

Abstract
Recent RNA interference screens have identified several proteins that are essential for store-operated Ca2+ influx and Ca2+ release-activated Ca2+ (CRAC) channel activity in Drosophila and in mammals, including the transmembrane proteins Stim (stromal interaction molecule)1,2 and Orai3–5. Stim probably functions as a sensor of luminal Ca2+ content and triggers activation of CRAC channels in the surface membrane after Ca2+ store depletion1,6. Among three human homologues of Orai (also known as olf186-F), ORAI1 on chromosome 12 was found to be mutated in patients with severe combined immunodeficiency disease, and expression of wild-type Orai1 restored Ca2+ influx and CRAC channel activity in patient T cells3. The overexpression of Stim and Orai together markedly increases CRAC current5,7–9. However, it is not yet clear whether Stim or Orai actually forms the CRAC channel, or whether their expression simply limits CRAC channel activity mediated by a different channel-forming subunit. Here we show that interaction between wild-type Stim and Orai, assessed by co-immunoprecipitation, is greatly enhanced after treatment with thapsigargin to induce Ca2+ store depletion. By site-directed mutagenesis, we show that a point mutation from glutamate to aspartate at position 180 in the conserved S1–S2 loop of Orai transforms the ion selectivity properties of CRAC current from being Ca2+-selective with inward rectification to being selective for monovalent cations and outwardly rectifying. A charge-neutralizing mutation at the same position (glutamate to alanine) acts as a dominant-negative non-conducting subunit. Other charge-neutralizing mutants in the same loop express large inwardly rectifying CRAC current, and two of these exhibit reduced sensitivity to the channel blocker Gd3+. These results indicate that Orai itself forms the Ca2+-selectivity filter of the CRAC channel.
Orai and Orai1 possess four hydrophobic stretches that are predicted to span the membrane. On the basis of strategies used previously for several different ion channels, we made point mutations to investigate the most conserved loop between putative transmembrane segments (Supplementary Fig. 1a) and examined properties of ion selectivity, current–voltage rectification and block that are intimately associated with a pore-forming subunit. Wild-type or mutant Orai proteins were overexpressed together with wild-type Stim in S2 cells; messenger RNA and protein expression were verified by RT–PCR and western blotting (Fig. 1a and Supplementary Fig. 1b). In addition, by co-immunoprecipitation of the epitope-tagged wild-type proteins, we evaluated whether Stim and Orai are associated with each other before and after Ca2+ store depletion. When cells were cultured without stimulation, only limited interaction was seen. However, Ca2+ store depletion triggered by thapsigargin, which inhibits the SERCA (sarco/endoplasmic reticulum Ca2+-ATPase) re-uptake pump, markedly increased Stim–Orai protein interaction when either protein was used to pull down the other by co-immunoprecipitation (Fig. 1a). This result shows that both transfected proteins are expressed and, moreover, that they interact, either directly or as part of a complex, after Ca2+ store depletion to initiate CRAC channel activation. During whole-cell recording on the co-transfected cells, dialysis of a Ca2+ chelator (BAPTA) into the cytoplasm activated a very large Ca2+-selective inward current, as shown previously5. Using a pipette solution that maintains normal Ca2+ store content to prevent spontaneous channel opening, we confirmed that addition of inositol-1,4,5-trisphosphate (IP3) inside or thapsigargin (TG) outside resulted in greatly augmented CRAC current (Fig. 1b).
Figure 1. Orai interacts with Stim and generates increased store-operated currents in S2 cells.
