

Abstract
Genome-wide analyses of protein binding sites generate large amounts of data; a ChIP dataset might contain 10,000 sites. Unbiased motif discovery in such datasets is not generally feasible using current methods that employ probabilistic models. We propose an efficient method, gadem, which combines spaced dyads and an expectation-maximization (EM) algorithm. Candidate words (four to six nucleotides) for constructing spaced dyads are prioritized by their degree of overrepresentation in the input sequence data. Spaced dyads are converted into starting position weight matrices (PWMs). gadem then employs a genetic algorithm (GA), with an embedded EM algorithm to improve starting PWMs, to guide the evolution of a population of spaced dyads toward one whose entropy scores are more statistically significant. Spaced dyads whose entropy scores reach a pre-specified significance threshold are declared motifs. gadem performed comparably with meme on 500 sets of simulated “ChIP” sequences with embedded known P53 binding sites. The major advantage of gadem is its computational efficiency on large ChIP datasets compared to competitors. We applied gadem to six genome-wide ChIP datasets. Approximately, 15 to 30 motifs of various lengths were identified in each dataset. Remarkably, without any prior motif information, the expected known motif (e.g., P53 in P53 data) was identified every time. gadem discovered motifs of various lengths (6–40 bp) and characteristics in these datasets containing from 0.5 to >13 million nucleotides with run times of 5 to 96 h. gadem can be viewed as an extension of the well-known meme algorithm and is an efficient tool for de novo motif discovery in large-scale genome-wide data. The gadem software is available at www.niehs.nih.gov/research/resources/software/GADEM/.
Key words: ChIP, de novo motif discovery, expectation-maximization, genetic algorithm, k-mer, spaced dyad
1. Introduction
Recently, genome-wide location analyses have been carried out for proteins such as OCT4 (Boyer et al., 2005), P53 (Wei et al., 2006), EREα (Carroll et al., 2006; Lin et al., 2007), c-Myc (Zeller et al., 2006), CTCF (Kim et al., 2007), FOXP3 (Zheng et al., 2007), NRSF (Johnson et al., 2007), STAT1 (Robertson et al., 2007), and for histone markers (Bernstein et al., 2006; Lee et al., 2006; Mikkelsen et al., 2007; Pan et al., 2007; Schones et al., 2008; Wang et al., 2008). One goal of these studies is to discover short functional elements such as cis-elements embedded in these sites that are a few hundreds to tens of thousands of nucleotides long. Computational tools for de novo motif discovery in such massive data are needed.
During the last decade or so, many de novo motif discovery methods have been developed (Bailey and Elkan, 1994; Buhler and Tompa, 2002; Down and Hubbard, 2005; Elemento et al., 2007; Eden et al., 2007; Hertz and Stormo, 1999; Linhart et al., 2008;Liu et al., 2001, 2002; Pavesi et al., 2001; Pevzner and Szu, 2000; Roth et al., 1998; Sinha and Tompa, 2002; Sumazin et al., 2005; Thijs et al., 2001; van Helden et al., 2000). These methods fall into two categories: word enumeration and local search. The performance of many algorithms has recently been assessed (Tompa et al., 2005). Word-enumeration techniques count the number of occurrences of a motif, defined as a string of letters {a,c,g,t and sometime with degenerate letters, e.g., y = c,t and r = a,g} of certain length (e.g., 6–20) in the sequence data. When no degenerate letters are used in the motif profile/model, a subsequence is considered an instance of the motif when the number of mismatches between the subsequence and the motif is less than a threshold. The motifs are then rank-ordered based on their overrepresentation, thus, these approaches guarantee the global optimum—e.g., producing motifs with the highest overrepresentation. Many methods in this group have been developed. For instance, Consensus (Hertz and Stormo, 1999) first uses each k-mer to form the first sequence to construct an alignment matrix and the matrix is further updated. The projection algorithm (Buhler and Tompa, 2002) projects every l-mer in the input data into a smaller space by hashing. Other methods in this category includes winnower (Pevzner and Szu, 2000), spaced dyads (van Helden et al., 2000; Li et al., 2002), Weeder (Pavesi et al., 2001), mitra (Eskin and Pevzner, 2002), YMF (yeast motif finder) (Sinha and Tompa, 2002), DWE (Sumazin et al., 2005), Drim (Eden et al., 2007), and fire (Elemento et al., 2007). Recently, Xie et al. (2007) enumerated a list of candidate k-mers (12–22 nucleotides) and counted the number of matching instances in a set of conserved noncoding elements in the human genome.
Perhaps more widely used approaches employ local search techniques such as EM and Gibbs sampling (for recent reviews, see Jensen et al. [2004] and van Nimwegen [2007]). Unlike word-enumeration, the local search techniques use the PWM as the motif model. Initially, these models are either pre-defined or randomly specified. The models are then updated by an iterative process until convergence. Local search methods include meme (Bailey and Elkan, 1994), AlignACE (Roth et al., 1998), BioProspector (Liu et al., 2001), motifSampler (Thijs et al., 2001), MDScan (Liu et al., 2002), Nestedmica (Down and Hubbard, 2005) and fdrMotif (Li et al., 2008).
