

Abstract
In our efforts to explore marine cyanobacteria as a source of novel bioactive compounds we discovered a statine unit-containing linear decadepsipeptide, grassystatin A (1), which we screened against a diverse set of 59 proteases. We describe the structure determination of 1 and two natural analogs, grassystatins B (2) and C (3), using NMR, MS, and chiral HPLC techniques. Compound 1 selectively inhibited cathepsins D and E with IC50s of 26.5 nM and 886 pM, respectively. Compound 2 showed similar potency and selectivity against cathepsins D and E (IC50s 7.27 nM and 354 pM, respectively), whereas the truncated peptide analog grassystatin C (3), which consists of two fewer residues than 1 and 2, was less potent against both but still selective for cathepsin E. The selectivity of compounds 1–3 for cathepsin E over D (20- to 38-fold) suggests that these natural products may be useful tools to probe cathepsin E function. We investigated the structural basis of this selectivity using molecular docking. We also show that 1 can reduce antigen presentation by dendritic cells, a process thought to rely on cathepsin E.
Although all proteases share in common the ability to cleave peptide bonds, their various regulatory roles have made them interesting targets for drug discovery. They are involved in such diverse processes as blood coagulation, the cell cycle, infection and neurodegenerative disorders, among others.1 However, because of the ubiquity of proteolytic signaling, potential therapeutic inhibitors must be selective in order to reduce the chance of off-target effects. Non-selective inhibition of metalloproteases is thought to be the reason for musculoskeletal side effects seen in early matrix metalloprotease (MMP) inhibitors that were evaluated for cancer treatment.2
Modified peptides have potential for use as protease inhibitors. Their peptidic nature may allow them to bind in a substrate like manner at the active site, or at other sites on the enzyme. Modifications made to the normal peptide structure could increase resistance to proteolytic cleavage (so that they act as inhibitors and not substrates), or confer on them more drug-like properties, such as increased lipophilicity. It is with this in mind that we have been engaged in a systematic search for protease inhibitors amongst natural products produced by marine cyanobacteria. This ancient group of organisms is known to produce a vast array of secondary metabolites. These are often lipophilic modified peptides that possess potent cytotoxicity.3 Such metabolites have presumably been optimized by millions of years of natural selection to be potent and specific to their intended target. In some cases, the ecological target may be protease enzymes. Already, we have identified several lyngbyastatins that potently inhibit the serine protease elastase.4 Cyanobacteria produce modified peptides through the non-ribosomal peptide synthetase (NRPS) pathway or through combinations of the NRPS and polyketide synthase (PKS) pathways.5 Both of these pathways are highly modular, presumably allowing evolution of bioactive compounds through combinatorial alterations. The modular architecture of these pathways has led several groups to pursue combinatorial biosynthesis of novel “non-natural” products.6
Herein, we describe the isolation, structure determination, and biological evaluation of three linear modified peptides, grassystatins A–C (1–3, Figure 1). All three contain a statine unit [(3S,4S)-4-amino-3-hydroxy-6-methylheptanoic acid, Sta], which was first described in the broad-spectrum natural aspartic protease inhibitor pepstatin A (Figure 2).7 In the latter, statine arises from a mixed NRPS/PKS pathway that condenses leucine and malonate units.8 The closest structural relatives among cyanobacterial natural products are tasiamide9 and tasiamide B10 (Figure 2). Tasiamide does not contain a statine unit and there are also some differences in configuration of several amino acid residues (Figure 2). To test aspartic and other protease inhibitory activity, we screened compound 1 against 59 diverse proteases and found selective inhibition of the aspartic proteases cathepsins D and E. Notably, compounds 1–3 discriminate between these two enzymes, while pepstatin A does not. We demonstrate the inhibition of cathepsins in a cellular system, and also the disruption of antigen presentation by dendritic cells (DCs), a process in which cathepsin E has been recently implicated.11
Figure 1.
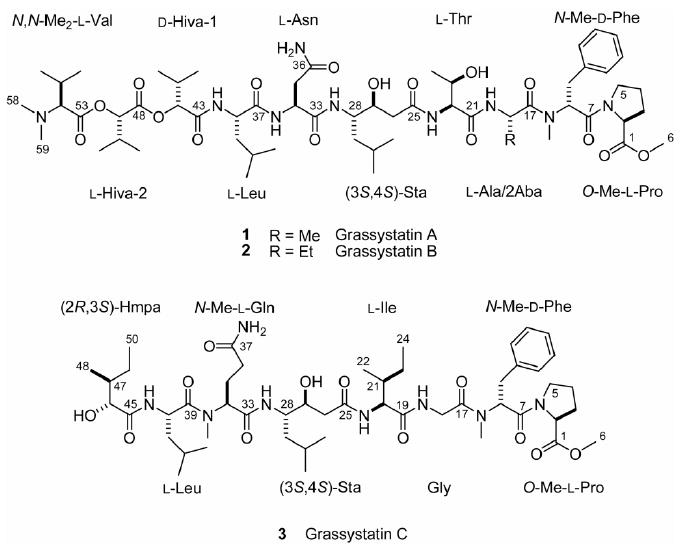
Structures of grassystatins A–C (1–3).
Figure 2.
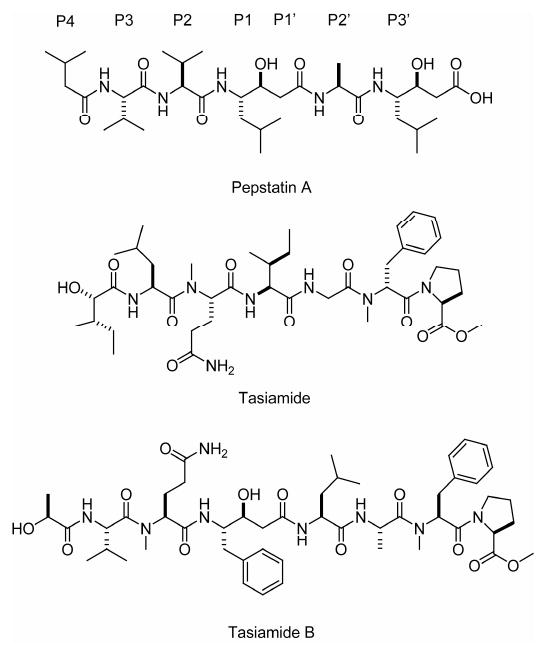
Structures of pepstatin A (including binding site nomenclature), tasiamide and tasiamide B.
Results
Isolation and Structure Determination
Samples of the cyanobacterium, identified as Lyngbya cf. confervoides, were collected off Grassy Key as described previously,12 and off Key Largo, Florida. The non-polar extract (MeOH–EtOAc 1:1) of each collection was subjected to silica chromatography and reversed phase HPLC to furnish 1 and 2. Compound 3 was found only in the Key Largo collection that previously yielded grassypeptolide.12
HRESI/APCIMS and NMR data for 1 suggested a molecular formula of C58H95N9O16 (m/z 1196.6812 for [M + Na]+, 1174.6988 for [M + H]+, 598.8455 for [M + H + Na]2+, and 587.8544 for [M + 2H]2+). Perusal of the 1H and 13C NMR spectra revealed that it was a depsipeptide (Table 1), with several exchangeable proton signals characteristic of amides (δH ∼6 to ∼8), α-protons (δH ∼4 to ∼5), and some deshielded signals in both the 1H and 13C NMR spectra indicative of methines adjacent to an ester linkage (δH/C 5.13/78.1 and 4.70/77.5). There were also several N-methyl signals (δH 3.01 and 2.30) and one O-methyl apparent (δH 3.72). In addition two conformers were present in the ratio 15:1.13 Analysis of the 1H NMR, 13C NMR, APT, COSY, edited HSQC, HMBC, ROESY and TOCSY spectra in CDCl3 of 1 (Table 1 and Supporting Information) revealed the presence of four proteinogenic amino acid units (Ala, Thr, Asn and Leu) and two hydroxyisovaleric acid (Hiva) moieties. In addition, O-Me-Pro, N-Me-Phe, N,N-Me2-Val and statine (Sta, C-25–C-32) were deduced. Given that there were two terminal groups (O-Me-Pro and N,N-Me2-Val), it was clear from the degree of unsaturation that the compound was linear (all 16 double bond equivalents were accounted for). The hydroxyl protons for Sta and Thr14 units were evident and thus precluded branching of the chain through ester linkages at these positions. The fragments Sta–Thr–Ala–N-Me-Phe–O-Me-Pro and N,N-Me2-Val–Hiva–Hiva–Leu–Asn were readily constructed with the help of HMBC and ROESY data. The continuous sequence of the two fragments was confirmed by ESIMS fragmentation (Figure 3). Without other evidence, however, it was still unclear whether the Sta and Asn units were joined through C-1 or C-4 of Asn (C-33 and C-36 in 1), as no correlations were observed through the NH2 group or from the NH or H-28 in the Sta unit. Collection of NMR data for 1 in DMSO-d6 (see Table S1, Supporting Information) revealed an extra HMBC correlation from one of the NH2 amide protons to the β-carbon of Asn, thus determining the chain proceeded through C-1.
Table 1.