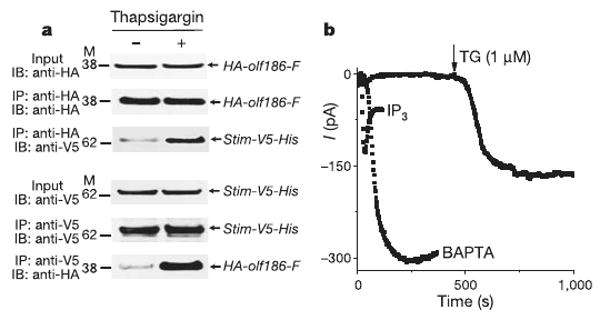
a, Representative co-immunoprecipitation of Stim and Orai (n = 3). Input: total cell lysate, showing an equal amount of samples prepared for immunoprecipitation. Cells were transfected with HA-olf186-F (Orai) and Stim-V5-His (Stim) constructs, as indicated. M refers to molecular weight markers (kDa). b, Activation of CRAC current (at −130 mV) by three methods: IP3 (10 μM IP3 added to high-Ca2+ pipette solution; current density −11.4 ± 3.1 pA pF−1, n = 5); thapsigargin (high-Ca2+ pipette solution and thapsigargin added to bath; −7.6 ± 2.5 pA pF−1, n = 2); or passive store-depletion (Ca2+-free pipette solution; −28.7 ± 1.8 pA pF−1, n = 27). All three methods produced substantially greater CRAC current density than passive store depletion in control cells (−2.5 ± 0.4 pA pF−1, n = 18).
Expression of an Orai mutant with a glutamate-to-aspartate mutation at position 180 (Orai(E180D)), again together with wild-type Stim, generated a current that developed with the normal kinetics of CRAC current during passive Ca2+ store depletion, but also exhibited marked alterations in ion selectivity and rectification. In contrast to the normal inward CRAC current, Orai(E180D) expression produced a very large outward current and a smaller inward current that developed in parallel (Fig. 2a). Current–voltage (I–V) curves of the Orai(E180D)-induced current revealed an outwardly rectifying shape with a reversal potential of 6.0 ± 0.4 mV (n = 14 cells), in contrast to the characteristic inwardly rectifying I–V curves with a reversal potential >50 mV of native or wild-type Orai-induced CRAC current (Fig. 2b).
Figure 2. Mutation E180D of Orai alters ion selectivity of the CRAC current.
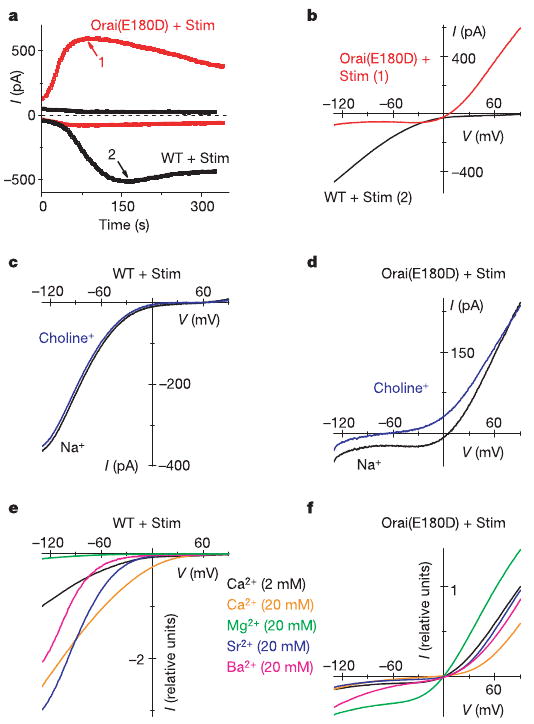
a, Time courses of inward current at −130 mV and outward current at 90 mV in representative cells overexpressing wild-type Orai (WT, black) or Orai(E180D) mutant (red). Ca2+-free internal solution. b, I–V curves at times indicated in a. c, I–V curves of wild-type Orai-induced current in Ca2 solution containing 160 mM Na+ and 2 mM Ca2+ and in choline external solution with 1.1 mM Na+ and 2 mM Ca2+. (Complete solution recipes are indicated in Supplementary Table 1.) d, Representative I–V curves for Orai(E180D) with the same solutions as c (n = 3). e, Divalent cation selectivity of wild-type Orai CRAC current. I–V curves are normalized to current values at −130 mV in Ca2+ solution. Test solutions contained 20 mM test divalent and 124 mM Na+. External solutions for e and f are labelled according to colour. f, I–V curves (not leak-subtracted) for Orai(E180D) with the same divalent cations, normalized to currents at 90 mV in Ca2+ solution.