One advantage of the local search methods is that initial motif models are iteratively updated. On the other hand, the search space can be very large. Consequently, one of the main challenges for the local search techniques is how to obtain starting positions for the local search algorithms. For instance, meme converts each subsequence of length, w, into a letter probability matrix and uses it as the starting point for its EM algorithm. Only one step of EM is carried out for each starting matrix. The resulting best models are subjected to full EM. Motifs with the strongest statistical significances (E-values) are reported. This approach almost guarantees good starting positions for the EM algorithm. However, examining all possible starting positions for various lengths of w's (e.g., 6–30) for a large dataset is computationally too costly to be practically useful. Here we present an efficient method that combines existing algorithms to identify good starting positions for an EM algorithm for unbiased motif discovery in large scale data sets.
Our method begins by counting the number of matching instances of all k-mers (k = 3, 4, 5, 6) in the data. For instance, there are 43 = 64 possible tri-nucleotides (3-mers). For each k, the k-mers are rank-ordered based on their overrepresentation. The top-ranked k-mers for all four k's are subsequently used as the words for the spaced dyads (Li et al., 2002; van Helden et al., 2000). The top-ranked words may be viewed as “seeds” for a motif. Unlike the word-enumeration methods, we do not count the numbers of matching instances of the spaced dyads in the data. Instead, we convert the spaced dyads into letter probability matrices (similar to meme), which in turn, are used as the initial models for a local search technique via an EM algorithm. Thus, one might regard our method as a hybrid of the word enumeration and local search techniques. Similar hybrid methods have been proposed. For instance, Eskin (2004) developed the mitra-pssm algorithm that combined an efficient branch and bound algorithm for finding consensus patterns and a local search algorithm.
A spaced dyad consists of two words separated by a certain number of spacers (unspecified bases), d. If we choose Nk top-ranked k-mers as the possible words and d spacers for the spaced dyads, the number of possible spaced dyads constructed from these building blocks can be large. In theory, there are (N3 + N4 + N5 + N6) × (d + 1) × (N3 + N4 + N5 + N6) possible spaced dyads when both words of the spaced dyads can independently come from any of the four k-mer groups and there are 0 to d spacers between the words. Subjecting all of them (after conversion to probability matrices) to EM is impractical. Therefore, we employ a genetic algorithm (GA) (Goldberg, 1989) to guide the formation of spaced dyads so that only a small fraction of the spaced dyads are examined while finding most or all motifs. A GA is very effective in searching high-dimensional space and has been used in many optimization/search problems. Earlier, Wei and Jensen (2006) proposed a GA-based approach, game, to evolve motifs from randomly generated starting motifs. We refer to our method as gadem (Genetic Algorithm guided formation of spaced Dyads coupled with EM for Motif identification).
2. Methods
2.1. Overview
gadem employs a genetic algorithm (GA) to guide the formation of a “population” of spaced dyads. Each spaced dyad is converted into a letter probability matrix, which in turn, serves as the starting PWM for an EM algorithm. The EM-optimized PWM is then used to scan for binding sites in the data. A subsequence of the length of the PWM is declared a binding site when the p-value of its PWM score is less than or equal to a pre-specified threshold (e.g., 2.5 × 10−5). The significance (E-value) of the alignment of the binding sites (referred to as a motif) is then computed and the logarithm of the E-value is used as the fitness score for the spaced dyad from which the motif is derived. A GA is used to “evolve” the spaced dyads in the population through several generations (e.g., five). The resulting unique motifs with fitness values less than or equal to a pre-specified cutoff are reported and corresponding binding sites in the original sequences were subsequently masked. The above procedure is repeated until no further motifs can be found. A workflow of the gadem algorithm is shown in Figure 1. Details are given below.
FIG. 1.

Flowchart of gadem algorithm. The algorithm is divided into three parts: formation of spaced dyads (left large box), GA (center large box) and motif declaration (right box). The three parts constitute one cycle of gadem. gadem automatically carries out several such cycles until no further motifs with E-values below a pre-specified threshold can be found. For each gadem cycle, the steps in the blue box are repeated for a user-specified number of generations (indexed by g, g = 0 at the beginning of GA), whereas the steps in the red and green boxes are carried out only once for each gadem cycle. gadem begins by enumerating the matching instances of all k-mers (k = 3, 4, 5, 6). For each k, the words are rank-ordered based on their z-scores. This results in four groups of top-ranked k-mers. A spaced dyad is formed by randomly choosing two words (a1 and a2) independently from any of the four groups and a randomly chosen width between 0 and d (e.g., d = 10). In the GA stage, a “population” indexed by r (e.g., 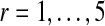 ) of such spaced dyads is generated. The r spaced dyads are converted into r position weight matrices (PWMs), θ. The PWMs are subjected to a user-specified number (e.g., 40) of steps of EM or until it converges. The score distribution of the integerized form of
) of such spaced dyads is generated. The r spaced dyads are converted into r position weight matrices (PWMs), θ. The PWMs are subjected to a user-specified number (e.g., 40) of steps of EM or until it converges. The score distribution of the integerized form of 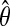 is computed. The same integerized
is computed. The same integerized 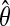 is also used to scan for binding sites in the data. A subsequence of length w is declared a binding sites when the p-value of its PWM score is below a threshold (e.g., ≤ 2.5 × 10−5). The entropy score of the aligned binding sites (motif) is computed and the logarithm of its statistical significance (E-value) is used as the fitness score for the spaced dyad from which the motif is derived. Next, all except the best performing spaced dyad(s) (with the lowest E-value) in the population are subjected to either mutation or crossover operations. This process (blue box) is repeated until the maximal number of generations (e.g., 5) has been reached.
is also used to scan for binding sites in the data. A subsequence of length w is declared a binding sites when the p-value of its PWM score is below a threshold (e.g., ≤ 2.5 × 10−5). The entropy score of the aligned binding sites (motif) is computed and the logarithm of its statistical significance (E-value) is used as the fitness score for the spaced dyad from which the motif is derived. Next, all except the best performing spaced dyad(s) (with the lowest E-value) in the population are subjected to either mutation or crossover operations. This process (blue box) is repeated until the maximal number of generations (e.g., 5) has been reached.
