41.5-fold; Figure 2B). Depending on the experimental conditions and status of the IMR-32 cells, we observed ARE activation by EKODE at 10 μM from 20- to 45-fold. At 32 μM, EKODE activated the ARE even more (110-fold; Figure 2B), but also demonstrated toxicity, while EKODE did not introduce significant toxicity at 10 μM.
Figure 2.
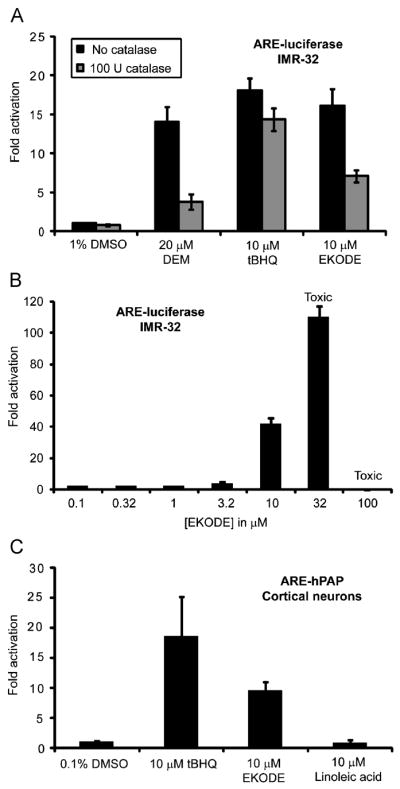
Reporter gene assays in IMR-32 cells and primary cortical cultures. (A) ARE activation in IMR-32 cells by diethyl maleate (DEM), tBHQ, and EKODE in the presence and absence of catalase. (B) Dose–response analysis of ARE activation by EKODE in IMR-32 cells. Concentrations ≥32 μM resulted in toxicity. (C) ARE activation in primary cortical cultures derived from ARE-hPAP transgenic mice. EKODE, but not linoleic acid, activates the ARE compared with control. Results are the means ± standard deviation (n = 4).
To test whether EKODE can also activate the ARE in a more differentiated cell type, we measured ARE activity in primary, dissociated murine neuronal cortical cultures derived from ARE-hPAP transgenic mice (Figure 2C). As observed in IMR-32 cells, EKODE activated the ARE in primary cells as well, although at half the efficacy of tBHQ, (9.6- vs 18.6-fold).
3.3 EKODE-mediated activation of endogenous ARE-regulated genes and glutathione levels in IMR-32 cells
To exclude the possibility that EKODE’s ARE activity is specific to the (artificial) reporter gene assay, we evaluated its effects on levels of endogenous gene products. NAD(P)H:quinone oxidoreductase 1 (NQO1), which prevents reduction of quinones that lead to radical species formation, is known to be strongly induced by ARE activators [20]. We first assessed NQO1 transcript levels by quantitative PCR (qPCR) after reverse transcription (RT) upon treatment of IMR-32 cells with EKODE for 8 h. At 10 μM, EKODE induced NQO1 by 15.1-fold (Figure 3A) as compared with 8.7-fold for tBHQ (data not shown). A lower EKODE concentration (1 μM) was significantly less effective in increasing NQO1 mRNA levels. In contrast, mRNA levels of transcription factor NRF2 were virtually unchanged. Next, we used immunoblot analysis to measure NQO1 protein levels upon 24 h of EKODE treatment. NQO1 levels were increased in IMR-32 cells treated with 10 μM but not 1 μM EKODE (Figure 3B). Analysis of NQO1 transcript and NQO1 protein levels in IMR-32 cells upon EKODE treatment paralleled the result from the ARE-luciferase reporter assay.
Figure 3.

