

Abstract
We have designed prodrugs that release NO on metabolism by Glutathione S-Transferases (GST). This design exploits the upregulation of GST in Acute Myeloid Leukemia (AML) cells. O2-(2,4-Dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl] diazen-1-ium-1,2-diolate (JS-K, a member of this class) has potent antileukemic activity. HL-60 myeloid leukemia cells were used for in vitro studies of the combination of JS-K with Daunorubicin (DAUNO), Cytarabine (ARA-C) or Etoposide (ETOP) using the median effect method to determine synergistic, antagonistic, or additive effects. Combinations of JS-K added simultaneously, 2 hours before or 2 hours after the other compounds were used. JS-K and DAUNO were antagonistic in all 3 drug sequences. JS-K and ETOP were also antagonistic but to a lesser degree. JS-K and ARA-C showed strong synergy. The combination index at the 50% fraction affected was 0.37 ± 0.23, 0.24 ± 0.27, and 0.15 ± 0.11 for simultaneous, JS-K first and ARA-C first additions, respectively. JS-K by itself induced DNA strand breaks at relatively high concentrations. However, at submicromolar concentrations, it significantly augmented ARA-C-induced DNA strand breaks. NMR spectroscopy revealed no evidence of chemical interaction between JS-K and the other chemotherapeutic agents. We conclude that ARA-C and JS-K have synergistic anti-leukemic activity and warrant further exploration in combination.
Introduction
The in vitro anti-leukemic activity of NO is well established (1-3). The main obstacle to the use of NO in vivo for the treatment of malignancies is its induction of vasodilatation with resultant hypotension (4). A solution to this problem is the use of enzyme-activated drugs that target NO delivery to leukemia cells. In an effort to use NO-based therapy for the treatment of Acute Myeloid Leukemia (AML), we have developed a class of such enzymatically activated NO-generating pro-drugs. These agents are built from the conjugation of an O2-aryl ring with a diazeniumdiolate moiety (Figure 1A). This design creates pro-drugs that hydrolyze only slowly to release NO spontaneously in solution. However, upon nucleophilic attack by free thiols (particularly glutathione), the diazeniumdiolate moiety is liberated and releases NO (5). The reaction is catalyzed by the glutathione S-transferases (GST) (Figure 1A) (5). This design exploits the upregulation of GST in AML cells (6,7), thus allowing a relatively selective delivery of NO to the malignant cells. Consequently, this drug design decreases side effects associated with systemic NO release.
Figure 1. Synthesis and metabolic activation of GST-activated NO donors.

A- Upon GST metabolism, the compounds are converted to the free anionic diazeniumdiolate moiety which spontaneously releases NO at physiologic pH. X = F or Cl-; EWG = Electron Withdrawing Group. B- Structure of JS-K.
After screening a library of arylated diazeniumdiolates and extensive lead optimization (8), we have identified O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl] diazen-1-ium-1,2-diolate, or JS-K (Figure 1B), as the most active anti-leukemic agent of this class of drugs (5,8). JS-K has potent anti-leukemic activity in vitro and in vivo. Its in vitro 50% growth inhibitory concentration (IC50) towards human myeloid leukemia HL-60 cells is 0.2 – 0.5 μM (5). In a subcutaneous leukemia xenograft model in NOD/SCID mice, JS-K inhibited HL-60 cell growth by more than 50% and induced extensive leukemia cell death in vivo (5). Importantly, at therapeutic doses, it did not induce hypotension, indicating delivery of NO to AML cells while circumventing the main side effect associated with NO-based therapy. Thus, JS-K establishes a new class of anti-leukemic agents.
Because of its unique mechanism of action, JS-K could potentially have synergistic interactions with anti-leukemic agents currently available in the clinic. Identifying such interactions will lead to the development of new drug combinations with significant impact on disease outcome. We therefore undertook the current study to determine the effect on leukemic cell growth of the combination of JS-K with other anti-leukemic agents, namely daunorubicin (DAUNO), etoposide (ETOP) and cytarabine (ARA-C). Results show strong synergy between JS-K and ARA-C.