2.2. Spaced dyads
A spaced dyad consists of two words that are separated by spacers (Li et al., 2002; van Helden et al., 2000). Let D denote a spaced dyad, D = a1 · nx · a2, where a1 and a2 are the first and second words of the dyad, n is a string of unspecified nucleotides, x is the number of them (width of spacer), 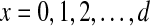 d is the pre-specified maximal width of the spacer (e.g., d = 10). We limit the words to 3–6 letters in length, consisting of only {a,c,g,t}. If one would enumerate all possible words and spacers, it generates (43 + 44 + 45 + 46) × 11 × (43 + 44 + 45 + 46) ≈ 3.3 × 108 spaced dyads. Evaluating all of them for large datasets is impractical and not necessary. We consider fewer but retain a broad range of possibilities by using a selected subset of the words in conjunction with GA.
d is the pre-specified maximal width of the spacer (e.g., d = 10). We limit the words to 3–6 letters in length, consisting of only {a,c,g,t}. If one would enumerate all possible words and spacers, it generates (43 + 44 + 45 + 46) × 11 × (43 + 44 + 45 + 46) ≈ 3.3 × 108 spaced dyads. Evaluating all of them for large datasets is impractical and not necessary. We consider fewer but retain a broad range of possibilities by using a selected subset of the words in conjunction with GA.
2.3. Top-ranked k-mers
We count all possible short DNA words (tri-, tetra-, penta-, and hexi-nucleotides; collectively called k-mers, k = 3, 4, 5, 6) in the input sequence data with self-overlapping ones discarded. For instance, “aacaa” in “aacaacaa” is only counted once. The k-mers are then rank-ordered by their z-scores, calculated as 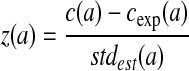 , where c(a) is the number of counts observed for k-mer, a. cexp(a) is the expected number of counts for a and stdest(a) is an estimate of the standard deviation of occurrences of a, based on the background {a,c,g,t} distribution estimated from the entire data, assuming independence between positions. The higher the z-score, the more likely the k-mer is enriched in the data and present in motif(s). Of course, the top-ranked words of different length will generally overlap. Treating them as unique words, however, allows flexible combinations of word length and spacer length to provide a rich set of spaced dyads as initial motifs for the EM. Let Nk be the number of top-ranked k-mers, we arbitrarily set Nk to at most 20, 40, 60, and 100 (only those with a z-score at or above 6.0 are considered), for k = 3, 4, 5, 6, respectively. These settings appear to favor a larger proportion of short k-mers over long k-mers. However, this bias is lessened by the high dependency (overlapping) among the top-ranked k-mers. This reduces the number of possible spaced dyads to (220 × 11 × 220 ≈ 5.3 × 105). Subjecting all (after being converted into PWMs, see below) to an EM algorithm is still computationally formidable. An intelligent method is needed to subject only a subset of them to EM without significantly compromising the results. For this reason, we adopt an effective stochastic search algorithm, GA, to guide formation of the spaced dyads.
, where c(a) is the number of counts observed for k-mer, a. cexp(a) is the expected number of counts for a and stdest(a) is an estimate of the standard deviation of occurrences of a, based on the background {a,c,g,t} distribution estimated from the entire data, assuming independence between positions. The higher the z-score, the more likely the k-mer is enriched in the data and present in motif(s). Of course, the top-ranked words of different length will generally overlap. Treating them as unique words, however, allows flexible combinations of word length and spacer length to provide a rich set of spaced dyads as initial motifs for the EM. Let Nk be the number of top-ranked k-mers, we arbitrarily set Nk to at most 20, 40, 60, and 100 (only those with a z-score at or above 6.0 are considered), for k = 3, 4, 5, 6, respectively. These settings appear to favor a larger proportion of short k-mers over long k-mers. However, this bias is lessened by the high dependency (overlapping) among the top-ranked k-mers. This reduces the number of possible spaced dyads to (220 × 11 × 220 ≈ 5.3 × 105). Subjecting all (after being converted into PWMs, see below) to an EM algorithm is still computationally formidable. An intelligent method is needed to subject only a subset of them to EM without significantly compromising the results. For this reason, we adopt an effective stochastic search algorithm, GA, to guide formation of the spaced dyads.
2.4. Genetic algorithm
2.4.1. Initialization
For each word (a1 or a2) in a spaced dyad, we first randomly choose a k, k = 3, 4, 5, 6, with equal probability, from which a word in the k-mer group will be selected. Next, a word from the Nk top-ranked k-mers is chosen with probability that is proportional to its z-score. Both a1 and a2 are chosen independently. The width, x, is randomly chosen between 0 to d with equal probability. A “population” of such spaced dyads (indexed by r, e.g., 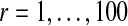 ) is generated. An example spaced dyad would be gggcnnnnnnntttgca, where a1 = gggc, x = 7, and a2 = tttgca.
) is generated. An example spaced dyad would be gggcnnnnnnntttgca, where a1 = gggc, x = 7, and a2 = tttgca.