EKODE induces endogenous ARE regulated genes and affects GSH levels in IMR-32 cells. (A) Effect of EKODE on NQO1 transcript levels as analyzed by quantitative real-time PCR (qPCR). IMR-32 cells were treated for 8 h, total RNA was isolated, reverse-transcribed to cDNA and subjected to TaqMan analysis. Results are the relative expression means ± standard deviation (n = 3). GAPDH was used as internal control for normalization. EKODE induced NQO1 expression at 1 and 10 μM. For comparison, NRF2 transcript levels remained unchanged. (B) Effect of EKODE on NQO1 protein levels as analyzed by immunoblot analysis. IMR-32 cells were treated with EKODE for 24 h, proteins were extracted, resolved by SDS-PAGE and subjected to immunoblot analysis for NQO1 (β-actin = control). EKODE greatly increased NQO1 protein levels at 10 μM. (C) Time-dependent changes of GSH levels in IMR-32 cells upon exposure with EKODE vs. vehicle (0.5% DMSO). Cells were treated with EKODE for the indicated times, harvested and resuspended in 5% SSA solution. Upon two freeze thaw (liquid N2/37 °C) cycles and centrifugation, the supernatant served as a source of cellular glutathione. Total glutathione (GSH, GSSG) was assessed using the Glutathione Assay Kit (Sigma). Results are the means ± standard deviation (n = 2).
As pointed out above, Michael acceptors at toxic concentrations can alkylate glutathione and deplete glutathione levels [21]. However, since glutathione biosynthetic genes are regulated by the ARE, activation of the ARE should have an opposing effect, contributing to an enhanced cellular antioxidant status. We assessed the effect of EKODE on levels of glutathione in a time-dependent manner at the nontoxic concentration at which EKODE activates the ARE (10 μM). While glutathione levels were reduced by about 22% relative to vehicle control after 2 h of exposure, longer incubation times (8 h, 16 h, 24 h) led to a steady increase of glutathione concentrations with a peak at 16 h (+61%) (Figure 3C). This trend has been observed with Michael acceptors at nontoxic concentrations, an effect originally attributed to enhanced cystine uptake [21] but better-explained by enhanced glutathione synthesis.
3.4 Mechanism of ARE activation by EKODE
The major transcription factor involved in ARE activation is NRF2 [17,22]. To test if EKODE requires NRF2 to exert its ARE-activating effect, we carried out two sets of experiments. First, we depleted IMR-32 cells of NRF2 using small interfering RNAs (siRNAs) specifically targeting the NRF2 transcript and cotransfected the ARE-luciferase reporter along with control constructs for normalization (actin-lacZ) and to monitor DNA transfection efficiency (CMV-GFP). We used siRNAs at 50 nM which reduced NRF2 levels by >80% as we previously validated by dose-response analysis [17]. After 48 h, the transfected cells were treated with EKODE for another 24 h and ARE activity was measured by luminescence detection. EKODE was unable to activate the ARE reporter in NRF2-depleted cells (Figure 4A). The activity of tBHQ was also completely abolished in this system (Figure 4A). In a second experiment, we cotransfected a dominant-negative Nrf2 with the reporter plasmids; again EKODE as well as tBHQ lacked ARE activity in those cells (data not shown).
Figure 4.