In a series of ion-substitution experiments, we evaluated the ion selectivity properties of current induced by Orai(E180D). Substitution of Na+ by choline while maintaining constant Ca2+ in the external solution—an experimental procedure that has little effect on native CRAC current10 or CRAC current with overexpressed wild-type Orai (Fig. 2c)—reduced the inward current and shifted the reversal potential to hyperpolarized potentials (Fig. 2d), indicating that the Orai(E180D)-induced inward current is carried predominantly by Na+ rather than by Ca2+. Next, different divalent cation species in the external solution were tested, resulting in a I–V pattern of wild-type Orai-induced CRAC current similar to native CRAC current described previously10. Ca2+, Sr2+ and Ba2+(20 mM in each case, while maintaining high external Na+) resulted in increased inward current (Fig. 2e), indicating that each of these divalent cations is permeant, whereas Mg2+ is relatively impermeant. In contrast, representative I–V curves of Orai(E180D)-induced current (shown in Fig. 2f with the same colour code for comparison with Fig. 2e) reveal an entirely different pattern. A tenfold increase of Ca2+ concentration ([Ca2+]) in the external solution significantly decreased the outward current and did little to the small inward current. Substitution of 2 mM external Ca2+ by 20 mM Sr2+ had little effect; Mg2+ increased the current; and Ba2+ significantly increased the inward current while decreasing the outward current (Supplementary Fig. 2a). These results indicate that divalent cations block the outward current (carried by Cs+) in the potency sequence Ca2+ > Ba2+ > Sr2+ > Mg2+, and the inward current (carried by Na+) in the potency sequence Ca2+ ≈ Sr2+ > Mg2+ > Ba2+. Together, these ion-substitution experiments indicate that the Orai(E180D) mutation transforms the CRAC channel from being selective for divalent cations to being a monovalent-selective channel that selects against, and indeed is blocked by, divalent cations.
CRAC channels share with voltage-gated Ca2+ channels the property of monovalent ion permeation when external divalent ions are removed11–15. Upon withdrawal of external divalent cations, Na+ carries significant current through CRAC channels, whereas Cs+ is far less permeant10,14,16,17. The monovalent CRAC current normally declines within 10 s, a process termed depotentiation15 that is due to removal of external Ca2+. To check whether the Orai(E180D) mutation alters monovalent CRAC current, both wild-type- and Orai(E180D)-induced CRAC currents were recorded in divalent-free Na+ and Cs+ test solutions. Three clear differences can be discerned (Fig. 3). First, the inward Na+ current was much larger in the Orai(E180D) mutant and did not depotentiate to the same extent as either wild-type Orai (Fig. 3a, b) or native CRAC current in S2 cells10. Second, in contrast to the relatively small Cs+ current density in wild-type Orai (−1.6 ± 0.6 pA pF−1, n = 4), Orai(E180D)-induced Cs+ current density was much larger (−191 ± 46 pA pF−1, n = 4), similar in amplitude to the Na+ current (−238 ± 43 pA pF−1, n = 9), and the large Cs+ current did not depotentiate (Fig. 3b). Third, the measured reversal potential in Na+external solution was 7.9 ± 0.3 mV (n = 9) for Orai(E180D), whereas it was 52.4 ± 0.9 mV (n = 5) for wild-type Orai (Fig. 3c, d). Calculated from reversal potentials, the average permeability ratio PNa/PCs changed from 8.0 for wild-type Orai to 1.4 as a result of the E180D mutation, indicating an increase in pore diameter or altered electrostatic interactions with cations. I–V curves for Orai(E180D)-induced current changed shape with varying external [Ca2+] (Fig. 3e), revealing voltage-dependent block (Fig. 3f) that causes outward rectification in physiological saline. This examination of ion selectivity properties shows that a conservative mutation of glutamate to aspartate at position 180 of Orai transforms the CRAC channel from being Ca2+-selective and inwardly rectifying to one that conducts Na+ or Cs+ and is outwardly rectifying due to voltage-dependent Ca2+ block.