NMR Spectral Data for Grassystatins A (1) and B (2) at 500 MHz (1H) and 150 MHz (13C) in CDCl3
| C/H no. | δH (J in Hz) | δC,a mult. | Grassystatin A (1) | HMBC b | ROESY | Grassystatin B (2) | ||
|---|---|---|---|---|---|---|---|---|
| 1H–1H COSY | δH (J in Hz) | δC,a mult. | ||||||
| O-Me-Pro | 1 | 172.49, s | 172.4, s | |||||
| 2 | 4.42, dd (7.1, 7.1) | 59.2, d | 3a, 3b | 1, 3, 4 | 3a, 3b, 5a, 5b | 4.42, dd (7.7, 6.8) | 59.0, d | |
| 3a | 2.18, m | 28.9, t | 2, 3b, 4a, 4b | 1, 2, 4, 5 | 2, 3b, 4b | 2.20, m | 28.7, t | |
| 3b | 1.84, m | 2, 3a, 4a, 4b | 1, 2, 4, 5 | 3a, 4a | 1.82, m | |||
| 4a | 1.94, m | 25.3, t | 3a, 3b, 4b, 5a, 5b | 1, 2, 3 | 3b, 4b, 5a, 5b | 1.95, m | 25.2, t | |
| 4b | 1.80, m | 3a, 3b, 4a, 5a, 5b | 1, 2, 3, 5 | 3a, 4a, 5a, 5b | 1.81, m | |||
| 5a | 3.46, m | 47.0, t | 4a, 4b, 5b | 1, 2, 3, 4 | 2, 4a, 4b, 5b, 8 | 3.42, m | 46.8, t | |
| 5b | 3.29, m | 4a, 4b, 5a | 3, 4, 7† | 2, 4a, 4b, 5a, 8 | 3.29, m | |||
| 6 | 3.72, s | 52.2, q | 3.72, s | 52.0, q | ||||
| N-Me-Phe | 7 | 168.1, s | 167.9, s | |||||
| 8 | 5.66, dd (9.2, 6.5) | 55.7, d | 9a, 9b | 7, 9, 10, 16, 17 | 2, 5a, 5b, 9a, 9b, 16 | 5.64, dd (8.7, 6.8) | 77.8, d | |
| 9a | 3.23, dd (−14.1, 6.5) | 34.7, t | 8, 9b | 7, 8, 10, 11/15 | 8, 9b, 11/15 | 3.22, dd (−14.2, 6.8) | 34.5, t | |
| 9b | 2.97, dd (−14.1, 9.2) | 8, 9a | 7, 8, 10, 11/15 | 8, 9a | 2.91, dd (−14.2, 8.7) | |||
| 10 | 136.7, s | 136.7, s | ||||||
| 11/15 | 7.20, m | 129.5, d | 9, 11/15, 13 | 8, 9a, 9b, 16, 18, 19 | 7.21, m | 129.4, d | ||
| 12/14 | 7.22, m | 128.2, d | 10, 12/14 | 7.22, m | 128.3, d | |||
| 13 | 7.15, m | 126.6, d | 11/15, | 7.16, m | 126.5, d | |||
| 16 | 3.01, s | 30.4, q | 8, 17 | 11/15, 18, 19 | 3.04, s | 30.4, q | ||
| Ala/Aba | 17 | 172.46, s | 171.7, s | |||||
| 18 | 4.72, dq (7.4, 7.2) | 45.5, d | 19, NH | 17, 19, 21 | 11/15, 16, 19, NH | 4.71, m | 50.3, d | |
| 19a | 0.86, d (7.2) | 17.0, q | 18 | 17, 18 | 1.39, m | 24.5, t | ||
| 19b | - | 1.20, m | ||||||
| 20 | - | 0.65, t (7.4) | 9.4, q | |||||
| NH | 7.41, d (7.4) | 18 | 21 | 18, 22 | 7.26, m | |||
| Thr | 21 | 170.2, s | Not obs. | |||||
| 22 | 4.31, dd (8.0, 7.3) | 58.3, d | 23, NH | 21 | 24 | 4.35, dd (7.8, 3.1) | 58.2, d | |
| 23 | 4.19, dq (7.3, 6.3) | 67.5, d | 22, 24 | 24 | 24 | 4.19, b | 67.2,c d | |
| 24 | 1.12, d (6.3) | 18.9, q | 23 | 22, 23 | 1.14, d (6.2) | 18.8, q | ||
| OH | ||||||||
| NH | 7.60, d (8.0) | 22 | 25 | 7.55, m | ||||
| Sta | 25 | 172.2, s | 172.1, s | |||||
| 26a | 2.53, dd (−14.0, 8.7) | 40.5, t | 26b, 27 | 25, 27, 28 | 2.54, dd (−14.0, 9.3) | 40.5, t | ||
| 26b | 2.39, dd (−14.0, 5.4) | 26a, 27 | 25, 27 | 27, 28 | 2.39, dd (−14.0, 5.4) | |||
| 27 | 4.02, dddd (8.7, 5.4, 5, 2.7) | 70.3, d | 26a, 26b, 28, OH | 26b, 29a, 29b, 31 | 4.02, m | Not obs. | ||
| 28 | 3.85 dddd (9.0, 6, 4.7, 2.7) | 51.8, d | 27, 29a, 29b, NH | 26b, 29a, 29b, 31, 32 | 3.82, m | Not obs. | ||
| 29a | 1.56, m | 39.9, t | 28, 29b, 30 | 28, 30, 31, 32 | 27, 28, 29b, 31, 32 | 1.55, m | 39.7,c t | |
| 29b | 1.36, ddd (−15.1, 10.2, 4.8) | 28, 29a, 30 | 28, 30, 31, 32 | 27, 28, 29a, 31, 32 | 1.37, m | |||
| 30 | 1.61, m | 24.7, d | 29a, 29b, 31, 32 | 28, 29, 31, 32 | 29b, 31, 32 | 1.55, m | 26.7, d | |
| 31 | 0.87, m | 23.1, q | 30 | 29, 30, 32 | 0.88, m | 22.8, q | ||
| 32 | 0.84, m | 22.0, q | 30 | 29, 30, 31 | 0.86, m | 21.9, q | ||
| OH | 4.63, br | 27 | ||||||
| NH | 7.18, m | 28 | 7.10, m | |||||
| Asn | 33 | 173.2,d s | 172.6,d s | |||||
| 34 | 4.82, ddd (8.0, 6.2, 4.5) | 50.3, d | 35a, 35b, NH | 33, 35, 36, 37 | 35a, 35b | 4.78, m | 49.9, d | |
| 35a | 2.79, dd (−15.0, 4.5) | 37.5, t | 34, 35b | 33, 34, 36 | 34 | 2.79, dd (−15.0, 4.3) | 37.1, t | |
| 35b | 2.73, dd (−15.0, 6.2) | 34, 35a | 33, 34, 36 | 34 | 2.68, dd (−15.0, 6.0) | |||
| 36 | 170.6,d s | 170.5,d s | ||||||
| NH2a | 6.42, br | 6.66, br | ||||||
| NH2b | 6.80, br | 6.23, br | ||||||
| NH | 7.61, d (7.6) | 34 | 38 | 7.53, m | ||||
| Leu | 37 | 171.7, s | 168.3, s | |||||
| 38 | 4.35, m | 52.6, d | 39a, 39b, NH | 37, 39, 40, 43 | 39a, 39b, 41, 42 | 4.28, m | 48.8, d | |
| 39a | 1.69, m | 40.2, t | 38, 39b, 40 | 37, 38, 40, 41, 42 | 38, 41, 42, NH | 1.69, m | 39.9, t | |
| 39b | 1.62, m | 38, 39a, 40 | 37, 38, 40, 41, 42 | 38, 41, 42, NH | 1.63, m | |||
| 40 | 1.65, m | 24.6, d | 39a, 39b, 41, 42 | 38, 39, 41, 42 | 38, 41, 42, NH | 1.64, m | 24.3, d | |
| 41 | 0.90, d (5.3) | 22.6, q | 40 | 39, 40, 42 | 0.92, m | 22.3, q | ||
| 42 | 0.86 | 22.2, q | 40 | 39, 40, 41 | 0.88, m | 22.8, q | ||
| NH | 7.05, d (5.8) | 38 | 39, 43 | 38, 39b, 44, 49 | 7.06, d (5.4) | |||
| Hiva-1 | 43 | 169.9, s | 170.1, s | |||||
| 44 | 5.13, d (3.3) | 78.1, d | 45 | 43, 45, 46, 47, 48 | 45, 46, 47, 51, NH (Leu) | 5.12, d (3.2) | 77.8, d | |
| 45 | 2.40, qqd (6.9, 6.9, 3.3) | 30.2, d | 44, 46, 47 | 46, 47 | 44, 46, 47 | 2.42, qqd (6.9, 6.9, 3.2) | 29.9,c d | |
| 46 | 0.95, d (6.9) | 19.2,e q | 45 | 44, 45, 47 | 0.96, d (6.9) | 18.98, q | ||
| 47 | 0.92, d (6.9) | 16.4, q | 45 | 44, 45, 46 | 0.94, d (6.9) | 16.2, q | ||
| Hiva-2 | 48 | 169.5, s | 169.4, s | |||||
| 49 | 4.70, d (5.9) | 77.5, d | 50 | 48, 50, 51, 52 | 44, 50, 51, 52, 54, 58/59, NH (Leu) | 4.68, d (5.9) | 77.5, d | |
| 50 | 2.20, qqd (6.8, 6.5, 5.9) | 29.8, d | 49, 51, 52 | 48, 49, 51, 52 | 49, 51, 52 | 2.20, qqd (6.8, 6.6, 5.9) | 29.5, d | |
| 51 | 1.04, d (6.5) | 17.7, q | 50 | 48, 49, 52 | 1.06, d (6.8) | 18.5, q | ||
| 52 | 1.06, d (6.8) | 18.8, q | 50 | 48, 49, 51 | 1.05, d (6.6) | 17.6, q | ||
| N,N-Me2-Val | 53 | 172.46, s | 172.3, s | |||||
| 54 | 2.82, d (10.7) | 73.8, s | 55 | 53, 55, 56, 57, 58/59 | 49, 55, 56, 57, 58/59 | 2.83, d (10.6) | 73.6, d | |
| 55 | 1.98, m | 27.5, d | 54, 56, 57 | 53, 54, 56, 57 | 54, 56, 57, 58/59 | 1.96, m | 27.3, d | |
| 56 | 0.88, m | 19.5, q | 55 | 54, 55, 57 | 0.99, d (6.6) | 18.90, q | ||
| 57 | 0.98, d (6.5) | 19.2,e q | 55 | 53, 54, 55, 56 | 0.88, m | 19.3, q | ||
| 58/59 | 2.30, s | 41.1, q | 58/59 | 49, 54 | 2.31, s | 40.9, q | ||
Multiplicity derived from APT and HMQC spectra.