Methods
Cells and culture conditions
HL-60 cells were from ATCC (Manassas, VA) and were cultured (1 × 105/ml) in RPMI-1640 media supplemented with 10% Fetal Bovine Serum at 37°C in a 5% CO2 humidified atmosphere.
Chemicals and drug treatments
JS-K was synthesized as previously described (9). DAUNO, ETOP and ARA-C were from Sigma (St. Louis, MO). In different experiments, JS-K was added simultaneously, 2 hours before or 2 hours after each one of the other agents. Three days after addition of the drugs to the cultures, cell viability was determined with the CellTiter 96 assay from Promega (Madison, WI) using the manufacturer's protocol.
Analysis of drug interactions
We used the Median Effect method of Chou and Talalay in order to analyze interactions between JS-K and the other chemotherapeutic agents (10). After generating growth curves with each agent individually, drugs were added to the cultures in increasing amounts while keeping the ratio of their concentrations constant. Analysis, modeling, and Combination Index (CI) calculations were done using the CalcuSyn software from Biosoft (Ferguson, MO). A CI equal to, smaller than, or greater than 1 indicates additive, synergistic or antagonistic interactions, respectively. The fraction affected (fa) was defined as the ratio of the cell growth of drug-treated cells to untreated controls. Experiments were set-up in triplicate.
Comet assay for DNA strand breaks
The alkaline comet assay for DNA strand breaks was performed using a modification of the method of Singh (11). HL-60 cells were treated with JS-K and/or ARA-C at the indicated concentrations for 24 hours. HL-60 cells were then harvested and mixed 1:10 with 0.8% low melting point agarose and layered onto comet slides (Trevigen, Gaithersburg, MD). The slides were placed in a cold lysing solution (Trevigen) and lysed for 1 hour at 4°C in the dark. After 30 min in alkaline solution (300 mM NaOH, 1 mM Na2EDTA, pH 12.1) for DNA unwinding, the slides were subjected to electrophoresis at 1 V/cm for 20 min. They were rinsed with water, dehydrated with 70 % ethanol and air dried, then stained with SYBR Gold (1:10,000, Invitrogen Corporation, Carlsbad, CA). Comets were observed with a Zeiss Axiovert 200 M fluorescence microscope and images were captured with an AxioCam Mrm digital camera. Typically, 50 sequentially observed cells were analyzed per sample. The comet tail intensities were quantified using the Labworks 4.0 software (UVP, Upland, CA). Results were expressed as average comet tail intensities (arbitrary units) ± SE. Statistical tests were carried out using GraphPad Instat version 3.00 (GraphPad Software, San Diego, CA).
Studies of the chemical interactions
In order to determine if there was a chemical interaction between JS-K and ARA-C, DAUNO, or ETOP, NMR experiments were designed to assess the stability of these combinations in dimethylsulfoxide-d6 (DMSO-d6) solutions. In these experiments, JS-K was mixed in DMSO-d6 with either ARA-C, DAUNO, or ETOP at the indicated concentrations and for the indicated periods of time. NMR spectroscopy was then used to identify any evidence of degradation or chemical change in the respective agents. The spectra were recorded on a 400 MHz Varian UNITY INOVA spectrometer; chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane as the internal standard.