2.4.2. Fitness evaluation
Fitness evaluation consists of several steps. First, the spaced dyads in the population are converted into initial PWMs. Second, the PWMs are iteratively updated by an EM algorithm using all or a subset of randomly selected sequences. Third, the updated PWMs are used to scan for binding sites in the entire data. Fourth, the relative entropy score of the aligned binding sites is computed and the logarithm of its statistical significance (E-value) is used as the fitness score for the spaced dyad. Details of each step are described below.
Step i. Spaced dyad to PWM. Each spaced dyad in the population is converted into a PWM in that the corresponding position in the matrix is assigned 1 and 0 otherwise. A value of 1 is assigned to each letter in a1 and a2, as well as to all cells in the matrix corresponding to the spacers. A small pseudo count (e.g., 0.01) is added to each cell containing zero. Each column is then standardized to sum to 1.0. Other assignments such as that from meme can also be considered.
Step ii. EM algorithm. We wish to find binding site locations and the base probabilities using only the sequence data and the initial PWM. We use an EM algorithm described in Lawrence and Reilly (1990) for this purpose. We conveniently assume that the positions within a sequence are mutually independent, i.e., a sequence follows a product of multinomial distributions. Details of the EM algorithm can be found in supplementary material and in Lawrence and Reilly (1990) and Li et al. (2008).
Applying EM to all sequences can be computationally costly. gadem allows all or only a randomly selected subset of sequences in the EM steps. For genome-wide data with thousands to tens of thousands sequences, a 25% to 50% sample should be adequate for obtaining a good estimate of the PWM.
Step iii. Binding site declaration. The EM derived PWM,  , is then log-transformed and multiplied by a scale factor, α, (e.g., α = 200) followed by rounding the real numbers to their closest integers [
, is then log-transformed and multiplied by a scale factor, α, (e.g., α = 200) followed by rounding the real numbers to their closest integers [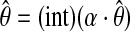 ]. We compute the exact score distribution of the integerized
]. We compute the exact score distribution of the integerized 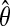 using the probability generating functions method of Staden (1989). The same integerized
using the probability generating functions method of Staden (1989). The same integerized 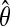 is subsequently used to scan for binding sites in the data. A subsequence is declared a binding site when the p-value of the observed PWM score sum is less than or equal to a threshold (e.g., 2.5 × 10−5).
is subsequently used to scan for binding sites in the data. A subsequence is declared a binding site when the p-value of the observed PWM score sum is less than or equal to a threshold (e.g., 2.5 × 10−5).
Step iv. Fitness evaluation—E-value. The binding sites (motif) are aligned and the log likelihood ratio (llr) score (Stormo, 2000) of the alignment is computed as follows,
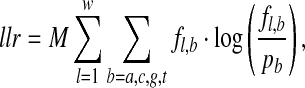 |
where M is the number of binding sites in the alignment, fl, b is the frequency of base b at position l of the alignment and pb is the background frequency of base b, computed from the entire data. Here again we assume that the letters in a sequence are independent and identically distributed (iid) multinomial random variables. These scores are not directly comparable for different M and w. To assess the significance of the llr score, one needs to compute its p-value, that is, the probability of observing an llr score or higher under the null hypothesis that the distribution of the letters in each column follows an independent multinomial distribution. The background {a,c,g,t} distribution is estimated from the entire data. Methods for computing the p-value of llr score have been proposed and discussed (Bailey and Gribskov, 1998; Hertz and Stormo, 1999; Nagarajan et al., 2005). gadem adopts the approach of Bailey and Gribskov (1998) as implemented in meme (Bailey and Elkan, 1994).
The logarithm of the approximated E-value (E-value = p-value × the size of the search space) is used as the fitness score for the spaced dyad from which the binding sites are derived. Accordingly, each spaced dyad in the population is assigned a fitness score.
2.4.3. “Genetic” operations
Once the entire population of spaced dyads (current generation) has been evaluated, a new population of spaced dyads (next generation) can be formed through “genetic” operations: “crossover” or “mutation.” The spaced dyads that resulted in unique motifs (see supplementary material) with fitness scores [log (E-values)] below a pre-specified cutoff are guaranteed to pass onto the next generation without genetic operations. Those typically constitute less than 10% of the spaced dyads. To fill in the remainder of the next generation, we first decide whether they come from mutation or crossover with equal probability. Next, one (two non-identical for crossover) spaced dyad(s) is selected from the entire current generation with probability proportional to its fitness score followed by the genetic operation. This process is repeated until the entire population is filled.
Mutation. Once a spaced dyad, D, is chosen for mutation, we first choose, with equal probability, which of its three components (a1, nx, a2) will be mutated. If a word (a1 or a2) is to be mutated, we first determine the source for the new word by randomly selecting a k, k = 3, 4, 5, 6. Once k is selected, a new word, a, is randomly chosen from the Nk top-ranked k-mers with probability proportional to its z-score. If the width of the spacer is to be mutated, we replace it with a randomly chosen new width between 0 and d. In the following example, a2 is replaced by a new word, aacaat, from the 6-mer (hexamer) group:
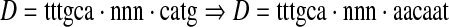 |
Crossover. Two non-identical spaced dyads are chosen with probabilities proportional to their fitness scores. One of the three components of the spaced dyads is randomly chosen and exchanged between the two spaced dyads. For example,
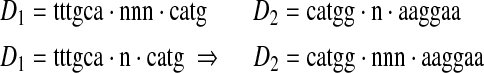 |
In this example, the spacers are exchanged between D1 and D2 whereas the words remain unchanged.