Protons showing long-range correlation to indicated carbon.
The chemical shift of these carbons was deduced by virtue of HMBC correlations, as their correlation(s) were not observed in the HMQC spectrum.
There is insufficient information to distinguish between carbons 33 and 36.
These carbons have the same chemical shift.
Figure 3.
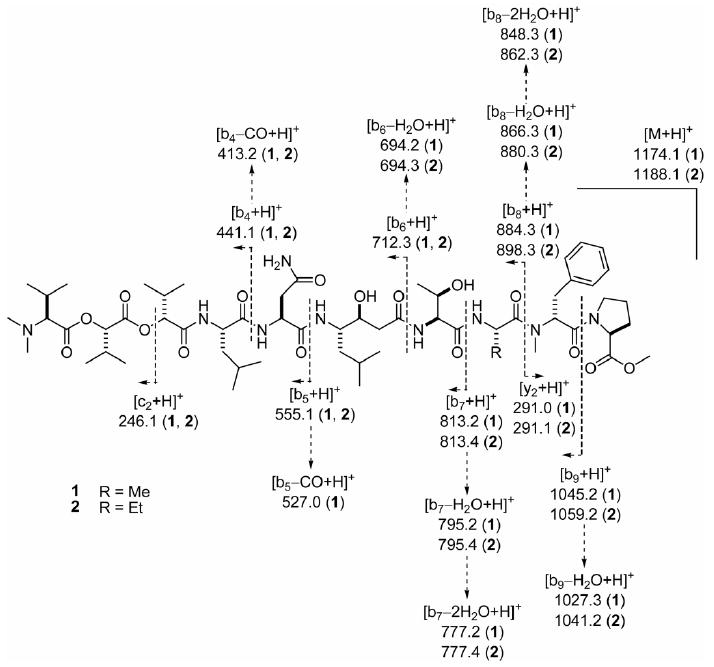
ESIMS fragmentation pattern of grassystatins A (1) and B (2).
A portion of 1 was hydrolysed (6 N HCl, 110 °C, 24 h) and analyzed by chiral HPLC-MS. This revealed the presence of l-Pro, N-Me-d-Phe, l-Ala, l-Thr, l-Asp,15 l-Leu and N,N-Me2-l-Val. In addition, peaks corresponding to both l- and d-Hiva were detected, indicating two units of opposite configuration were present. To assign their order, another portion of 1 was subjected to base hydrolysis (0.5 N NaOH/MeOH 1:1, rt, 72 h) to selectively hydrolyze the ester bonds and liberate the two terminal units (Hiva-2 and N,N-Me2-Val). Chiral analysis of the base hydrolysate indicated the presence of l-Hiva only, thus determining the configuration shown for 1 (Figure 1).
To establish the configuration of the Sta, a portion of the acid hydrolysate of 1 was derivatized with l-FDLA and subjected to modified Marfey's analysis.16 Peaks corresponding to both (3S,4S)- and (3R,4S)-Sta-l-FDLA were detected, probably due to epimerization at C-3 resulting from dehydration/rehydration. An attempt to confirm the relative configuration of this unit in situ by J-based analysis17 failed, probably because the small H-27–H-28 coupling (2.7 Hz) precluded measurement of heteronuclear coupling constants by HETLOC18,19 across this bond. It was recently shown that the relative configuration of statine and statine-like units derived from other amino acids can be easily determined by examination of the coupling constants of the α-methylene signals.20 The downfield H-2a signal (H-26a) shows a large coupling to H-27 and the upfield H-26 (H-26b) shows a small coupling to H-27 (8.7 and 5.4 Hz, respectively), thus indicating that the configuration is 3S,4S.
HRESI/APCIMS of 2 suggested a molecular formula of C59H97N9O16 (m/z 1226.6687 for [M + K]+, 1210.6936 for [M + Na]+, 1188.7119 for [M + H]+, and 605.8516 for [M + H + Na]2+), and the 1H NMR spectrum indicated a striking similarity to 1, including the same conformational ratio. Examination of the 1H NMR, COSY, HMQC, HMBC, ROESY and TOCSY spectra of 2 (Table 1 and Supporting Information) revealed the presence of the same units found in 1, except for 2-amino-butyric acid (Aba) in place of Ala. The close similarity of proton and carbon chemical shifts between 1 and 2 indicated that 2 had the same sequence and relative configuration as 1. Compounds 1 and 2 exhibited very similar optical rotation ([α]20D −4.4 and −5.0 respectively, c 0.08 and 0.1, both in MeOH), indicating that they have the same absolute configuration. The sequence of 2 was confirmed by ESIMS fragmentation (Figure 3).
The m/z peak at 1009.5941, for [M + Na]+, in the HRESI/APCIMS and NMR data for 3 suggested a molecular formula of C50H82N8O12. Analysis of the 1H NMR spectrum suggested that the compound was a peptide (amide signals at δH 6–8, α-proton signals at δH ∼4–5.5) with at least two conformers, the most prominent of which exists in the ratio of 2.45:1. Aromatic signals (δH 7.2–7.3), putative N-methyl singlets (δH 3.090, 3.087, 3.05 and 2.77) and an O-methyl singlet (δH 3.75) were also observed. Analysis of the 1H NMR, COSY, edited HSQC, HMBC, ROESY and TOCSY spectra of 3 recorded in CDCl3 revealed the presence of four proteinogenic α-amino acids (Pro, Gly, Ile, Leu), two N-methylated α-amino acids (N-Me-Phe, N-Me-Gln), one hydroxy acid (2-hydroxy-3-methylpentanoic acid, Hmpa), and Sta (C-25–C-32, Table 2). The sequence N-Me-Phe–Gly–Ile–Sta–Gln–Leu–Hmpa could be determined by HMBC analysis. A ROESY correlation between H-5a and H-8 allowed the joining of O-Me-Pro to N-Me-Phe. ROESY data also confirmed the previously determined HMBC sequence (Table 2). It was unambiguously established that C-5 of N-Me-Gln (C-37 in 3) was the primary amide carbon, by virtue of the HMBC correlation of H-28 to C-33 and correlations from H-34 and H3-38 to C-39. Additionally, there was a [M − 128]+ peak at m/z 858.5322 in the HRESI/APCIMS which was consistent with loss of O-Me-Pro (calcd for C44H72N7O10, 858.5341). By default, an OH group was proposed at C-46, and this was supported by the proton chemical shift at this position (δH 4.15), which suggested OH rather than an acyloxy group. The sequence was further confirmed by ESIMS fragmentation (Figure 4).
Table 2.