Figure 3. Monovalent current in the absence of divalent ions exhibits altered ion selectivity in the Orai(E180D) mutant.
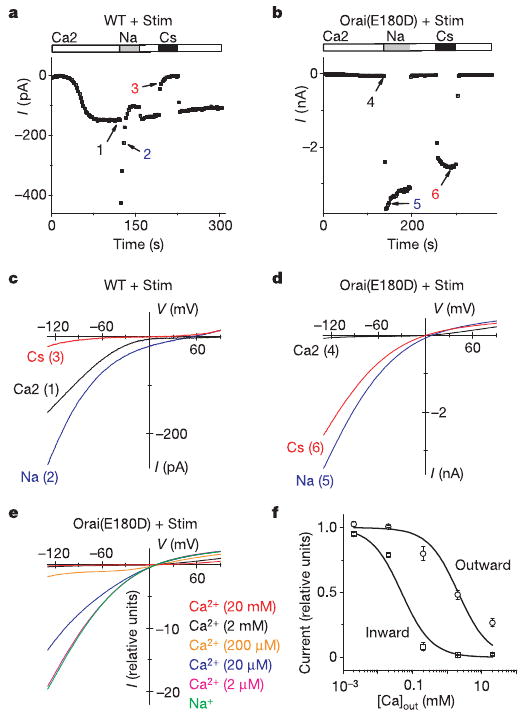
a, Time course of inward current in a cell expressing wild-type Orai. Bars indicate external solution exchange. b, Same as a but for Orai(E180D). c, I–V curves for wild-type Orai at times indicated in a. d, I–V curves for Orai(E180D) at times indicated in b. e, I–V curves of Orai(E180D)-induced current in the presence of varying external [Ca2+]. f, [Ca2+] dependence of Orai(E180D) CRAC current at −130 (squares) and 90 mV (circles), scaled to the current in divalent-free solution. Ca2+-dependent block (mean ± s.e.m.) was fitted by the function y = 1/(1 + [Ca2+]/IC50), where IC50 is the calculated half-blocking Ca2+ concentration for inward current at −130 mV (48 ± 13 μM; n = 13, 8, 4, 6 and 2 cells for 20, 2, 0.2, 0.02 and 0.002 mM free external [Ca2+], respectively) and outward current at 90 mV (2.05 ± 0.78 mM; n = 13, 8, 4, 5 and 3 cells for 20, 2, 0.2, 0.02 and 0.002 mM free external [Ca2+], respectively).
Expression of the charge-neutralizing Orai(E180A) mutant completely abolished native CRAC current (Fig. 4a), indicating that alanine-containing subunits prevent ion conduction in hetero-multimeric CRAC channels by a dominant-negative action. In contrast, expression of Orai(D184A), Orai(D186A) or Orai(N188A) mutants with Stim resulted in increased CRAC current with unaltered I–V shape (Fig. 4b) and normal monovalent Na+ current that exhibited depotentiation and low Cs+ permeability (data not shown). Figure 4c summarizes the effect of each Orai mutant tested on CRAC current density. Expressed by itself or together with Stim, only Orai(E180A) potently inhibited native CRAC current. The inward current density and I–V shape in cells expressing Orai(D184A), Orai(D186A) and Orai(N188A) mutants did not differ significantly from current observed in cells expressing wild-type Orai. Orai(E180D) was unique in producing very large outward currents as a result of altered ion selectivity.
Figure 4. Charge-neutralizing mutations in the S1–S2 loop.