Both mutation and crossover create a new population of spaced dyads with the best performing unique ones from the previous generation unaltered in the population. The new generation of spaced dyads is subject to fitness evaluation through steps i–iv described above.
2.5. Motif declaration
One may let the GA evolve for many generations so that the majority of the spaced dyads in the population converge to a single solution as in the classical GA (Goldberg, 1989). However, this would result in only one “best” spaced dyad—one motif, from a complete GA cycle. Many cycles would be needed if more than one motif is sought. This classic approach is computationally expensive and perhaps unnecessary. We observed that many unique motifs remain unchanged from generation to generation. Thus, we limit the number of GA generations to only a few (e.g., five to 10) and consider all resulting motifs with E-values below a pre-specified cutoff of interest.
To see if the resultant motif can be extended, for each of the identified sites, gadem extracts 10 preceding and 10 following bases around the site. This results in a temporary motif of length of 20 + w, where w is the length of the original motif. The information content (in the scale of 2 bits) at each position in the extended motif, 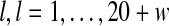 , is computed as follows,
, is computed as follows,
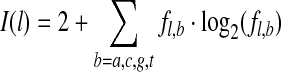 |
The alignment is then trimmed from both ends one base at a time and stopped when one of the following arbitrary conditions is met: 1) I(l) ≥ 0.5 at any single position, or 2) I(l) ≥ 0.3 at any two consecutive positions; 3) I(l) ≥ 0.2 at any three consecutive positions. Next, all binding sites of all motifs are masked by uninformative ‘N's in the input data.
The above steps 2.2 to 2.5 complete one cycle of gadem. A typical cycle may produce 1 to 10 motifs depending on the number of spaced dyads in a population. A new gadem cycle begins with identifying the top-ranked words in the sequences with the newly identified motifs masked (Fig. 1). gadem stops when no motifs with a log (E-value) below the threshold in one complete gadem cycle.
3. Data
We downloaded six genome-wide human ChIP data sets from the UCSC genome browser (http://genome.ucsc.edu) using custom tracks. The repetitive elements in the data were not masked. The datasets are briefly described below.
•Boyer et al. (2005) carried out genome-wide location analysis of OCT4, SOX2, and NANOG binding sites in human ES cells. At least 603 OCT4 loci were identified with an average length of 695 bp.
•Wei et al. (2006) applied the ChIP-PET technique to human HCT116 cells and identified at least 542 high confidence P53 binding sites with an average length of 1187 bp.
•Carroll et al. (2006) applied ChIP-chip on MCF-7 breast cancer cells and identified 3665 estrogen receptor α (EREα) binding sites. The average length of the sequences is 771 bp.
•Lin et al. (2007) independently applied ChIP-PET technique to MCF-7 cells and identified 1234 high confidence EREα binding sites. The average length of the sequences is 1315 bp.
•Kim et al. (2007) identified 13,721 CCCTC-binding factor (CTCF) binding sites in human IMR90 cells with an average length of 815 bp.
•Robertson et al. (2007) profiled STAT1 DNA association using ChIP and massively parallel sequencing in two interferon-γ-stimulated and unstimulated human HeLa S3 cells. Many putative STAT1-binding regions in IFN-γ simulated cells were identified. Only those regions with peak heights greater or equal to 20 and are with 8 kb in length were considered in this analysis. A total of 9834 regions were identified.
4. Results
4.1. De novo discovery of known motifs
We tested gadem on six genome-wide ChIP datasets of various sizes. The number of sequences in these datasets ranges from 542 to 13,721 totaling ∼0.5 to ∼13.5 millions of nucleotides. The size of these data sets presents a challenge for current de novo motif discovery programs. We believe that no other existing local search methods are feasible for such large datasets without limiting motif search profiles.
Although the exact locations of the protein binding sites in these ChIP sequences are unknown, the characteristics of the binding sites are known. Hence, these datasets serve as good test cases for gadem to see if it can identify known motifs (e.g., P53 motif in P53 ChIP data) without specifying what they look like beforehand. We set the minimal and maximal numbers of unspecified nucleotides between the words in the spaced dyads to 0 and 10, respectively. This allows gadem to initially search for motifs of lengths between 6(3 + 0 + 3 = 6) and 22(6 + 10 + 6 = 22) in each dataset. The final motif lengths are determined at the post-processing steps through base extension and trimming. For each dataset, we carried out two independent gadem runs with the same parameters but different random seeds. For the two large datasets (CTCF and STAT1), we randomly selected 20% of the sequences and subjected them to an EM algorithm to derive the EM-optimized PWMs. In the final motif declaration step, the EM-optimized PWMs are used to scan for binding sites in the entire dataset.
Between 15 and 30 motifs were identified in each ChIP set depending on its size. The characteristics of the motifs are diverse and their lengths vary from 6 to 40. Most of the motifs are unknown and do not match any in the TRANSFAC (Knüppel et al., 1994) and JASPAR (Sandelin et al., 2004) databases. Methods for comparing motif similarity are available (Schones et al., 2005). Most of the long and highly abundant motifs correspond to retroelements. The results from all twelve runs are provided in supplementary materials.
Remarkably, the expected known motifs in all six datasets were identified in all twelve runs (two for each of the six datasets). Some of them were identified in the first generation of the first gadem cycle whereas others were identified after a few gadem cycles. These motifs have different characteristics. gadem was able to identify all of them (Table 1)without any prior motif information, e.g., specifying their lengths or consensus sequences. These results suggest that the combination of a genetic algorithm and an EM algorithm and the usage of spaced dyads with the top-ranked words as their “seeds” are efficient for motif discovery. We believe that, gadem, a de novo motif discovery tool, is capable of identifying motifs in datasets of different sizes.