NMR Spectral Data for Grassystatin C (3) at 600 MHz in CDCl3
| C/H No. | Conformer 1a | Conformer 2a | 1H–1H COSY | Key HMBCc | Key ROESY | |||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC,b mult. | δH (J in Hz) | δC,b mult. | |||||
| O-Me-Pro | 1 | 173.1, s | 173.1, s | |||||
| 2 | 4.35, dd (7.9, 5.4) | 58.8, d | 4.35, dd (7.9, 5.4) | 58.8, d | 3a, 3b | 1 | ||
| 3a | 2.02, m | 28.8, t | 2.02, m | 28.8, t | 2, 3b, 4a, 4b | 1 | ||
| 3b | 1.82, m | 1.82, m | 2, 3a, 4a, 4b | 1 | 6 | |||
| 4a | 1.88, m | 24.8, t | 1.88, m | 24.8, t | 3a, 3b, 4b, 5a, 5b | |||
| 4b | 1.72, m | 1.72, m | 3a, 3b, 4a, 5a, 5b | |||||
| 5a | 3.45, m | 46.7, t | 3.45, m | 46.7, t | 4a, 4b, 5b | H-8 | ||
| 5b | 3.05, m | 3.05, m | 4a, 4b, 5a | |||||
| 6 | 3.72, s | 52.3, q | 3.72, s | 52.3, q | 1 | 3b | ||
| N-Me-Phe | 7 | 168.6, s | 168.6, s | |||||
| 8 | 5.45, dd (8.8, 6.4) | 56.2, d | 5.40, dd (8.8, 6.3) | 56.4, d | 9a, 9b | 16, 17 | H-5a, 16 | |
| 9a | 3.27, m | 35.4, t | 3.27, m | 35.4, t | 8, 9b | 7 | 16 | |
| 9b | 2.83, m | 2.83, m | 8, 9a | 7 | 16 | |||
| 10 | 136.7, s | 136.7, s | ||||||
| 11/15 | 7.21, m | 129.3, d | 7.21, m | 129.3, d | 12/14 | |||
| 12/14 | 7.25, m | 128.5, d | 7.25, m | 128.5, d | 11/15, 13 | |||
| 13 | 7.19, m | 126.8, d | 7.19, m | 126.8, d | 12/14 | |||
| 16 | 3.057, s | 30.0, q | 3.061, s | 30.0, q | 8, 17 | 8, 9a, 9b, 18a, 18b | ||
| Gly | 17 | 168.8, s | 168.8, s | |||||
| 18a | 4.11, dd (−17.4, 4.7) | 41.2, t | 4.23, dd (−17.1, 5.5) | 41.1, t | 18b, NH | 17, 19 | 16 | |
| 18b | 3.94, dd (−17.4, 3.7) | 3.82, dd (−17.1, 3.8) | 18a, NH | 17, 19 | 16 | |||
| NH | 7.49, dd (4.7, 3.7) | 7.70, dd (5.5, 3.8) | 18a, 18b | 19 | 20 | |||
| Ile | 19 | 171.8, s | 171.8, s | |||||
| 20 | 4.50, dd (8.0, 5.7) | 57.9, d | 4.56, dd (9.2, 4.4) | 57.8, d | 21, NH | 19, 25 | NH (Gly) | |
| 21 | 2.01, m | 36.6, d | 2.13, m | 36.5, d | 20, 22, 23b | |||
| 22 | 0.91, m | 15.7, q | 0.93, m | 11.8, q | 21 | |||
| 23a | 1.48, m | 24.3, t | 1.46, m | 24.3, t | 23b, 24 | |||
| 23b | 1.12, m | 1.12, m | 22, 23a, 24 | |||||
| 24 | 0.88, m | 11.7, q | 0.88, m | 11.7, q | 23a, 23b | |||
| NH | 7.25, m | 7.40, d (8.9) | 20 | 25 | 26a, 26b | |||
| Sta | 25 | 172.6, s | 173.0, s | |||||
| 26a | 2.48, dd (−13.9, 9.3) | 38.7, t | 2.56, dd (−13.3, 9.6) | 38.2, t | 26b, 27 | 25 | NH (Ile) | |
| 26b | 2.35, m | 2.39, m | 26a, 27 | 25 | NH (Ile) | |||
| 27 | 3.92, m | 72.3, d | 3.95, m | 73.7, d | 26a, 26b, 28, OHd | |||
| 28 | 3.98, m | 52.0, d | 4.07, m | 52.5, d | 27, 29, NH | 33 | ||
| 29 | 1.35, m (2H) | 38.9, t | 1.35, m (2H) | 38.9, t | 28, 30 | |||
| 30 | 1.45, m | 24.7, d | 1.45, m | 24.7, d | 29, 31, 32 | 38 | ||
| 31 | 0.88, m | 23.7, q | 0.84, m | 23.3, q | 30 | |||
| 32 | 0.83, m | 21.2, q | 0.83, m | 21.2, q | 30 | |||
| OH | 4.78, b | 4.78, b | 27d | |||||
| NH | 6.49, b | 7.92, d (8.5) | 28 | 33 | 34 | |||
| N-Me-Gln | 33 | 170.2, s | 170.2, s | |||||
| 34 | 5.09, m | 55.8, d | 5.07, m | 59.5, d | 35a, 35b | 33, 38, 39 | 38, 40, NH (Sta) | |
| 35a | 2.36, m | 23.7, t | 2.60, m | 25.9, t | 34, 35b, 36a, 36b | 38 | ||
| 35b | 1.85, m | 1.64, m | 34, 35a, 36a, 36b | 33, 37 | 38 | |||
| 36a | 2.30, m | 32.5, t | 2.51, m | 33.1, t | 35a, 35b, 36b | 37 | ||
| 36b | 2.14, m | 2.22, ddd (−14.1, 8.5, 5.6) | 35a, 35b, 36a | 37 | 38 | |||
| 37 | 175.0, s | 175.0, s | ||||||
| 38 | 3.02, s | 30.3, q | 2.74, s | 29.1, q | 34, 39 | 30, 32, 34, 35a, 35b, 36b, 40, 41b, 43/44 | ||
| NH2a | 6.45, b | 6.43, b | NH2b | |||||
| NH2b | 6.15, b | 6.14, b | NH2a | |||||
| Leu | 39 | 173.5, s | 172.3, s | |||||
| 40 | 4.85, m | 47.9, d | 4.87, m | 46.9, d | 41a, 41b, NH | 39 (w), 45 (w) | 34, 38 | |
| 41a | 1.67, m | 40.8, t | 1.78, m | 41.0, t | 40, 41b, 42 | 39 | ||
| 41b | 1.32, m | 1.53, m | 40, 41a, 42 | 39 | 38 | |||
| 42 | 1.66, m | 24.7, t | 1.66, m | 24.7, t | 41a, 41b, 43, 44 | |||
| 43 | 0.97, m | 21.2, q | 0.94, m | 21.9, q | 42 | 38 | ||
| 44 | 0.93, m | 23.4, q | 0.97, m | 23.2, q | 42 | 38 | ||
| NH | 7.27, m | 7.19, m | 40 | 45 | ||||
| Hmpa | 45 | 174.3, s | 175.1, s | |||||
| 46 | 4.11, m | 74.1, d | 4.11, m | 74.1, d | 47 | 45 | ||
| 47 | 1.83, m | 38.5, d | 1.83, m | 38.5, d | 46, 48, 49a, 49b | |||
| 48 | 0.80, d (6.8) | 12.8, q | 0.76, d (6.8) | 12.4, q | 47 | |||
| 49a | 1.45, m | 26.2, t | 1.45, m | 26.2, t | 47, 49b, 50 | |||
| 49b | 1.31, m | 1.31, m | 47, 49a, 50 | |||||
| 50 | 0.92, m | 11.8, q | 0.92, m | 11.8, q | 49a, 49b | |||
| OH | ||||||||
Conformers 1 and 2 were the most prominent, and they were evident in approximately a 2.45:1 ratio.
Multiplicity derived from APT and HMQC spectra.
Protons showing long-range correlation to indicated carbon.
Correlation apparent for conformer 2 only.
Figure 4.
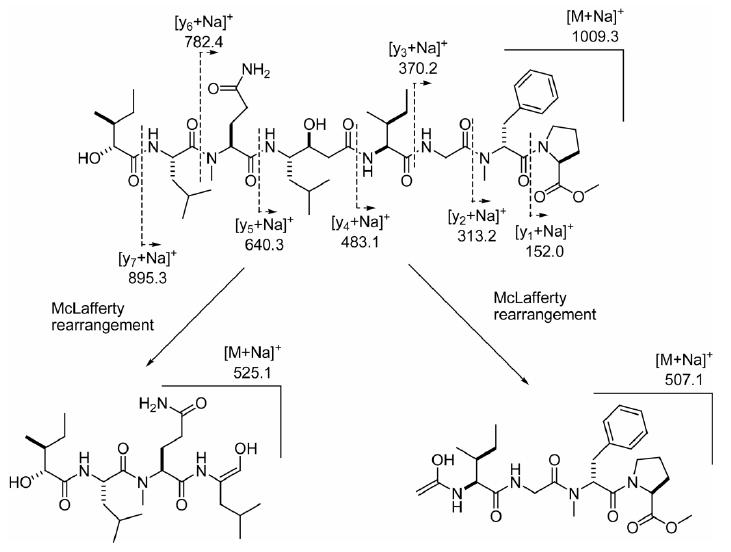
ESIMS fragmentation pattern for grassystatin C (3).
A portion of 3 was hydrolysed (6 N HCl, 110 °C, 24 h) and analyzed by chiral HPLC-MS. Peaks corresponding to l-Pro, N-Me-d-Phe, l-Ile, N-Me-l-Glu,21 and l-Leu were detected. The four stereoisomers of Hmpa eluted very closely together, but a putative assignment of (2R,3S)-Hmpa was made. This was later confirmed by analysis of the hydrolysate by conventional chiral HPLC with a different column, under conditions where the four stereoisomers eluted further apart (see Experimental Section). A portion of the hydrolysate was then derivatized with l-FDLA as with 1, and once again, two peaks were detected corresponding to (3R,4S)-Sta-l-FDLA and (3S,4S)-Sta-l-FDLA. The further downfield of the CH2 protons at C-26 showed a large coupling constant to H-27 (9.3 Hz), indicating that the configuration of this unit is 3S,4S.20
Protease Profiling and Cellular Activity Studies
The structures of compounds 1–3, in particular the presence of the statine unit and similarity to pepstatin A, led us to suspect that these compounds may be aspartic protease inhibitors. To test activity and to probe selectivity for certain aspartic and other proteases, we screened 1 against a panel of proteases to identify inhibitory activity at 10 μM (Figure 5 and Table S2, Supporting Information). It was found to be active against a subset of aspartic proteases – cathepsin D, cathepsin E. The only other proteases with compromised activities were the metalloproteases ADAM9, ADAM10 and TACE (Figure 5). Subsequent validation of these hits revealed that the greatest activity was against cathepsin E (IC50 886 pM). Compounds 1–3 all showed selectivity for cathepsin E over cathepsin D compared to pepstatin A (Table 3).
Figure 5.

Protease screen treated with grassystatin A (1), 10 μM. Values represent % enzyme activity compared to solvent control, and additionally represented by a continuous color scale (0% red, 100% green).
Table 3.
IC50s of Grassystatins A–C (1–3) against Aspartic and Metalloproteases Identified in the Primary Screen
| Protease | Grassystatin A (1) | Grassystatin B (2) | Grassystatin C (3) | Pepstatin Aa | GM6001a |
|---|---|---|---|---|---|
| Cathepsin D | 26.5 ± 5.4 nM | 7.27 ± 0.90 nM | 1.62 ± 0.3 μM | 173 ± 9.9 pM | |
| Cathepsin E | 886 ± 135 pM | 354 ± 192 pM | 42.9 ± 1.7 nM | 181 ± 8.5 pM | |
| ADAM9 | 46.1 ± 21.7 μMb | 85.5 ± 4.0 μMb | >100 μMb | 56.3 ± 6.4 nM | |
| ADAM10 | >100 μMb | 87.2 ± 17.1 μMb | >100 μMb | 263 ± 9.2 nM | |
| TACE | 1.23 ± 0.21 μMb | 2.23 ± 0.23 μMb | 28.6 ± 3.2 μMb | 13.1 ± 1.8 nM |
IC50s of positive controls.
These IC50s were calculated from the later part of the progress curves, as time-dependent inhibition was evident (see text).