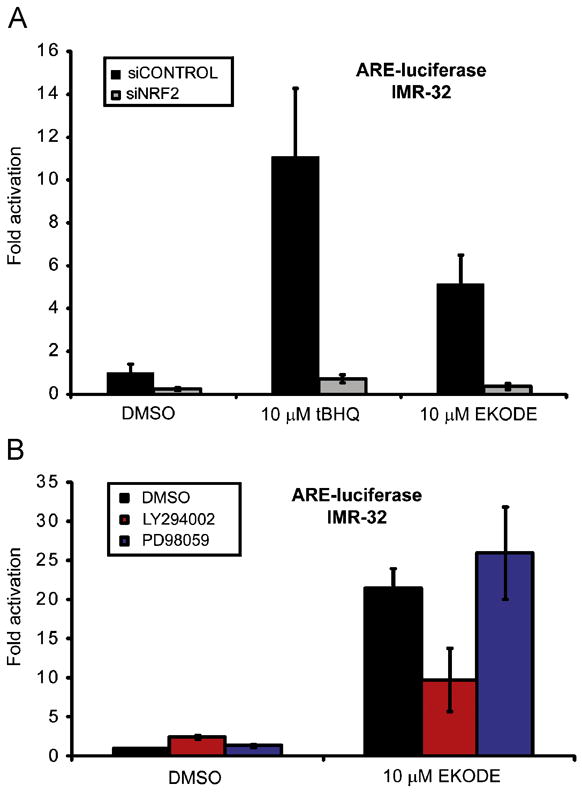
Mechanism of ARE activation in IMR-32 cells. (A) Effect of siRNAs targeting NRF2 on ARE activation by EKODE and tBHQ (10 μM each) in IMR-32 cells. The ARE-luciferase reporter was cotransfected with siRNAs targeting NRF2 or control siRNAs (50 nM), along with actin-lacZ (for normalization) and CMV-GFP (to monitor DNA transfection efficiency). After 48 h, luciferase and β-galactosidase activities were measured (n = 4). EKODE was unable to activate the ARE in IMR-32 cells depleted of NRF2. (B) Effect of PI3K and MAPK inhibitors on ARE activation by EKODE and tBHQ in IMR-32 cells. IMR-32 cells were transfected with ARE-luciferase and actin-lacZ reporters and 24 h later pre-treated with PI3K inhibitor LY294002 (25 μM) and MEK1 inhibitor PD98059 (50 μM) before EKODE addition. Inhibitor concentrations used were previously shown to be effective in this cell type. Luminescence was recorded 24 h later and normalized ARE activities are displayed. As shown, EKODE requires PI3K activity for full ARE activity, however, acts independent of the MAPK pathway. Results are the means ± standard deviation (n = 4).
Kinase mediated signaling has been implicated in ARE activation; particularly the PI3K and MAPK pathways [11,23]. To determine the signaling pathways that mediate EKODE’s ability to induce the NRF2–ARE target genes, we tested the effects of the PI3K inhibitor LY294002 and the MEK1 inhibitor PD98059 at concentrations that were previously shown to be effective in IMR-32 cells. EKODE did not fully activate the ARE reporter in cells that were pre-treated with LY294002, while PD98059 had no effect (Figure 4B).
4. Discussion
The ARE is a cis-acting enhancer element in the promoter of many phase II detoxification and antioxidant genes. The transcription factor that governs the expression of ARE-regulated genes is Nrf2, which is usually kept in the cytoplasm by the repressor protein Keap1 [22]. Keap1 contains highly reactive sulfhydryl groups and acts a cellular sensor that recognizes electrophilic inducers [24]. In response to oxidative stress, glutathione depletion, or mediators such as tert-butylhydroquinone (tBHQ), second messenger systems including PI3K promote the translocation of the transcription factor NRF2 to the nucleus, where it binds to the ARE and coordinates transcription of a collection of cytoprotective and detoxification genes including heme oxygenase-1 [25], glutathione-S-transferases (GSTs) [26], and NAD(P)H:quinone oxidoreductase 1 [27]. Induction of these enzymes by the ARE contributes to protection from a variety of toxins in a multitude of cell types [28–32].
We showed that EKODE, an endogenous product of linoleic acid oxidation, can activate this protective mechanism. Our results suggest that EKODE is a stronger ARE activator than many other conjugated enone-type Michael acceptors that result from fatty acid oxidation. This activity was not cell type specific as we could clearly show activity in human neuroblastoma cells (IMR-32) as well as in primary cortical cells. EKODE’s effect was not an artifact of the reporter assay system; EKODE also induced transcription of endogenous ARE-regulated genes: NQO1 mRNA levels were increased upon EKODE treatment concomitant with an increase of NQO1 protein levels in IMR-32 cells.
The observed increase in glutathione levels can be explained by enhanced synthesis as a result of inducing ARE-regulated genes that encode enzymes involved in glutathione biosynthesis. Partial blockade of its effects by catalase suggests that EKODE acts by a mechanism analogous to that of diethyl maleate, a pro-oxidant that depletes cellular glutathione at toxic concentrations and also increases glutathione levels at nontoxic concentration [21]. Either a small initial depletion of GSH triggers the ARE activation or, more likely, EKODE alkylates KEAP1 sulfhydryl groups(s), leading to release of NRF2, transcription factor translocation and ARE activation.