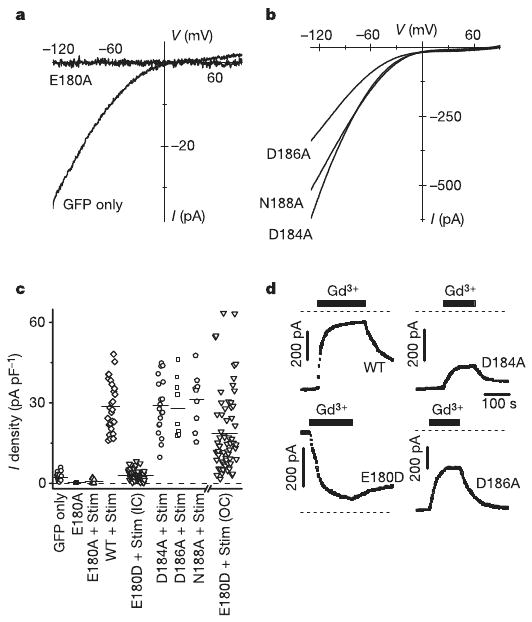
a, I–V curves in a cell expressing the Orai(E180A) mutant (without Stim) compared to control. b, I–V curves for Orai(D184A), Orai(D186A) and Orai(N188A) mutants. c, CRAC current density in transfected S2 cells. Each point represents the maximal CRAC current density (pA pF−1) in a single cell, plotted as absolute values (from left to right): GFP-transfected control; Orai(E180A) (P = 1.7 × 10−4 relative to control); Orai(E180A) plus Stim (P = 8.2 × 10−3); wild-type Orai plus Stim; Orai(E180D) plus Stim, inward current; Orai(D184A) plus Stim; Orai(D186A) plus Stim; Orai(N188A) plus Stim; and Orai(E180D) plus Stim, outward current. d, Suppression of CRAC current by 5 nM Gd3+ in wild-type Orai and the D184A, E180D and D186A Orai mutants. Bars indicate time of Gd3+ application; dashed lines indicate the zero-current level.
Gd3+ very effectively suppresses native and wild-type Orai-induced CRAC current5. In addition to ion selectivity properties, block by Gd3+ was evaluated in all mutants (Fig. 4d). Two charge-neutralizing mutations, aspartate to alanine at positions 184 and 186, and, to a lesser degree, mutation of asparagine to alanine at position 188, significantly reduced the potency of Gd3+ block (Fig. 4d and Supplementary Fig. 2b). As shown previously5, wild-type Orai CRAC current was potentiated by 2-aminoethyldiphenyl borate (2-PB) at a low concentration (5 μM) and inhibited at a high (20 μM) concentration (Supplementary Fig. 3a), similar to effects of 2-APB on native CRAC current in Jurkat and in S2 cells10,18. The effects of 2-APB on Orai(E180D)-induced CRAC current were complex but still showed potentiation and inhibition (Supplementary Fig. 3b). The time course of Orai(E180D) outward CRAC current showed an immediate decrease preceding potentiation by 5 μM 2-APB, and an immediate increase preceding inhibition during 20 μM application (n = 3). The pharmacological analysis provides support for the conclusion that the S1–S2 loop is involved in ion selectivity and block by Gd3+, and may contribute to the complex effects of 2-APB on CRAC current.
Our results demonstrate that thapsigargin-triggered store depletion dynamically strengthens an interaction between Stim and Orai, supporting a model for CRAC channel activation in which Stim serves as the Ca2+ sensor to detect store depletion and as the messenger to activate CRAC channels in the plasma membrane. More importantly, we conclude that Orai is a bona fide ion channel, based on the following: (1) RNA-interference-mediated knockdown of Orai expression suppresses thapsigargin-dependent Ca2+ influx and CRAC channel activity; (2) overexpression of Orai with or without Stim augments CRAC currents that exhibit biophysical properties identical to native CRAC current; and (3) mutations of negatively charged residues within the putative pore region of Orai significantly alter ion selectivity, current rectification and affinity to a charged channel blocker without altering channel activation kinetics. The marked alteration of these properties by a targeted point mutation provides definitive evidence that Orai embodies the pore-forming subunit of the CRAC channel.