Of the metalloproteases, only TACE inhibition was validated in the second round of assays (Table 3). IC50s against ADAM9 and ADAM10 were in the high micromolar range or above 100 μM. The IC50s of TACE inhibition were in the low micromolar range, and, in contrast to the dose-dependent but time-independent inhibition of cathepsins (Figure 6A and Figure S1A, Supporting Information), analysis of the progress curves revealed concentration- and time-dependent inhibition (Figure 6B and Figure S1B, Supporting Information).
Figure 6.
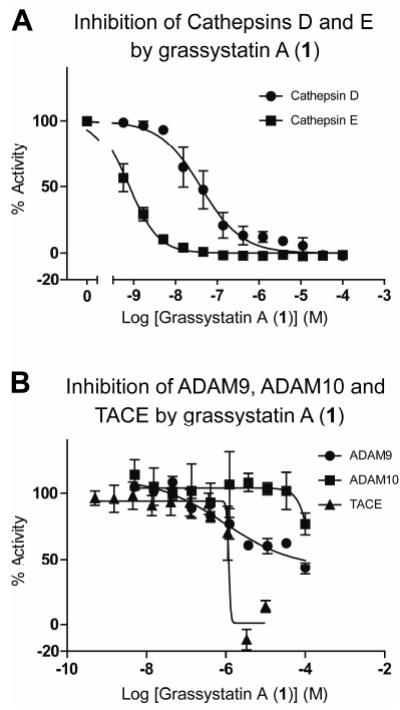
Dose-response curves for grassystatin A (1) against A) cathepsins D and E, and B) ADAM9, ADAM 10 and TACE. % Activity figures are derived for initial reaction slope for A) and end-slope for B), due to apparent time-dependent inhibition (see Figure S1, Supporting Information).
To assess whether grassystatin A (1) was able to inhibit cellular enzymes, we investigated the effect of 1 on MCF7 cell lysate, using the same fluorogenic substrate used in previous experiments involving cathepsins D and E (see Figure 7A and Experimental Section). MCF7 cells express both cathepsins D and E,22, 23 and they have previously been used to assess the cell-permeability of pepstatin A derivatives using enzymatic experiments.24 However, relative expression levels of cathepsin D and E in this cell line have not been directly compared. The apparent IC50s of inhibition in this system were ∼0.5 μM for 1 and ∼5 nM for pepstatin A. For both compounds, this represents roughly a 20-fold decrease in potency compared to experiments with purified cathepsin D (and cathepsin E in the case of pepstatin A), but for 1 a 564-fold decrease in potency compared to experiments with purified cathepsin E.
Figure 7.

Activities of grassystatin A (1) and pepstatin A against MCF7 cellular proteases as determined with a cathepsin D/E substrate (see text). A) MCF7 cell lysate was directly treated with grassystatin A (1) or pepstatin A. B) MCF7 cells were treated with grassystatin A (1) or pepstatin A for 1 h. Cells were lysed and the protease activity of the lysates assessed.
*Denotes significance of P < 0.05 using a two-tailed t test. Data points are shown ± SD.
If one examines the relative potency of 1 and pepstatin A in experiments using purified cathepsins D and E (Table 3), it is apparent that the IC50 of 1 is 153-fold higher than pepstatin A for cathepsin D, and against cathepsin E the IC50 of 1 is 4.9-fold higher than pepstatin A. In the MCF7 lysate experiment (Figure 7A), 1 exhibits a 100-fold difference in IC50 compared to pepstatin A (∼0.5 μM versus ∼5 nM, respectively). This fact, together with similar (20-fold) decrease in potency of pepstatin A and grassystatin A (1) of protease inhibition in MCF7 lysate compared with purified cathepsin D, suggests that cathepsin D predominates in MCF7 lysate, as the potency difference is closer to 153- than 4.9-fold. Pepstatin A exhibits reduced potency in MCF7 lysate compared to both purified cathepsins D and E, as mentioned previously. This suggests the presence of other enzymes in the lysate which can cleave the substrate, that are either not inhibited by pepstatin A, or only at high concentrations of this compound.
We then treated intact living MCF7 cells with both compound 1 and pepstatin A, using a protocol previously used to study pepstatin A conjugates (see Figure 7B).24 Following treatment, cells were washed and treated with trypsin, which has previously been shown to reduce surface bound inhibitors which can interfere with subsequent enzymatic assays.25 After lysis, the enzymatic activity of the lysate was measured as before with a fluorogenic substrate. Consistent with previous reports that pepstatin A has poor cell-permeability, its apparent IC50 (∼5 μM) is now almost 29,000-fold higher than with purified cathepsins D and E and 1,000-fold higher than with MCF7 lysate. Conversely, the apparent IC50 for compound 1 (also ∼5 μM) is almost 200-fold higher than with purified cathepsin D and 10-fold higher than with MCF7 lysate (see Figure 7B). The selectivity of pepstatin A is of course different to that of compound 1 (vide supra), and the subcellular targeting of 1 is unknown, including local concentration at the site of cathepsin D/E localization. Another uncertainty is the stability of 1 and its route of metabolism within cells and in lysates. However, collectively these results show that the barrier of the cell membrane reduces the apparent potency of pepstatin A to a much greater extent than that of compound 1 (1000- versus 10-fold), and they suggest that 1 can successfully enter the cell to inhibit cellular enzymes. Results of subsequent experiments (vide infra) supported this notion.
Investigation of the Effect of Grassystatin A (1) on Antigen Presentation
Cathepsin E is thought to have a functional role in the proteolysis of antigenic peptides, which are subsequently presented as antigens on the surface of antigen presenting cells (APCs) in the major histocompatability complex (MHC) class II pathway.24, 26 Antigen presentation to T cells stimulates their proliferation and the release of certain inflammatory cytokines (vide infra).27 We therefore investigated the effect of compound 1 on human peripheral blood mononuclear cells (PBMCs). PBMCs are a mixture containing various APCs (dendritic cells, B cells and macrophages) and T cells. We examined the effect of 1 on T cells using flow cytometry to gate for CD3+ lymphocytes (T cells). 10 μM 1 was able to significantly reduce T cell proliferation in response to exogenous antigen (Tetanus toxin C-fragment, TTc, Figure 8A). In the same experiment, T cell viability was unaffected (data not shown).
Figure 8.
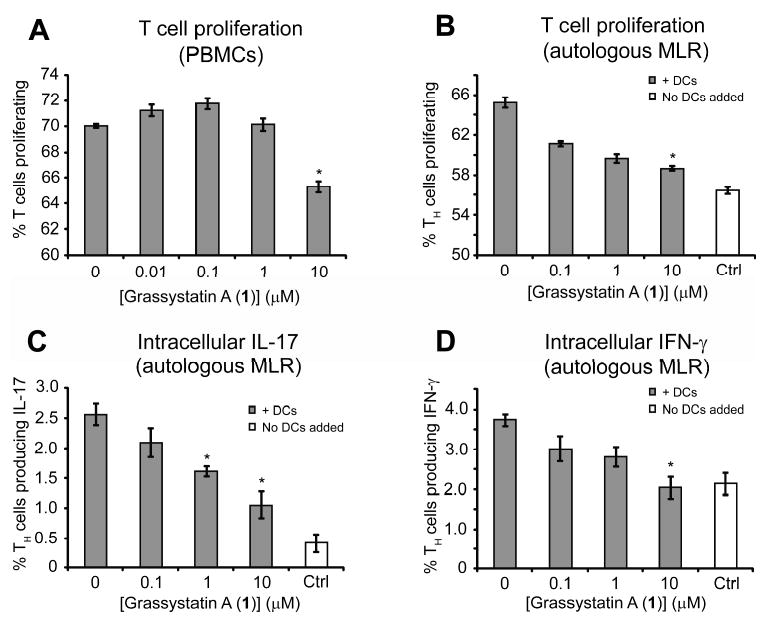
Downregulation of antigen presentation of T cells and TH cells after treatment with grassystatin A (1) on activated PBMC and DC. A) Downregulation of the activation of CD3+ T cells on whole PBMC after treatment with different concentrations of 1. B) Downregulation of TH activation (proliferation) by the addition of different concentrations of 1. C, D) Effect of 1 on the production of intracellular IFNγ (C) and IL-17 (D) by TH cells induced by autologous activated DC. “Ctrl” refers to T cells that did not have DCs added to them. *Denotes significance of P < 0.05 using a two-tailed t test. Data points are shown ± SEM.
We then investigated the effect of 1 on the interaction between monocyte derived dendritic cells (DCs) and CD4+ T cells (T helper cells, TH), in a mixed lymphocyte reaction (MLR). The first experiments were autologous MLRs, where DCs and T cells came from the same human donor. DCs were chosen for this study as they are the most potent antigen presenting cells and have much higher cathepsin E expression than other APCs.28 The main targets of antigen presentation are TH cells, which go on to orchestrate the ensuing immune response. We therefore decided to use an enriched population of these cells as the responders in our assay. Differentiated DCs were cultured in the presence of antigen (TTc), phorbol 12-myristate 13-acetate (PMA) and TH cells for 5 days. Compound 1 was able to reduce T cell proliferation in a dose dependent manner (Figure 8B). TTc and PMA alone (i.e., in the absence of DCs) were unable to increase T cell proliferation, and thus this effect of 1 is dependent on DCs. In these experiments, 1 was also able to inhibit upregulation of interleukin-17 (IL-17) (Figure 8C) and interferon-γ (IFN-γ) (Figure 8D) in response to antigen presentation.
To determine whether 1 had any effect on T cell recognition of foreign MHC II proteins, we carried out the same experiment with DCs and TH cells from different donors. We found that 1 had no effect on DC stimulated proliferation in an allogeneic MLR (data not shown). This is likely because T cells were recognizing non-self MHC II proteins on the surface of DCs.29 Even though proliferation was not reduced, we still observed a significant downregulation of IL-17 and IFN-γ production (Figures 9A and 9B, respectively).