Our experiments using NRF2-specific siRNAs, a dominant-negative Nrf2 construct, and a pharmacological inhibitor of PI3K activity demonstrated that NRF2 and PI3K mediate EKODE activity. These results are consistent with the hypothesis that the reported PI3K requirement for the protective effects of linoleic acid [10] could result from ARE activation by this metabolite of linoleic acid. Both NRF2 and PI3K were recently shown to be required for ARE activation by cDNAs coding for sequestosome 1 and dipeptidylpeptidase 3, which exert protective effects upon overexpression [17].
EKODE is not the only fatty acid derivative known to activate phase II enzymes; certain members of the J2 series of prostaglandins, in particular the presumably more electrophilic 15-deoxy-Δ12,14-PGJ2, can up-regulate detoxification genes such as GST [18]. Activity of these eicosanoids is also attributed to their enone functionality which provides an electrophilic carbon for attack by nucleophiles [33]. Prostaglandin J2 (PGJ2)-based cyclopentenone derivatives of arachidonic acid have shown neuroprotective effects in various models [34,35]. PGJ2 also showed ARE activation in our reporter assay system using IMR-32 cells, but to a lesser degree than EKODE at the same concentration. At the concentration where EKODE activated the ARE without inducing toxicity, PGJ2 was toxic; however, PGJ2 at 3.2 μM was not toxic and activated the ARE to a similar extent as EKODE did at 10 μM. Circulating levels of EKODE are higher than those of the J2 prostaglandins, EKODE is found in human plasma at concentrations between 10−9 and 5 × 10−7 mol/L [36], so the relevance of linoleic acid derivatives in intact animals may exceed that of eicosanoids.
EKODE stimulates aldosterone production by rat adrenal cells [7], and aldosterone can be pathogenic, in part by contributing to oxidative stress. Stimulation of antioxidant enzyme synthesis by EKODE might lessen the damaging effects of aldosterone and other pressors by “conditioning” cells against otherwise detrimental insults. Omega-6 fatty acids are generally portrayed as contributing to pathology, but the ARE-inducing properties of the oxidized derivative of linoleic acid may partially redeem their bad reputation.
Oxidation products of common fatty acids, like the parent acids themselves, display both helpful and harmful properties. Only when synthesis of these products can be safely inhibited in vivo will we learn the different roles of the derivatives and their precursors. Meanwhile, our data show that research into the biological properties of PUFAs must include consideration of their oxidation products.
Supplementary Material
Acknowledgments
This work was supported through awards to T.L.G. from the Office of Research and Development, Biomedical Laboratory R&D Service, Department of Veterans Affairs; the American Heart Association; and NHLBI grant HL076238; and to H.L. from the American Heart Association (Scientist Development Grant), the Evelyn F. and William L. McKnight Brain Institute of the University of Florida and NCI grant R21CA133681. J.T.K. thanks the NIEHS for funding under training grant T32 ES07015 and NRSA F32 ESO13463. We thank Professor J. Johnson for the ARE reporter construct and for providing the ARE reporter mice, Dr. J. Alam for the dominant-negative Nrf2 vector and Dr. Y. Liu for technical assistance.