Table 1.
Expected Known Motifs Identified in the Six ChIP Datasets by gadem
| ChIP data | Number of bases | Number of sites identified | Motif logo from the identified sites |
|---|---|---|---|
| OCT4 | 419,114 | 327 | |
| P53 | 643,217 | 583 | |
| EREα (Lin) | 1,622,241 | 692 | |
| EREα (Carroll) | 2,823,788 | 1695 | |
| CTCF | 11,187,200 | 11,383 | |
| STAT1 | 13,476,426 | 5601 |
In all runs, we set the population size and the number of generations to 100 and 5, respectively. The minimal and maximal numbers of unspecified nucleotides between the two words in spaced dyads were set to 0 and 10, respectively. EM was run 20 iterations or until convergence. The p-value for PWM score cutoff was set to 2.5 × 10−5 for all data sets except for OCT4 (5 × 10−5). The log (E-value) cutoff is set to 0 for all data sets. For ChIP sequences that are longer than 15 kb, only the first 15 kb were used in the analyses. The number of sites listed is the average from two independent runs. For STAT1, two similar motifs that resemble the IFN-stimulated response element (ISRE) motif were also identified (not shown here). Individual results for all motifs in all six datasets are provided in tables in Supplementary Materials (See online Supplementary Materials at www.liebertonline.com.)
4.2. Reproducibility
gadem employs a stochastic algorithm, GA, as its search algorithm; therefore, it does not guarantee identical results from run to run. To test its reproducibility, we carried out two independent runs for each ChIP dataset with the same parameters but different random seeds. In most cases, near identical motifs were obtained, especially for the abundant ones. However, the number of sites in each motif varied from run to run. For most of the data sets, the numbers of identified sites in the expected known motifs were similar between the two runs with differences in 3–10% range. The results from two independent runs for all six data sets are provided in supplementary material. A larger population size and/or additional GA generations produced similar results.
4.3. Computational efficiency
gadem is computationally efficient. For small real ChIP datasets such as OCT4 and P53, gadem identified 15–25 motifs in 6–10 h. For large datasets, e.g., CTCF, gadem identified 30 motifs in ∼96 h.
To compare gadem's computational efficiency with several competing tools on real ChIP datasets, we tested gadem, fire (Elemento et al., 2007), game (Wei and Jensen, 2006), meme (Bailey and Elkan, 1994), Nestedmica (Down and Hubbard, 2005), and Weeder (Pavesi et al., 2001) on the OCT4 ChIP dataset (Boyer et al., 2005). The OCT4 dataset contains only ∼419 kb, several orders of magnitudes smaller than many of the current ChIP datasets. The parameters used for each tool in this comparison are provided in supplementary text (Section 4). gadem identified 14 motifs of various lengths including the OCT4 motif in ∼5 h. game only identified one motif (but not the OCT4) after 120 h of run and the job was stopped. meme finished in ∼144 h and found 11 motifs including OCT4. Nestedmica was still running without outputs after 240 h. Weeder searches for motifs of lengths 6 to 12 bp. It identified the 12-bp core of the OCT4 motif in ∼19 h. fire was fast (<15 min) and found the 9-bp core of the OCT4 motif. It is worth pointing out that the tested tools that employ local search techniques produce multiple unique motifs whereas the tools using word enumeration technique produce fewer motifs of a few fixed lengths. However, we emphasize that some of tools such as meme and Nestedmica can be run on multiple processors and would have been faster if done so. Nonetheless, we believe that as a de novo motif discovery tool, gadem is more computationally efficient that its competitors. Since gadem uses a GA as its search algorithm, a parallelized version would also be considerably faster.
4.4. Simulation
To assess gadem's performance on small datasets, we compare it with three competing tools (meme and game) on 500 simulated “ChIP” datasets (Li et al., 2008). Briefly, we simulated 66 sequences, each of which was 250 bp. For each sequence, a location within the sequence was randomly chosen and the 20-bp segment to the 3′ end of the site was replaced by a randomly selected site from the 66 experimentally identified P53 binding sites without replacement (no binding sites were used twice). Each simulated sequence contains exactly one of the 66 known P53 binding sites. We independently generated 500 such datasets.
We then ran all four tools on these datasets using identical or similar parameters whenever possible. For each dataset, we monitored the numbers of true positives (TP), false negatives (FN), and false positives (FP) as defined in Tompa et al. (2005). The sensitivity (Sn), positive predictive value (PPV), the average site performance (ASP), and the performance coefficient (PC) all at the site level were computed as also described in Tompa et al. (2005). gadem performed comparably with meme in all four categories (supplementary Table s1).game employs Gibbs sampling as the local search tool; and, on average, it identified more binding sites than both meme and gadem. As a result, game had higher Sn but lower PPV compared to meme and gadem. For all 500 datasets, the summary results are provided in supplementary text.
5. Discussion
Genome-wide location analysis of protein binding sites generates a large amount of sequence data with potentially tens of millions of nucleotides. To our knowledge, existing de novo motif discovery methods that utilize a local search technique would be prohibitively time-consuming to apply to datasets of this scale. Unbiased methods that can identify motifs of various lengths (e.g., 6–30 bp) are needed. gadem might be regarded as the first attempt to achieve this goal.