Results
Drug interaction studies
Figure 2 shows the growth curves for each agent added alone to HL-60 cells. The IC50 for DAUNO, ARA-C, ETOP, and JS-K alone were 0.07, 0.18, 0.79, and 0.30 μM, respectively. This shows that JS-K compares favorably in its anti-leukemic activity with other established chemotherapeutic agents. For each drug combination, the concentration ratio chosen was an approximation of the ratio of their IC50's. JS-K was studied in combination with DAUNO at a fixed DAUNO:JS-K concentration ratio of 1:5. JS-K and DAUNO were antagonistic in all 3 drug addition sequences. The Combination Index (CI) at the 50% fraction affected (fa) level was 12.5 ± 1.6, 10.7 ± 0.41, and 7.2 ± 0.46 (average ± standard error of the mean) for simultaneous, JS-K first and DAUNO first additions, respectively (Table 1). JS-K and ETOP were studied at a fixed concentration ratio of ETOP:JS-K of 2:1. Again, JS-K and ETOP were antagonistic in all 3 drug addition sequences, but to a lesser extent than the combination of DAUNO and JS-K. The CI at the 50% fa level was 3.03 ± 0.26, 2.2 ± 0.33, and 1.7 ± 0.08 for simultaneous, JS-K first and ETOP first additions, respectively (Table 1). JS-K and ARA-C were studied at a fixed concentration ratio of ARA-C:JS-K of 1:1 and showed strong synergy in all 3 drug addition sequences. When JS-K and ARA-C were added simultaneously, the CI at the 50% fa level was 0.37 ± 0.23 (Table 1). The CI at the 50% fa level when JS-K was added 2 hours before ARA-C was 0.24 ± 0.27 (Table 1). The strongest synergy was observed when ARA-C was added to cultures 2 hours before JS-K, reflected by a CI at the 50% fa level of 0.15 ± 0.11 (Table 1). Figure 3 shows the growth curves for the combination of JS-K and ARA-C when the latter was added to the cultures 2 hours before the former.
Figure 2. Comparison of the in vitro anti-leukemic activity of different agents.
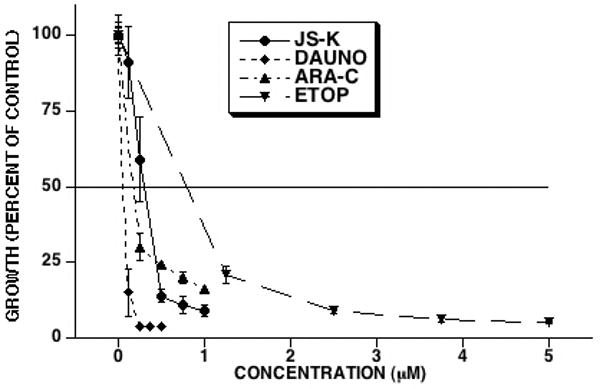
HL-60 cells were cultured with JS-K, DAUNO, ETOP, or ARA-C at the indicated concentrations. Three days after culture initiation cell growth was determined. JS-K inhibited leukemia cell growth with an IC50 of 0.30 μM, comparing favorably with other chemotherapeutic agents (averages and SEM of 3 separate experiments).
Table 1. CI Values for the Combination of JS-K with Each Chemotherapeutic Agent (Average ± SEM of 3 different experiments).
| Compound | Simultaneous addition | JS-K added first | JS-K added second |
|---|---|---|---|
| DAUNO | 12.5 ± 1.6 | 10.7 ± 0.41 | 7.2 ± 0.46 |
| ETOP | 3.03 ± 0.26 | 2.2 ± 0.33 | 1.7 ± 0.082 |
| ARA-C | 0.37 ± 0.23 | 0.24 ± 0.27 | 0.15 ± 0.11 |
Figure 3. Effect of the combination of JS-K and ARA-C on leukemia cell growth in vitro.
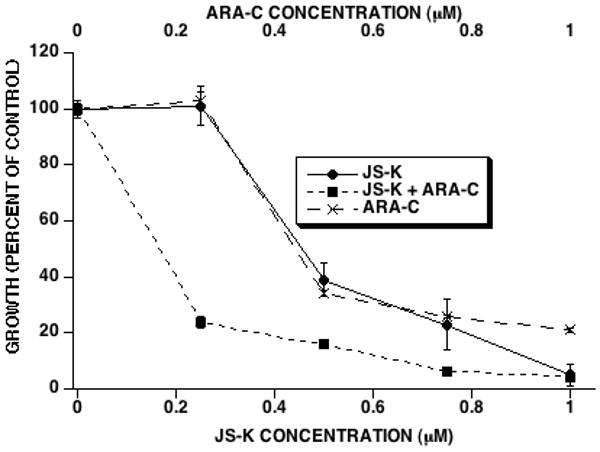
HL-60 cells were cultured with ARA-C and JS-K (JS-K added 2 hours after ARA-C) at a fixed concentration ratio of 1:1 for 3 days whereupon cell growth was determined. JS-K and ARA-C showed synergy in inhibiting HL-60 cell growth (averages and SEM of 3 separate experiments).