The consensus sequence within the S1–S2 loop, 179VEVQLDxD186, contains the critical glutamate (bold) shown here to control ion selectivity properties of the CRAC channel, and two aspartates (underlined) that may help to attract Gd3+ (and Ca2+) towards the pore. It is not similar to pore sequences found in other channels. Unlike the pore regions of voltage-gated Ca2+ (CaV) channels, which contain a relatively long loop and a ring of critical glutamates from different domains that form a high-affinity Ca2+-binding site19, the putative pore sequence of Orai is very short, and the key residue for ion selectivity (E180) is adjacent to the putative S1 segment. Nevertheless, because withdrawal of external divalent ions reveals permeability to monovalent cations in both CaV11,12 and CRAC channels10,14,16,17, it is possible that the CRAC channel ion-selectivity filter is also formed by a ring of glutamates and that the mechanism of Ca2+ permeation is similar, although the single-channel conductance and maximum permeant ion size of the CRAC channel selectivity filter are smaller than that of the CaV channel10,17,20,21. Negatively charged side chains also contribute to Ca2+ selectivity of TRPV6; in this instance, aspartate (at position 541) is proposed to coordinate with Ca2+ ions and line the selectivity filter in a ring structure formed by four subunits22. The CRAC channel may be a multimer that includes several identical Orai subunits, as a non-conducting pore mutant (E180A) exerts a strong dominant-negative action on native CRAC current. Biochemical approaches and cysteine-scanning mutagenesis should be useful to elucidate better the unique pore architecture of the CRAC channel.
Note added in proof: While in proof, our attention was drawn to another paper reporting complementary findings regarding the control of ion selectivity in the CRAC channel23. In our study, the critical glutamate residue is 180 in the Orai protein encoded by the Invitrogen S2 cell complementary DNA (GenBank accession number DQ503470, as described5). The corresponding residue is 178 in the Drosophila genome database (accession number AY071273), and 106 in the human Orai1 homologue (accession number BC015369).
Methods
Molecular cloning and mutagenesis
Generation of pAc5.1/EGFP, pAc5.1/D-STIM, pAc5.1/D-STIM-V5-His and pAc5.1/olf186-F (GenBank accession number DQ503470) were as described5. Orai pore mutants were made by exchanging the corresponding codons (E180A, GAG to GCG; E180D, GAG to GAC; D184A, GAT to GCT; D186A, GAT to GCT; N188A, AAT to GCT; Supplementary Fig. 1) using the QuikChange site-directed mutagenesis kit (Stratagene). The pAc5.1/HA-olf186-F clone was made by adding the haemagglutinin tag via PCR and re-cloned into the XhoI and NotI sites of pAc5.1/V5-His B expression vector. Resulting clones were confirmed by sequencing. Description of the primers and conditions for cloning are available upon request. Both HA-olf186-F and Stim-V5-His were verified for normal function by whole-cell recording.
Whole-cell recording
Cells transfected (Amaxa) with Orai and Stim constructs were identified by fluorescence of co-transfected GFP. Patch-clamp experiments were performed at room temperature in the standard whole-cell recording, using conditions similar to those reported previously10. The membrane capacitance of S2 cells selected for recording was 11.6 ± 0.4 pF (n = 131 cells). Membrane potentials were corrected for a liquid junction potential of 10 mV between the pipette and bath solution. The series resistance (2–7 MΩ) was not compensated. The membrane potential was held at −10 mV, and 220-ms voltage ramps from −130 to 90 mV alternating with 220-ms pulses to −130 mV were delivered every 2 s. Only cells with high input resistance (>2 GΩ) were selected for recording. External and pipette solution recipes are listed in Supplementary Table 1. Analysed data are presented as mean ± s.e.m.