EM and Gibbs sampling are the two major algorithms used in de novo motif discovery tools that employ local search techniques. Like meme and projection, gadem also employs an EM algorithm. However, gadem obtains the starting positions for EM differently. meme considers each subsequence of length w as the possible starting position whereas the projection algorithm projects l-mers into random k-mer subspaces (k < l). Both approaches use subsequences (length w in meme and l in projection, both w and l are motif width) in the input data to directly derive the starting positions for EM. On the other hand, gadem obtains its starting positions from spaced dyads (also length w) that are initially arbitrarily constructed from top-ranked k-mers (k < w) in the data, later through a genetic algorithm (GA). game utilizes a GA to guide the selection of subsequences of length w for constructing PWMs with an embedded Gibbs sampling algorithm to improve them whereas gadem employs a GA to “evolve” the spaced dyads from which the starting PWMs are derived and further improved by an EM algorithm.
Why does gadem work? gadem combines the two techniques used in de novo motif discovery: word-enumeration and local search. In gadem, word-enumeration generates the initial motif profiles, spaced dyads in this case, that are subsequently refined by a local search technique, an EM algorithm (Redner and Walker, 1984). Enumerating all possible spaced dyads is not possible. Instead, gadem uses only the over-represented words (tri-, tetra-, penta-, and hexamers) in the input sequence data as the words for the spaced dyads. These over-represented words may be considered as motif “seeds.” This way, gadem avoids examining the majority of the spaced dyads that are deemed not to produce motifs. For the remaining spaced dyads, gadem employs an efficient search algorithm, GA, to intelligently sample a subset of them followed by refinement by an EM algorithm.
EM can converge from any starting position to an optimum, although not necessarily the global optimum. The consistency of the EM solution (PWM) and the number of cycles needed to reach a solution depends on the quality of the starting position (Supplementary text, part 5). We showed that the combination of the top-ranked k-mers and the GA helps identify embedded motifs in simulated “ChIP” datasets, possibly through facilitating the selection of good starting positions for the EM algorithm, thus allowing the EM to more quickly reach a good candidate PWM.
Subjecting all sequences to an EM algorithm can be computationally costly. To make it feasible for large datasets, gadem allows users to run the EM algorithm on only a subset of randomly selected sequences. For most of the genome-wide data, a small subset still contains several hundreds to thousands of sequences. Thus, the maximal likelihood estimates of the PWM should be fairly accurate.
gadem declares a subsequence a binding site when the p-value of its PWM score is below or equal to a pre-specified threshold. The score distribution is determined under the convenient assumption that each column is iid multinomial random variables under the null (background). The choice for the threshold is arbitrary and errors in multiple testing are not fully corrected. Although this choice may be reasonable, different thresholds will likely result in somewhat different numbers of binding sites. Nonetheless, one might consider different runs with different thresholds, as currently, the same threshold applies to all motifs regardless of their lengths. Alternatively, one might consider an approach that takes false discovery rate into account (Li et al., 2008). However, such an approach is computationally too costly to apply to genome-wide data.
gadem adopts the method (Bailey and Gribskov, 1998) from meme to approximate the statistical significance of the relative entropy score of an alignment (motif). Instead of determining the distribution of the sum of the entropy scores from all w columns (Stormo, 1999; Najarajan et al., 2005), meme first determines the significances of the individual column entropy scores. Next, the probability that the product of w independent, uniform [0,1] random variables is determined. meme may over-estimate p-values by 10 to 100 fold (Najarajan et al., 2005), however, this systematic bias should not have much effect on the ranks of the motifs. Moreover, the approach from meme is computationally more efficient and is practical for large data sets.
gadem can be used to identify motifs of various lengths. However, short motifs (e.g., 6 bp) embedded in long sequences might be missed as their entropy scores may not reach the pre-specified significance threshold. Methods utilizing word enumeration techniques such as Weeder (Pavesi et al., 2001) may work better for those motifs.
Since neither GA nor EM guarantee convergence to a global maximum from a single starting value, multiple independent runs are recommended. The best motifs (lowest E-value) from individual runs might be considered as the most authoritative. For all six datasets tested, similar motifs were obtained from run to run, although the number of sites in each motif varies slightly.
6. Conclusion
We introduce a novel method, gadem. gadem uses a local search tool, EM, to update its models. The initial models are derived from spaced dyads that use the over-represented words (lengths 3–6) estimated from the entire sequences as its “seeds”. gadem employs a GA to guide the formation of the spaced dyads from those seeds. When tested on six transcription factor/insulator ChIP datasets totaling ∼0.5 to ∼1.35 million nucleotides, the expected known motifs were identified in all datasets without specifying what the motifs look like or their lengths. gadem can be viewed as an extension of the well-known meme algorithm as both use a similar EM algorithm and gadem adopts meme's procedure for computing motif significance. gadem is computationally efficient and easy to use. gadem represents a novel de novo motif discovery tool that can be applied to large scale sequence data for unbiased motif discovery.
Supplementary Material
Acknowledgments
We thank David Umbach and Grace Kissling for discussions and careful reading of the manuscript. We thank Clare Weinberg and Shyamal Peddada for discussions. We thank the reviewers for their thought provoking comments. We thank Robert Bass for creating the gadem web site and the Computational Biology Facility at NIEHS for computing time. This research was supported by Intramural Research Program of the NIH, National Institute of Environmental Health Sciences to L.L. (ES101765-05).