Alkaline comet assay
JS-K induces DNA strand breaks in multiple myeloma cells (12). We therefore sought to determine if it had the same effect on leukemia cells alone or in combination with ARA-C. At a concentration of 0.5 μM, JS-K alone did not induce significant DNA strand break damage (Figure 4). However, at a concentration of 10 μM, there was evidence of extensive DNA damage probably resulting from high levels of nitric oxide release (not shown). ARA-C alone induced significant DNA strand break damage at concentrations of 0.125 and 0.250 μM (P<0.001; Figure 4). We then studied the effect of the combination of JS-K and ARA-C on DNA strand breaks in HL-60 cells. In these experiments, both agents were added simultaneously at a 1:1 concentration ratio to HL-60 cells for 24 hours, at which time DNA strand break analysis was done. This concentration ratio was chosen because it was the ratio we used to study JS-K and ARA-C interactions in our previous experiments. There was significant and dose-dependent induction of DNA strand breaks by the drug combination (Figure 4). At concentrations of 0.125 or 0.250 μM of each drug, the combination of JS-K and ARA-C induced significantly higher levels of DNA damage than at concentrations of 0.125 or 0.250 μM ARA-C alone (P<0.001; Figure 4).
Figure 4. Comet assay to study the effect of JS-K and ARA-C on HL-60 cell DNA.

A- HL-60 cells were treated with the indicated concentrations of each agent for 24 hours whereupon the comet assay for DNA strand breaks was conducted as described in the text. At a concentration of 0.5 μM, JS-K by itself did not induce significant DNA damage. ARA-C induced DNA damage at submicromolar concentrations. B- JS-K and ARA-C were added simultaneously to HL-60 cells at the indicated concentrations of each drug. After 24 hours, the comet assay was conducted as detailed in the text. JS-K significantly augmented DNA damage induced by ARA-C (images are representative of 3 independent experiments). C – DNA single-strand breaks were quantified on the basis of fluorescence intensity of the single-cell comet tails after gel electrophoresis. Results are expressed as average ± SE of 50 comets per sample and are representative of 3 separate experiments. Cells treated with ARA-C alone had significantly (P<0.001) greater level of DNA damage compared with untreated controls. Lower concentrations of JS-K and ARA-C combined resulted in significantly (P<0.001) more DNA strand break damage than higher doses of ARA-C alone.
Chemical interaction studies
One possible explanation for the observed effects of the drug combinations used is chemical modification of each individual agent by the other. In order to determine if that was the case, NMR spectroscopy was used to determine possible chemical interactions between JS-K and the three other chemotherapeutic agents. We started by studying the interaction between JS-K and ARA-C. In these experiments, ARA-C was dissolved in DMSO-d6 containing an equimolar amount of JS-K. The solution was then allowed to stand at room temperature protected from light for 4 hours, whereupon the 1H NMR spectrum was recorded. Sixty-eight hours later, the spectrum was re-recorded. The aromatic regions of the two spectra, shown in Figure 5, are identical, with no new peaks having appeared during that time, nor was there any loss in intensity of the peaks observed. As the spectra demonstrate, there was no evidence of chemical interaction between the two drugs, nor any sign of decomposition in the solution. Similar experiments were carried out with JS-K and ETOP. A solution of ETOP and JS-K in DMSO-d6 was incubated for 4 days at room temperature. Even after this extended period, no interaction or decomposition was observed (data not shown). Likewise, similar experiments conducted between DAUNO and JS-K showed no interaction or decomposition after a 24-hour incubation at room temperature (data not shown).
Figure 5. Analysis of potential chemical interaction between JS-K and ARA-C.

NMR spectroscopy experiments were conducted as detailed in the text in order to determine whether there was any chemical interaction between JS-K and ARA-C. A- NMR spectrum of JS-K and ARA-C incubated together for 4 hours. B- NMR spectrum of the same solution after incubation at room temperature for 72 hours.