Co-immunoprecipitation
Two days after transfection, 5 × 106 cells were treated with either Ringer's solution or zero-Ca2+ Ringer's solution plus 1 μM thapsigargin using solutions described6 for 15 min at room temperature. Cells transfected with Stim-V5-His only or HA-olf186-F only were used as controls. Cell extracts were then prepared using RIPA lysis buffer (Upstate) according to the manufacturer's instructions. The extracts were pre-cleared with protein A/G beads (Pierce) and protein concentration was determined, using the Pierce BCA Protein Reagent Kit. The cell lysate was then diluted to approximately 1 mg ml−1 total protein with PBS and mixed with either anti-HA monoclonal antibodies (1 μg per 100 μg total protein, Santa Cruz) or anti-V5 monoclonal antibodies (1 μg per 100 μg total protein, Invitrogen) for 4 h at 4 °C. Equal amounts of samples were mixed with either normal mouse IgG or vehicle as negative controls. Protein A/G beads were subsequently added (1 μl per 1 μg IgG) to rock overnight at 4 °C. Proteins were eluted by boiling in 4× sample buffer (Invitrogen). Samples were resolved by SDS–PAGE and analysed by standard western blotting techniques. Anti-HA monoclonal antibodies (Santa Cruz) were used at a dilution of 1:200. Anti-V5 monoclonal antibodies (Invitrogen) were used at a dilution of 1:2,500. Proteins were detected by developing with the SuperSignal (Pierce) detection system. Samples treated with normal mouse IgG or vehicle showed no detectable signal for both Stim-V5-His and HA-olf186-F (data not shown), indicating that the immunoprecipitation and co-immunoprecipitation are specific.
Supplementary Material
Acknowledgments
We thank L. Forrest for assistance in cell culture and G. Chandy for use of molecular reagents in his laboratory. This work was supported by a grant from the National Institutes of Health (M.D.C.), by a fellowship from the George E. Hewitt Foundation (S.L.Z.), and by a Scientist Development Grant from the American Heart Association (Y.Y.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions A.V.Y. was responsible for all patch-clamp experiments and analysis. S.L.Z. was responsible for all molecular biology and biochemistry experiments, with the assistance of W.J. Y.Y. and O.S. performed RT–PCR. M.D.C. provided advice and overall direction, and supervised project planning and execution.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
References
- 1.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 4.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang SL, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercer JC, et al. Large store-operated calcium-selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor. Stim1 J Biol Chem. 2006 June 28; doi: 10.1074/jbc.M604589200. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peinelt C, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nature Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soboloff J, et al. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 10.Yeromin AV, Roos J, Stauderman KA, Cahalan MD. A store-operated calcium channel in Drosophila S2 cells. J Gen Physiol. 2004;123:167–182. doi: 10.1085/jgp.200308982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almers W, McCleskey EW. Non-selective conductance in calcium channels of frog muscle: calcium selectivity in a single-file pore. J Physiol (Lond) 1984;353:585–608. doi: 10.1113/jphysiol.1984.sp015352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hess P, Tsien RW. Mechanism of ion permeation through calcium channels. Nature. 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- 13.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol (Lond) 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepple-Wienhues A, Cahalan MD. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys J. 1996;71:787–794. doi: 10.1016/S0006-3495(96)79278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozak JA, Kerschbaum HH, Cahalan MD. Distinct properties of CRAC and MIC channels in RBL cells. J Gen Physiol. 2002;120:221–235. doi: 10.1085/jgp.20028601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakowski D, Parekh AB. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current ICRAC in rat basophilic leukemia cells. Pflugers Arch. 2002;443:892–902. doi: 10.1007/s00424-001-0775-8. [DOI] [PubMed] [Google Scholar]
- 18.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol (Lond) 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellinor PT, Yang J, Sather WA, Zhang JF, Tsien RW. Ca2+ channel selectivity at a single locus for high-affinity Ca2+ interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- 20.Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voets T, Janssens A, Droogmans G, Nilius B. Outer pore architecture of a Ca2+-selective TRP channel. J Biol Chem. 2004;279:15223–15230. doi: 10.1074/jbc.M312076200. [DOI] [PubMed] [Google Scholar]
- 23.Prakriya M, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006 August 20; doi: 10.1038/nature05122. advance online publication. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.