Figure 9.
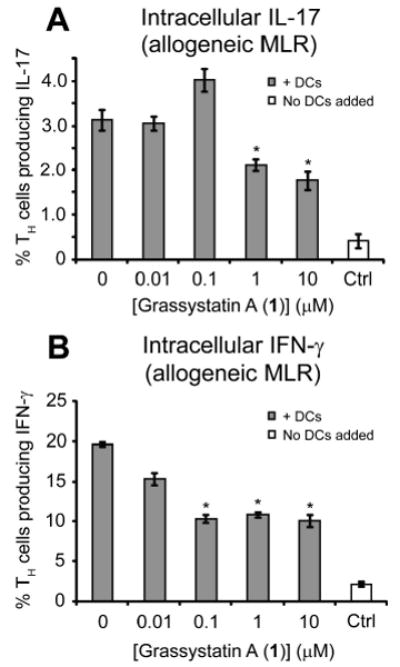
Effect of 1 on the production of A) intracellular IFNγ and B) IL-17 by TH cells induced by allogeneic activated DC in an MLR. “Ctrl” refers to T cells that did not have DCs added to them. *Denotes significance of P < 0.05 using a two-tailed t test. Data points are shown ± SEM.
Molecular Docking
To gain some insight into the structural basis for the selectivity of the grassystatins for cathepsin E over D, compounds 1 and 3 were docked into these two enzymes (Figure 10). For both proteins, compounds 1 and 3 were successfully docked using AutoDock Vina 1.0,30 with the ligand treated as fully flexible (see Experimental Section). To be consistent with the mode of cathepsin inhibition by pepstatin A, input structures of the ligands had all amide bonds trans, except the proline amide, for which separate cis and trans structures were produced. For cathepsin D, the crystal structure of pepstatin A bound to this enzyme was used for docking (PDB code 1LYB).31 We were able to successfully re-dock pepstatin A into this structure (Figure S2A, Supporting Information), prior to docking our ligands. For cathepsin E, homology modeling was used to obtain an appropriate starting structure, because the only crystal structure published is of an early activation intermediate (PDB code 1TZS, see Experimental Section).32 For the homology modeling, the structure of mature porcine pepsin (PDB code 4PEP),33 a protein with highly homologous primary sequence, was used as a template for the primary sequence of cathepsin E, taken from 1TZS (see Experimental Section). Pepstatin A was successfully docked into the resulting structure (Figure S2B, Supporting Information) before docking of the 1 and 3 was attempted.
Figure 10.
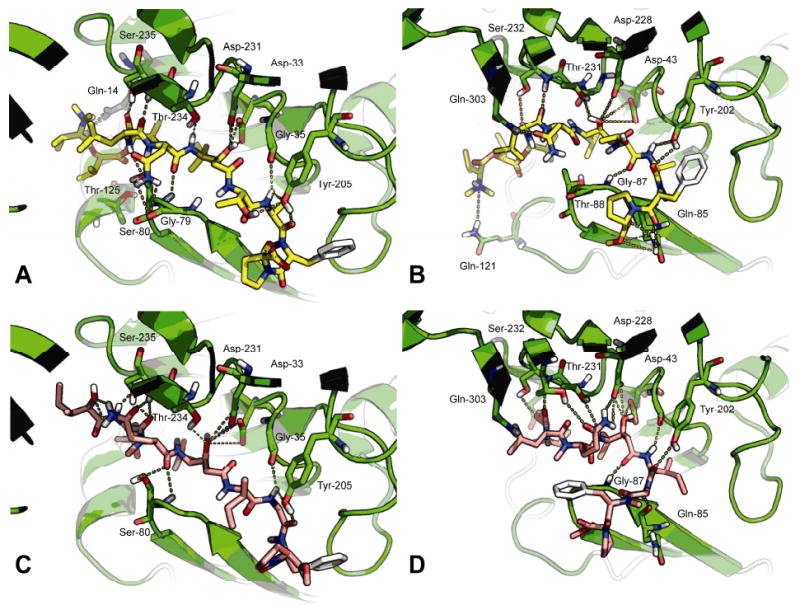
Docked structures of grassystatins A (1) and C (3) with cathepsins D and E. For each the protein is shown in green, and possible hydrogen bonds are shown as dotted yellow lines. A) Docked conformation of grassystatin A (1, yellow) with cathepsin D. B) Docked conformation of grassystatin A (1) with cathepsin E. C) Docked conformation of grassystatin C (3, pink) with cathepsin D. D) Docked conformation of grassystatin C (3) with cathepsin E. For docked conformations of pepstatin A, see Figure S2, Supporting Information.
Many of the putative hydrogen bond interactions suggested by the crystal structure of pepstatin A bound to cathepsin D (PDB code 1LYB)31 are also present in the model of compound 1 bound to this enzyme (Figure 10A). The reduced affinity of 1 versus pepstatin A may be due to the presence of a polar residue at P2 (Val in pepstatin A and Asn in 1). Cathepsin D has an established preference for hydrophobic residues in this position, although it is somewhat tolerant of polar residues here.34 In our docked conformation of 1, the Asn side chain is curled down in order to interact with Ser-80 in the flap, and to avoid the hydrophobic residues Met-307 and Met-309. The docked structure of grassystatin A (1) in cathepsin E (Figure 10B) shows this unit interacting with the polar residue Gln-303, which replaces Met-307 in cathepsin D. This could be one reason for an increased affinity for cathepsin E versus D. Another factor could be the numerous hydrogen bond interactions possible between the O-Me-Pro unit of 1 with Gln-85 in cathepsin E (Figure 10B). It is probably not possible to form so many hydrogen bonds with the equivalent residue in cathepsin D (His-77).
Grassystatin C (3) was less potent than grassystatins A and B (1 and 2, respectively) against both cathepsins D and E. One potential reason for this is the absence of the terminal N,N-Me2-Val, which could act as either a hydrogen bond donor (if protonated) or acceptor (if unprotonated). N,N-dimethyl groups have in the past been used to improve the affinity of ligands that interact with acidic groups, such as a key Asp residue in the H1 receptor (the native ligand histamine instead has a primary amine).35 In cathepsin D, there are several polar residues within reach of this unit (Tyr-10, Gln-14, Thr-125, Lys-130, Gln-258 and Gln-260). The situation is similar with cathepsin E, where there is a number of polar residues in the same pocket (Tyr-20, Glu-24, Glu-27, Glu-121, Asp-125, Glu-256 and Tyr-257). In the docked structure of 1 shown in Figure 10B, the basic nitrogen of N,N-Me2-Val is close to Gln-121. In the same run, another similar structure was produced, where the basic nitrogen was close to Tyr-20 (not shown). Thus, this unit may serve to anchor the inhibitor in the correct position within the binding cleft. Indeed, we observed that when docking into the same protein structure, more spurious structures36 were produced for grassystatin C (3) than for grassystatin A (1). In these situations, because 3 has fewer rotatable bonds than 1, it is unlikely that the search parameters would be insufficient for 3 and not 1.
Discussion
Consistent with the notion that natural products are compounds pre-optimized by evolution to act on specific biological targets, the marine cyanobacterium Lyngbya confervoides has yielded potent and selective protease inhibitors. Grassystatins A–C (1–3) and the related compound tasiamide B10 (Figures 1 and 2) all contain a statine or statine-like unit with the same configuration (3S,4S). This is also the same configuration as the statine units in pepstatin A. It has previously been shown that the configuration of C-3 in the central statine is important to pepstatin A's inhibitory activity against aspartic proteases.37
Like pepstatin A, 1–3 inhibited cathepsin D and E, although they were selective for cathepsin E (∼20 to ∼38 fold), while pepstatin A did not discriminate between these proteases. In pepstatin A, the central statine unit is the pharmacophore of inhibition that binds to cathepsin D at the P1–P1′ site (see Figure 2 for sites).31 If the binding mode is the same for 1–3, then the units flanking the statine unit confer the differential activity for cathepsin D and E. Cathepsin D strongly favors hydrophobic amino acids in the P2 position, compared to polar units such as Asn in 1 and 2, or N-Me-Gln in 3,38 which are more tolerated by cathepsin E.34 This could explain why these compounds are less potent inhibitors against cathepsin D compared to pepstatin A, which has valine at P2 (Figure 2). Both cathepsins D and E allow polar (but not charged) units at position P2, and hydrophobic units such as leucine are also allowed.34, 39 Therefore, the change from Thr in 1 and 2 to Ile in 3 may not account for its lower activity. The putative hydrogen bond between Asn-NH and Ser-80-OH may be particularly important to binding and this interaction is not possible in 3 because the α-nitrogen of Gln is methylated. Compound 3 does not possess terminal units N,N-Me2-l-Val–l-Hiva. The basic nitrogen of N,N-Me2-Val is probably able to interact with acidic residues in both cathepsins D and E (see Results). It has previously been shown that occupation of the S5 subsite of cathepsin E with Lys increases substrate turnover.34 Occupation of this site by positively charged residues may therefore be key to inhibitor binding.
Three metalloproteases in the ADAM family were identified in the primary screen of compound 1 (ADAM9, ADAM10 and TACE). Only one of the hits (TACE), however, could be replicated in a dose-response assay (Table 3). Inhibition of TACE by compounds 1–3 was concentration- and time-dependent (Figure 6B), with IC50s of 1.23, 2.23 and 28.6 μM, respectively. A slow onset of inhibition indicates slow binding of the inhibitor, and is apparent by a noticeable curve in the progress curve of the reaction within a timescale where the uninhibited reaction is still linear.40 Statine-based slow-binding inhibitors of aspartic proteases have been described.41 There are several examples of slow-binding inhibitors of zinc metalloproteases, for example the antihypertensive drugs captopril and enalapril are both slow-binding inhibitors of angiotensin converting enzyme (ACE).42 There are also some slow-binding inhibitors of MMPs.43 The reason for slow-binding in these cases may be the expulsion of a tightly bound, catalytically active water molecule from the active site.40 With slow binding inhibitors, the onset of inhibition depends on the preincubation time of the test compound with the enzyme. In the large scale screen, this time may have been longer than desirable, leading to an apparent lower IC50 for compound 1 against ADAM9 and ADAM10.