Disclosure Statement
No competing financial interests exist.
References
- Bailey T.L. Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bailey T.L. Gribskov M. Methods and statistics for combining motif match scores. J. Comput. Biol. 1998;5:211–221. doi: 10.1089/cmb.1998.5.211. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E. Mikkelsen T.S. Xie X., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler J. Tompa M. Finding motifs using random projections. J. Comput. Biol. 2002;9:225–242. doi: 10.1089/10665270252935430. [DOI] [PubMed] [Google Scholar]
- Carroll J.S., et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Down T.A. Hubbard T.J. NestedMICA, sensitive inference of over-represented. Nucleic Acids Res. 2005;33:1445–1453. doi: 10.1093/nar/gki282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., et al. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemento O., et al. A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell. 2007;28:337–350. doi: 10.1016/j.molcel.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskin E. Pevzner P.A. Finding composite regulatory patterns in DNA sequences. Bioinformatics. 2002;18:354–363. doi: 10.1093/bioinformatics/18.suppl_1.s354. [DOI] [PubMed] [Google Scholar]
- Eskin E. From profiles to patterns and back again, a branch and bound algorithm for finding near optimal motif profiles. RECOMB 2004. 2004:115–124. [Google Scholar]
- Goldberg D.E. Genetic Algorithms in Search, Optimization and Machine Learning. Kluwer Academic Publishers; Boston, MA: 1989. [Google Scholar]
- Hertz G.Z. Stormo G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Jensen S.T., et al. Computational discovery of gene regulatory binding motifs, a Bayesian perspective. Statist. Sci. 2004;18:188–204. [Google Scholar]
- Johnson D.S., et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kim T.H., et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knüppel R., et al. TRANSFAC retrieval program, a network model database of eukaryotic transcription regulating sequences and proteins. J. Comput. Biol. 1994;1:191–198. doi: 10.1089/cmb.1994.1.191. [DOI] [PubMed] [Google Scholar]
- Lawrence C.E. Reilly A.A. An expectation maximization EM algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins. 1990;7:41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- Lee T.I., et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. Identification of the binding sites of regulatory proteins in bacterial genomes. Proc. Natl. Acad. Sci. USA. 2002;99:11772–11777. doi: 10.1073/pnas.112341999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., et al. fdrMotif, identifying cis-elements by an EM algorithm coupled with false discovery rate control. Bioinformatics. 2008;24:629–636. doi: 10.1093/bioinformatics/btn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.Y., et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:867–885. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart C., et al. Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome Res. 2008;8:1180–1189. doi: 10.1101/gr.076117.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., et al. BioProspector, discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. 2001;6:127–138. [PubMed] [Google Scholar]
- Liu X.S., et al. An algorithm for finding protein-DNA binding sites with applications to chromatin-immuno-precipitation microarry experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- Mikkelsen T.S., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan N., et al. Computing the P-value of the information content from an alignment of multiple sequences. Bioinformatics. 2005;21:i311–i318. doi: 10.1093/bioinformatics/bti1044. [DOI] [PubMed] [Google Scholar]
- Pan G., et al. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Pavesi G., et al. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics. 2001;17:S207–S214. doi: 10.1093/bioinformatics/17.suppl_1.s207. [DOI] [PubMed] [Google Scholar]
- Pevzner P.A. Sze S.H. Combinatorial approaches to finding subtle signals in DNA sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2000;8:269–278. [PubMed] [Google Scholar]
- Redner R.A. Walker H.F. Mixture densities maximum likelihood and EM algorithm. SIAM Rev. 1984;26:195–239. [Google Scholar]
- Robertson G., et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Roth F.P., et al. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Sandelin A., et al. JASPAR, an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones D.E., et al. Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics. 2005;21:307–313. doi: 10.1093/bioinformatics/bth480. [DOI] [PubMed] [Google Scholar]
- Schones D.E., et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. Tompa M. Discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res. 2002;30:5549–5560. doi: 10.1093/nar/gkf669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.D., et al. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc. Natl. Acad. Sci. USA. 2005;102:1560–1565. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. Methods for calculating the probabilities of finding patterns in sequences. Comput. Appl. Biosci. 1989;5:89–96. doi: 10.1093/bioinformatics/5.2.89. [DOI] [PubMed] [Google Scholar]
- Stormo G.D. DNA binding sites, representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Sumazin P., et al. DWE, discriminating word enumerator. Bioinformatics. 2005;21:31–38. doi: 10.1093/bioinformatics/bth471. [DOI] [PubMed] [Google Scholar]
- Thijs G., et al. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics. 2001;17:1113–1122. doi: 10.1093/bioinformatics/17.12.1113. [DOI] [PubMed] [Google Scholar]
- Tompa M., et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- van Helden J., et al. Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res. 2000;28:1808–1818. doi: 10.1093/nar/28.8.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nimwegen E. Finding regulatory elements and regulatory motifs, a general probabilistic framework. BMC Bioinformatics. 2007;8:S4. doi: 10.1186/1471-2105-8-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.L., et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Wei Z. Jensen S.T. GAME, detecting cis-regulatory elements using a genetic algorithm. Bioinformatics. 2006;22:1577–1584. doi: 10.1093/bioinformatics/btl147. [DOI] [PubMed] [Google Scholar]
- Xie X., et al. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc. Natl. Acad. Sci. USA. 2007;104:7145–7150. doi: 10.1073/pnas.0701811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller K.I., et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA. 2006;103:17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.