References
- 1.Horrobin DF, editor. Omega-6 Essential Fatty Acids: Pathophysiology and Roles in Clinical Medicine. Wiley-Liss; New York: 1990. [Google Scholar]
- 2.Hennig B, Meerarani P, Ramadass P, Watkins BA, Toborek M. Fatty acid-mediated activation of vascular endothelial cells. Metabolism. 2000;49:1006–1013. doi: 10.1053/meta.2000.7736. [DOI] [PubMed] [Google Scholar]
- 3.Fagot-Campagna A, Balkau B, Simon D, Warnet JM, Claude JR, Ducimetiere P, Eschwege E. High fatty acid concentration: an independent risk factor for hypertension in the Paris Prospective Study. Int J Epidemiol. 1998;27:808–813. doi: 10.1093/ije/27.5.808. [DOI] [PubMed] [Google Scholar]
- 4.Turpeinen AM, Basu S, Mutanen M. A high linoleic acid diet increases oxidative stress in vivo and affects nitric oxide metabolism in humans. Lipids. 1999;34(Suppl):S291–292. doi: 10.1007/BF02562321. [DOI] [PubMed] [Google Scholar]
- 5.Greene JF, Hammock BD. Toxicity of linoleic acid metabolites. In: Honn KV, Marnett LJ, Nigam S, Serham CN, Dennis EA, editors. Eicosanoids and other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury. Academic/Plenum Press; New York: 1999. pp. 471–477. [Google Scholar]
- 6.Buchanan MR. Linoleic acid metabolites in health and disease. Adv Exp Med Biol. 1999;469:463–469. doi: 10.1007/978-1-4615-4793-8_68. [DOI] [PubMed] [Google Scholar]
- 7.Goodfriend TL, Ball DL, Raff H, Bruder ED, Gardner HW, Spiteller G. Oxidized products of linoleic acid stimulate adrenal steroidogenesis. Endocr Res. 2002;28:325–330. doi: 10.1081/erc-120016804. [DOI] [PubMed] [Google Scholar]
- 8.Saraswathi V, Wu G, Toborek M, Hennig B. Linoleic acid-induced endothelial activation: role of calcium and peroxynitrite signaling. J Lipid Res. 2004;45:794–804. doi: 10.1194/jlr.M300497-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Beeharry N, Lowe JE, Hernandez AR, Chambers JA, Fucassi F, Cragg PJ, Green MH, Green IC. Linoleic acid and antioxidants protect against DNA damage and apoptosis induced by palmitic acid. Mutat Res. 2003;530:27–33. doi: 10.1016/s0027-5107(03)00134-9. [DOI] [PubMed] [Google Scholar]
- 10.Beeharry N, Chambers JA, Green IC. Fatty acid protection from palmitic acid-induced apoptosis is lost following PI3-kinase inhibition. Apoptosis. 2004;9:599–607. doi: 10.1023/B:APPT.0000038039.82506.0c. [DOI] [PubMed] [Google Scholar]
- 11.Lee JM, Hanson JM, Chu WA, Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem. 2001;276:20011–20016. doi: 10.1074/jbc.M100734200. [DOI] [PubMed] [Google Scholar]
- 12.Gardner HW, Crawford CG. Degradation of linoleic acid hydroperoxides by a cysteine • FeCl3 catalyst as a model for similar biochemical reactions. III. A novel product, trans-12,13,epoxy-11-oxo-trans-9-octadecenoic acid, from 13-L(S)-hydroperoxy-cis-9,trans-11-octadecadienoic acid. Biochim Biophys Acta. 1981;665:126–133. doi: 10.1016/0005-2760(81)90240-x. [DOI] [PubMed] [Google Scholar]
- 13.Lin D, Zhang J, Sayre LM. Synthesis of six epoxyketooctadecenoic (EKODE) isomers, their generation from nonenzymatic oxidation of linoleic acid, and their reactivity with imidazole nucleophiles. J Org Chem. 2007;72:9471–9480. doi: 10.1021/jo701373f. [DOI] [PubMed] [Google Scholar]
- 14.Moehlenkamp JD, Johnson JA. Activation of antioxidant/electrophile responsive elements in IMR-32 human neuroblastoma cells. Arch Biochem Biophys. 1999;363:98–106. doi: 10.1006/abbi.1998.1046. [DOI] [PubMed] [Google Scholar]
- 15.Lee JM, Moehlenkamp JD, Hanson JM, Johnson JA. Nrf2-dependent activation of the antioxidant responsive element by tert-butylhydroquinone is independent of oxidative stress in IMR-32 human neuroblastoma cells. Biochem Biophys Res Commun. 2001;280:286–292. doi: 10.1006/bbrc.2000.4106. [DOI] [PubMed] [Google Scholar]