Consistent with previously reported experiments using pepstatin A,24, 26a, b, 26d we found that 1 was able to reduce antigen-stimulated T cell proliferation in PBMCs (Figure 8A) and antigen presentation by DCs to TH cells (Figure 8B). Concurrently, we found that 1 reduced production of IL-17 and IFN-γ by T cells (Figures 8C and 8D, respectively). IFN-γ is a pro-inflammatory molecule and the signature cytokine produced by TH1 cells that, among other effects, activates macrophages.27 These cells are strongly involved in cellular immunity against cancer and intracellular pathogens such as viruses, but are also involved in the etiology of transplant rejection.44 IL-17 is another pro-inflammatory cytokine, produced by a recently described subset of T cells, TH17 cells.45 TH17 cells and IL-17 have been implicated in a number of autoimmune and allergic diseases such as rheumatoid arthritis and asthma.45 Because of their involvement in proinflammatory disorders and the association of such pathologies with the activation of T cells by antigen presentation, we decided to investigate the contribution of 1 to the modulation or downregulation of pro-inflammatory cytokines.
Exogenous antigens are internalized by APCs and proteolytically cleaved within endosomes, before they are presented on the cell surface bound to MHC class II proteins.27 The invariant chain (Ii) is a chaperone that prevents endogenous peptides from binding to MHC class II proteins while they are transported from the endoplasmic reticulum to endosomes.46 Ii undergoes several cleavage steps both before and after entering the endosome. Its removal allows antigens to bind MHC II for subsequent presentation. Cysteine proteases, such as cathepsin S, have a well established role in Ii cleavage.47 There have been conflicting reports, however, of whether an aspartic protease-dependent cleavage is also required for MHC II maturation. Marić et al.48 found that aspartic protease inhibitors reduced cleavage of MHC II-Ii complexes in B-lymphoblastoid cells. Similarly, Zhang et al.26d found that pepstatin A induced an accumulation of Ii-containing cleavage intermediates both in a murine B cell line and live mice. Two other reports26b, c found that pepstatin A could inhibit presentation of antigens when cells (murine microglia and human DCs) were treated with intact antigens, but not when treated with pre-cleaved epitopic peptides. Recently, Costantino et al.49 have presented results suggesting that the role of different enzymes in Ii cleavage is highly variable and there is a large degree of redundancy. Our own results suggest that 1 is not able to inhibit MHC II-Ii cleavage, as DCs treated with 10 μM 1 in the allogeneic (but not autologous) MLR were still able to stimulate T cell proliferation. Furthermore, 1 was able to downregulate pro-inflammatory cytokines in both types of assays. This indicates that either presentation of TTc is inhibited in both cases, with inhibition of cytokine production being a consequence of this, or that 1 has a direct effect on cytokine expression. While further studies are needed, it opens the possibility for this compound to be used in the study of antigen presentation and also of the role of cathepsin E in inducing pro-inflammatory responses.
In conclusion, we believe that compound 1 could prove to be a valuable probe for the study of cathepsin E function. Previously, only one inhibitor selective for cathepsin E was known, a protein from the roundworm Ascaris lumbricoides.50 Because this inhibitor is not widely available, studies into the function of cathepsin E have had to rely on non-selective inhibitors such as pepstatin A or cathepsin D knockout animals/cells.51 To the best of our knowledge, grassystatins A–C (1–3) are the only other natural protease inhibitors that are selective for cathepsin E over D.
Experimental Section
General Experimental Procedures
Optical rotation was measured on a Perkin-Elmer 341 polarimeter. UV was measured on a SpectraMax M5 (Molecular Devices) and IR data obtained on a Bruker Vector 22 instrument. The purities of compounds 1–3 were determined to be greater than 95% by HPLC, MS and NMR. 1H and 2D NMR spectra in CDCl3 for 1 and 2 were recorded on a Bruker 500 MHz spectrometer. 13C and APT spectra for 1 were recorded on a Bruker 600 MHz Avance Spectrometer. 1H and 2D NMR spectra in CDCl3 for 3, and also 1H and 2D NMR spectra in DMSO-d6 for 1 were collected on a Bruker Avance II 600 MHz spectrometer using a 1-mm triple-resonance high-temperature superconducting cryogenic probe.52 Spectra were referenced to residual solvent signals [δH/C 7.26/77.0 (CDCl3) and δH/C 2.49/39.5 (DMSO-d6)]. HMQC and HSQC experiments were optimized for 145 Hz, and HMBC experiments were optimized for 7 Hz. HRESI/APCIMS data were recorded on an Agilent LC-TOF mass spectrometer equipped with an APCI/ESI multimode ion source detector in positive ion mode. LC-MS data were obtained using an API 3200 (Applied Biosystems) equipped with a Shimadzu LC system. ESIMS fragmentation data were obtained on an API 3200 by direct injection with a syringe driver. Flow cytometry was carried out on a FACSCalibur flow cytometer using CellQuest software (BD Biosciences, Heidelberg, Germany). Figures of docked ligands were prepared using PyMol.
Extraction and Isolation
Samples of Lyngbya confervoides were collected off Grassy Key on May 26, 2004 and fractionated as previously described.53 The silica gel fraction eluting with 100% methanol was purified by preparative reversed-phase HPLC (Phenomenex Luna 10u C18 AXI, 100 × 21.2 mm, 10.0 mL/min; UV detection at 220 and 254 nm), using a MeOH–H2O linear gradient (60–100% over 30 min, then 100% MeOH for 10 min), to give impure 1 and 2 at tR 24.3 and 24.9 min, respectively. These were purified using a different column (Phenomenex Ultracarb 5u ODS (30), 250 × 10.0 mm, 2.0 mL/min; UV detection at 220 and 254 nm) using the same linear gradient to furnish compound 1, tR 33.8 min (5.7 mg), and 2, tR 35.1 min (1.3 mg).
Samples of the same species were collected off Key Largo on May 8, 2003. A voucher specimen is maintained at the Smithsonian Marine Station. The freeze-dried organism was extracted with EtOAc–MeOH (1:1) to afford the non-polar extract, which was directly fractionated by silica gel column and eluted with increasing concentrations of isopropanol in CH2Cl2. The fraction eluting with 100% isopropanol (665.8 mg) was subjected to preparative reversed-phase HPLC [column, Luna C18(2) 100A AXI, 10 μm (100 × 21.20 mm), Phenomenex; flow rate 10.0 mL/min; detection by UV at 220 and 254 nm] using a linear MeOH–H2O gradient (60–100% MeOH over 30 min, then 100% MeOH for 5 min). In addition to previously isolated compounds, a minor peak eluted at tR 17.2 min, which was then deconvoluted using different conditions [column, ODS-AQ (10 × 250 mm), YMC; flow rate, 2.0 mL/min; detection by UV at 220 and 254 nm] using the same linear MeOH–H2O gradient, to furnish pure 3, tR 27.3 min (1.0 mg).
Grassystatin A (1)
Colorless amorphous solid; [α]20D −4.4 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 206 (4.93), 258 (4.03), 324 (3.2); IR (film) νmax 3291 (br), 3068 (w), 3054 (w), 3019 (w), 2955, 2937, 2925, 2914, 2851, 2360, 2342, 1733, 1646, 1540, 1457, 1374 (w), 1265, 1109 (w), 1023 (w), 896 (w), 739; NMR data, 1H NMR, 13C NMR, APT, COSY, HMQC, HMBC, ROESY, TOCSY in CDCl3, see Table 1, 1H NMR, COSY, edited HSQC, HMBC, ROESY in DMSO-d6, see Table S1, Supporting Information; HRESI/APCIMS m/z [M + Na]+ 1196.6812 (calcd for C58H95N9O16Na, 1196.6794), [M + H]+ 1174.6988 (calcd for C58H96N9O16 1174.6975), [M + H + Na]2+ 598.8455 (calcd for C58H96N9O16Na, 598.8436), [M + 2H]2+ 587.8544 (calcd for C58H97N9O16, 587.8527).
Grassystatin B (2)
Colorless amorphous solid; [α]20D −5.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 202 (4.47), 266 (2.96), 320 (2.45); IR (film) νmax 3276 (br), 3079 (w), 3054 (w), 3017 (w), 2961, 2927, 2874, 2360, 2342, 1752, 1732, 1690, 1627, 1549, 1493 (w), 1463 (w), 1436 (w), 1389 (w), 1369 (w), 1267 (w), 1207 (w), 1179 (w), 1124 (w), 1023 (w); NMR data, 1H NMR, COSY, HMQC, HMBC, ROESY, TOCSY in CDCl3, see Table 1; HRESI/APCIMS m/z [M + K]+ 1226.6687 (calcd for C59H97N9O16K, 1226.6690), [M + Na]+ 1210.6936 (calcd for C59H97N9O16Na, 1210.6951), [M + H]+ 1188.7119 (calcd for C59H98N9O16, 1188.7131), [M + H + Na]2+ 605.8516 (calcd for C59H98N9O16Na, 605.8515).