Discussion
We show in this study that JS-K and ARA-C have significant synergistic anti-leukemic activity in vitro when used in combination. On the other hand, JS-K seemed to be antagonistic with DAUNO and ETOP. JS-K is unique in that it belongs to a totally new class of chemotherapy agents. Besides delivery of a toxin (NO) to leukemia cells, it is likely to affect cell growth by modifying proteins through arylation reactions at critical thiol groups. Our previous work suggests that arylation pathways do indeed contribute to JS-K's antileukemic activity (8). Furthermore, work done with the JS-K analog O2-[2,4-dinitro-5-[4-(N-methylamino)benzoyloxy] phenyl] 1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate or PABA/NO shows that protein glutathionylation also plays a role in the antineoplastic activity of this agent (13). However, this has not yet been established with JS-K. JS-K was also shown to trigger apoptosis in leukemia and multiple myeloma cells through activation of both the intrinsic and extrinsic apoptosis pathways (12,14). Additionally, JS-K was shown to induce DNA strand breaks in multiple myeloma cells (12). Interestingly, JS-K acted synergistically with the proteasome inhibitor bortezomib to inhibit multiple myeloma cell growth (12).
The reason for the drug interactions we observed is unknown. Clearly, the 4 agents under study here have very different mechanisms of action. Chemical reaction between JS-K and DAUNO or ETOP is unlikely to explain the observed antagonism since results were similar if the drugs were added to cultures simultaneously or sequentially. This is confirmed by the NMR spectroscopy experiments that we conducted and showed no evidence of interaction between JS-K and either drug, even after extended periods of incubation.
The observed synergy between ARA-C and JS-K is quite interesting. Here too, chemical interaction between the 2 agents does not seem to occur and therefore would not explain the observed results. ARA-C is metabolized intracellularly in 3 successive phosphorylation reactions to yield the triphosphate derivative ARA-CTP (15). The latter prevents DNA synthesis by inhibiting DNA polymerases and, through misincorporation into the DNA molecule, causes premature chain termination. Unlike multiple myeloma cells, JS-K induced DNA strand breaks in HL-60 cells only when added at the relatively high concentration of 10 μM. However, at submicromolar concentrations, it clearly augmented DNA damage induced by ARA-C. This may explain the strong synergy observed between the two compounds. The mechanism behind this synergistic interaction is not clear at this point. However, one could speculate about a potential role for NO released by JS-K. Indeed, NO has been shown to induce DNA strand breaks (16). Furthermore, NO inhibits several proteins that play critical roles in DNA repair. These include O6-methylguanine-DNA-methyltransferase (17), poly(ADP-RIBOSE) polymerase (18), DNA ligase (19), and Fapy-DNA glycosylase (20). It is also possible that JS-K induces arylation (and therefore inhibition) of enzymes involved in DNA repair. Consequently, the observed synergy may be due to the induction of DNA damage by ARA-C with concurrent inhibition of DNA repair enzymes by JS-K. More work is needed to further define the mechanism of synergy between the two drugs.
In conclusion, JS-K is a promising new anti-leukemic agent with great potential for clinical development. The combination of ARA-C and JS-K is likely to be very active for the treatment of AML and therefore warrants further exploration.
Acknowledgments
Financial support: Translational Research Award from the Leukemia and Lymphoma Society and NIH Grants R01 CA84496 and RO1 CA129611 (P.J.S). National Cancer Institute contract No. N01-CO-12400. Supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Author contributions: Paul J. Shami: designed research, analyzed and interpreted data, wrote manuscript.
Anna E. Maciag: performed research, reviewed and edited manuscript.
Jordan K. Eddington: performed research.
Vidya Udupi: performed research.
Ken M. Kosak: performed research.
Joseph E. Saavedra: designed, synthesized and provided JS-K. Performed research. Reviewed and edited manuscript.
Larry K. Keefer: designed JS-K. Reviewed and edited manuscript.