- 16.Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Kern JT, Walker JR, Johnson JA, Schultz PG, Luesch H. A genomic screen for activators of the antioxidant response element. Proc Natl Acad Sci USA. 2007;104:5205–5210. doi: 10.1073/pnas.0700898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawamoto Y, Nakamura Y, Naito Y, Torii Y, Kumagai T, Osawa T, Ohigashi H, Satoh K, Imagawa M, Uchida K. Cyclopentenone prostaglandins as potential inducers of phase II detoxification enzymes. 15-Deoxy-Δ12,14-prostaglandin J2-induced expression of glutathione S-transferases. J Biol Chem. 2000;275:11291–11299. doi: 10.1074/jbc.275.15.11291. [DOI] [PubMed] [Google Scholar]
- 19.Talalay P, De Long MJ, Prochaska HJ. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci USA. 1988;85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinkova-Kostova AT, Fahey JW, Talalay P. Chemical structures of niotinamide quinine oxidoreductase I (NQO1) Methods Enzymol. 2004;382:423–448. doi: 10.1016/S0076-6879(04)82023-8. [DOI] [PubMed] [Google Scholar]
- 21.Bannai S. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J Biol Chem. 1984;259:2435–2440. [PubMed] [Google Scholar]
- 22.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 23.Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, Kong AN. Role of mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem. 1999;274:27545–27552. doi: 10.1074/jbc.274.39.27545. [DOI] [PubMed] [Google Scholar]
- 24.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang M-I, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE) Mol Med. 1995;1:827–837. [PMC free article] [PubMed] [Google Scholar]
- 26.Prestera T, Talalay P. Electrophile and antioxidant regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA. 1995;92:8965–8969. doi: 10.1073/pnas.92.19.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem. 1992;267:15097–15104. [PubMed] [Google Scholar]
- 28.Li J, Lee JM, Johnson JA. Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J Biol Chem. 2002;277:388–394. doi: 10.1074/jbc.M109380200. [DOI] [PubMed] [Google Scholar]
- 29.Kelly VP, Ellis EM, Manson MM, Chanas SA, Moffat GJ, McLeod R, Judah DJ, Neal GE, Hayes JD. Chemoprevention of aflatoxin B1 hepatocarcinogenesis by coumarin, a natural benzopyrone that is a potent inducer of aflatoxin B1-aldehyde reductase, the glutathione S-transferase A5 and P1 subunits, and NAD(P)H:quinone oxidoreductase in rat liver. Cancer Res. 2000;60:957–969. [PubMed] [Google Scholar]
- 30.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konwinski RR, Haddad R, Chun JA, Klenow S, Larson SC, Haab BB, Furge LF. Oltipraz, 3H-1,2-dithiole-3-thione, and sulforaphane induce overlapping and protective antioxidant responses in murine microglial cells. Toxicol Lett. 2004;153:343–355. doi: 10.1016/j.toxlet.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 33.Satoh T, Furuta K, Tomokiyo K, Namura S, Nakatsuka D, Sugie Y, Ishikawa Y, Hatanaka H, Suzuki M, Watanabe Y. Neurotrophic actions of novel compounds designed from cyclopentenone prostaglandins. J Neurochem. 2001;77:50–62. doi: 10.1046/j.1471-4159.2001.t01-1-00229.x. [DOI] [PubMed] [Google Scholar]
- 34.Satoh T, Baba M, Nakatsuka D, Ishikawa Y, Aburatani H, Furuta K, Ishikawa T, Hatanaka H, Suzuki M, Watanabe Y. Role of heme oxygenase-1 protein in the neuroprotective effects of cyclopentenone prostaglandin derivatives under oxidative stress. Eur J Neurosci. 2003;17:2249–2255. doi: 10.1046/j.1460-9568.2003.02688.x. [DOI] [PubMed] [Google Scholar]
- 35.Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proc Natl Acad Sci USA. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43:358–363. doi: 10.1161/01.HYP.0000113294.06704.64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.