Conflict of interest: Paul J. Shami is a founder of and holds stock in JSK Therapeutics Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Magrinat G, Mason SN, Shami PJ, Weinberg JB. Nitric oxide modulation of human leukemia cell differentiation and gene expression. Blood. 1992;80:1880–1884. [PubMed] [Google Scholar]
- 2.Shami PJ, Moore JO, Gockerman JP, Hathorn JW, Misukonis MA, Weinberg JB. Nitric oxide modulation of the growth and differentiation of freshly isolated acute non-lymphocytic leukemia cells. Leuk Res. 1995;19:527–533. doi: 10.1016/0145-2126(95)00013-e. [DOI] [PubMed] [Google Scholar]
- 3.Shami PJ, Sauls DL, Weinberg JB. Schedule and concentration-dependent induction of apoptosis in leukemia cells by nitric oxide. Leukemia. 1998;12:1461–1466. doi: 10.1038/sj.leu.2401131. [DOI] [PubMed] [Google Scholar]
- 4.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 5.Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK. JS-K, a Glutathione S-transferase-activated nitric oxide donor with potent anti-neoplastic activity. Mol Cancer Ther. 2003;2:409–417. [PubMed] [Google Scholar]
- 6.Baines P, Cumber P, Padua RA. Multidrug resistance in leukaemia. Baillieres in Clin Hematol. 1992;5:943–960. doi: 10.1016/s0950-3536(11)80053-3. [DOI] [PubMed] [Google Scholar]
- 7.Sargent JM, Williamson C, Hall AG, Elgie AW, Taylor CG. Evidence for the involvement of the glutathione pathway in drug resistance in AML. Adv Exp Med Biol. 1999;457:205–209. doi: 10.1007/978-1-4615-4811-9_22. [DOI] [PubMed] [Google Scholar]
- 8.Shami PJ, Saavedra JE, Bonifant CL, Chu J, Udupi V, Malaviya S, Carr BI, Kar S, Wang M, Jia L, Ji X, Keefer LK. Antitumor activity of JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl] diazen-1-ium-1,2-diolate] and related O2-aryl diazeniumdiolates in vitro and in vivo. J Med Chem. 2006;49(14):4356–4366. doi: 10.1021/jm060022h. [DOI] [PubMed] [Google Scholar]
- 9.Saavedra JE, Srinivasan A, Bonifant CL, Chu J, Shanklin AP, Flippen-Anderson JL, Rice WG, Turpin JA, Davies KM, Keefer LK. The secondary amine/nitric oxide complex ion R2N[N(O)NO] - as nucleophile and leaving group in SNAr reactions. J Org Chem. 2001;66:3090–3098. doi: 10.1021/jo0016529. [DOI] [PubMed] [Google Scholar]
- 10.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enz Reg. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 11.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 12.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. JS-K, a GST-activated nitric oxide generator, induces DNA double strand breaks, activates DNA damage response pathways, and induces apoptosis in human multiple myeloma cells. Blood. 2007;110:709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Townsend DM, Findlay VJ, Fazilev F, Ogle M, Fraser J, Saavedra JE, Ji X, Keefer LK, Tew KD. A glutathione S-transferase π-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69:501–508. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Udupi V, Yu M, Malaviya S, Saavedra JE, Shami PJ. JS-K, a nitric oxide prodrug, induces cytochrome c release and caspase activation in HL-60 myeloid leukemia cells. Leuk Res. 2006;30:1279–1283. doi: 10.1016/j.leukres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Chu E, Mota AC, Fogarasi MC. Antimetabolites. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Cancer Principles and Practice of Oncology. 6th. New York, NY: Lippincott Williams and Wilkins; 2001. pp. 388–415. [Google Scholar]
- 16.Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Scie USA. 1992;89:3030–3034. doi: 10.1073/pnas.89.7.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laval F, Wink DA. Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA-methyltransferase. Carcinogenesis. 1994;15:443–7. doi: 10.1093/carcin/15.3.443. [DOI] [PubMed] [Google Scholar]
- 18.Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Inhibition of poly(ADP-ribose) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Radic Biol Med. 2003;35:1431–8. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Graziewicz M, Wink DA, Laval F. Nitric oxide inhibits DNA ligase activity: potential mechanisms of NO-mediated DNA damage. Carcinogenesis. 1996;17:2501–5. doi: 10.1093/carcin/17.11.2501. [DOI] [PubMed] [Google Scholar]
- 20.Wink DA, Laval J. The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis. 1994;15:2125–9. doi: 10.1093/carcin/15.10.2125. [DOI] [PubMed] [Google Scholar]