Grassystatin C (3)
Colorless amorphous solid; [α]20D −21.9 (c 0.04, MeOH); UV (MeOH) λmax (log ε) 203 (4.74), 260 (2.89), 320 (2.17); IR (film) νmax 3307 (br), 3078 (w), 3054 (w), 3016 (w), 2659, 2927, 2904, 2874, 2361, 2340, 1742, 1635, 1531, 1462, 1442, 1410, 1368, 1285 (w), 1199 (w), 1047 (w); NMR data, 1H NMR, COSY, edited HSQC, HMBC, ROESY, TOCSY in CDCl3, see Table 2; HRESI/APCIMS m/z [M + Na]+ 1009.5941 (calcd for C50H82N8O12Na, 1009.5950), [M − 128]+ 858.5322 (calcd for C44H72N7O10, 858.5341).
Acid Hydrolysis and Chiral Amino Acid Analysis by LC-MS and HPLC
A sample of 1 (100 μg) was treated with 6 N HCl at 110 °C for 24 h. The hydrolysate was concentrated to dryness, reconstituted in 100 μL H2O and then analyzed by chiral HPLC [column, Chirobiotic TAG (4.6 × 250 mm), Supelco; solvent, MeOH–10 mM NH4OAc (40:60, pH 5.23): flow rate, 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)]. l-Thr, l-Leu, l-Pro, N,N-Me2-l-Val and N-Me-d-Phe eluted at tR 7.2, 9.0, 14.4, 27.0 and 45.4 min, respectively. The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acids were as follows: l-Thr (7.2; 120→74), l-allo-Thr (7.5), d-Thr (8.6;), d-allo-Thr (11.9), l-Pro (14.4; 116→70), d-Pro (39.5), l-Leu (9.0; 132→86), d-Leu (20.6), N-Me-l-Phe (25.0; 180→134), N-Me-d-Phe (45.4), N,N-Me2-l-Val (27.0; 146→100), and N,N-Me2-d-Val (69.8). The assignment of l-Thr was confirmed by co-injection of the hydrolysate with l-allo-Thr and l-Thr. The MS parameters used were as follows: DP 31.0, EP 8.0, CE 17.3, CXP 3.1, CUR 35, CAD Medium, IS 4500, TEM 750, GS1 65, GS2 65. l-Ala was also detected in positive ion mode, at tR 8.0, but with slightly different MS conditions. The retention times (tR, min; MRM ion pair, parent→product) of the authentic standards were as follows: l-Ala (8.0; 90→44), d-Ala (14.6). The MS parameters used were as follows: DP 21.0, EP 8.0, CE 15.0, CXP 5.0, CUR 50, CAD Medium, IS 4500, TEM 750, GS1 65, GS2 65. Asp was only detected weakly in positive ion mode and consequently negative ion mode was used with the same LC conditions. l-Asp eluted at tR 6.1 min, indicating that the configuration of the Asn unit was l. The retention times (tR, min; MRM ion pair, parent→product) of the authentic standards were as follows: l-Asp (6.1; 132→88), d-Asp (6.8). The MS parameters used were as follows: DP −30.0, EP −5.0, CE −18.5, CXP −13.0, CUR 30, CAD High, IS −4500, TEM 750, GS1 65, GS2 65.
A sample of 3 was treated with 6 N HCl at 110 °C for 24 h. The hydrolysate was concentrated to dryness, reconstituted in 100 μL H2O and then analyzed by chiral HPLC [column, Chirobiotic TAG (4.6 × 250 mm), Supelco; solvent, MeOH–10 mM NH4OAc (40:60, pH 5.33): flow rate, 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)]. N-Me-l-Glu, l-Ile, l-Leu, l-Pro and N-Me-d-Phe eluted at tR 6.0, 8.3, 8.6, 13.3 and 41.7 min, respectively. The retention times (tR, min; MRM ion pair, parent→product) of the authentic standards were as follows: N-Me-l-Glu (6.0; 162→44), N-Me-d-Glu (15.8), l-Ile (8.3; 132→86), l-allo-Ile (8.5), d-allo-Ile (19.6), d-Ile (22.2), l-Leu (8.6; 132→86), d-Leu (19.8), l-Pro (13.3; 116→70), d-Pro (35.2), N-Me-l-Phe (23.2; 180→134), and N-Me-d-Phe (41.7). To further separate the isobaric Ile and Leu units, different LC conditions were employed [column, Chirobiotic TAG (4.6 × 250 mm), Supelco; solvent, MeOH–10 mM NH4OAc (90:10, pH 5.65); flow rate, 0.5 mL/min; detection by MS (MRM scan)]. l-Ile and l-Leu eluted at tR 12.3 and 13.1 min, respectively. The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acid standards were as follows: l-Ile (12.3; 132→86), l-allo-Ile (13.4), d-allo-Ile (57.5), d-Ile (70.5), l-Leu (13.1; 132→86), and d-Leu (51.7). The MS parameters used were as follows: DP 31.0, EP 8.0, CE 17.3, CXP 3.1, CUR 35, CAD Medium, IS 4500, TEM 750, GS1 65, GS2 65.
Hmpa in the hydrolysate of 3 was detected in negative ion mode [column, Chirobiotic TAG (4.6 × 250 mm), Supelco; solvent, MeOH–10 mM NH4OAc (40:60, pH 5.35); flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan)]. The MS parameters used were as follows: DP −35.0, EP −8.0, CE −17.9, CXP −1.7, CUR 40, CAD Medium, IS −4500, TEM 750, GS1 65, GS2 65. (2R,3S)-Hmpa from the hydrolysate eluted at tR 6.4 min. The retention times (tR, min; MRM ion pair, parent→product) of the authentic standards were as follows: (2S,3R)-Hmpa (6.0; 131→85), (2S,3S)-Hmpa (6.2; 131→85), (2R,3S)-Hmpa (6.4; 131→85), (2R,3R)-Hmpa (7.0; 131→85). The hydrolysate was examined under different HPLC conditions in order to confirm this assignment [column, Chiralpak MA (+) (4.6 × 50 mm), Daicel Chemical Industries, Ltd.; solvent, 2 mM CuSO4–CH3CN (85:15); flow rate, 1.0 mL/min; detection by UV absorption at 254 nm]. (2R,3S)-Hmpa from the hydrolysate eluted at tR 15.4 min. The retention times (tR, min) of the authentic standards were as follows: (2R,3S)-Hmpa (15.4), (2R,3R)-Hmpa (17.9), (2S,3R)-Hmpa (22.7), (2S,3S)-Hmpa (27.5). Under these conditions, all other units eluted at tR <6.5 min.
Base Hydrolysis to Determine Configuration of Hiva Units
The acid hydrolysate of 1 was analysed by chiral HPLC [column, Chirobiotic TAG (4.6 × 250 mm), Supelco; solvent, MeOH–10 mM NH4OAc (60:40, pH 5.63); flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan)]. Both l-Hiva and d-Hiva were detected at tR 6.0 and 6.4 min, respectively. The retention times (tR, min; MRM ion pair, parent→product) of the authentic standards were as follows: l-Hiva (6.0; 117→71), d-Hiva (6.4). A sample of 1 (100 μg) was suspended in 80 μL MeOH–0.5 N NaOH (1:1) and left to stand at room temperature for 72 h. The solution was neutralized by the addition of 20 μL 1 N HCl, and was then analysed by chiral HPLC-MS as before. Only l-Hiva was detected at tR 6.0 min. The retention times (tR, min; MRM ion pair, parent→product) of the authentic standards were as follows: l-Hiva (6.0; 117→71), d-Hiva (6.4). The MS parameters used were as follows: DP −30.0, EP −3.0, CE −17.3, CXP −2.0, CUR 45, CAD Medium, IS −4500, TEM 650, GS1 50, GS2 25.
Modified Marfey's Analysis to Determine Configuration of Statine Units
Samples of both 1 and 3 (35 μg) were subjected to acid hydrolysis, derivatized with l-FDLA as described previously, and analyzed by reversed-phase HPLC [column, Alltima HP C18 HL (4.6 × 250 mm), 5 μm, Alltech; flow rate, 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan, 468→408)], using a linear gradient of MeOH in H2O (both containing 0.1% HCOOH, 40–100% MeOH over 50 min). Two peaks, corresponding to (3S,4S)-Sta-l-FDLA and (3R,4S)-Sta-l-FDLA, were observed in both samples in a 1:1 ratio at tR 35.5 and 35.9 min, respectively. The retention times (tR, min) of the authentic standards were as follows: (3S,4S)-Sta-l-FDLA (35.5), (3R,4S)-Sta-l-FDLA (35.9), (3S,4S)-Sta-d-FDLA [corresponding to (3R,4R)-Sta-l-FDLA, 45.7], (3R,4S)-Sta-d-FDLA [corresponding to (3S,4R)-Sta-l-FDLA, 46.4]. The MS parameters used were as follows: DP −60.0, EP −7.0, CE −28.0, CXP −7.4, CUR 40, CAD High, IS −4500, TEM 750, GS1 40, GS2 40.
Protease Inhibition Screen
Compound 1 was added into the reaction buffer containing enzyme by acoustic droplet ejection (Echo 550, Labcyte Inc., Sunnyvale, CA) such that the final concentration was 10 μM. After incubation at room temperature for 10–15 min, the substrate was added, after which fluorescence at each relevant Ex/Em wavelength was measured every 5 min for 2 h. The substrate alone in the reaction buffer served as background. The activity of 1 was evaluated by obtaining % enzyme activity relative to the slope of no inhibitor control. Each enzyme assay was performed in duplicate by Reaction Biology Corp. (Malvern, PA). For individual buffers, substrates and positive controls, see Table S2.
Protease Inhibition Assays to Determine IC50 Values
Assays for 1 and 2 were carried out in the same way as in the protease screen, using 3-fold serial dilutions in DMSO, starting at 10 μM and 100 μM respectively, with 10 different concentrations of each. Compound 3 was insoluble in DMSO, and thus a dilution series in EtOH was used. For 1–3, a 3-fold dilution series starting at 100 μM was used, with 10 different concentrations. Assays were carried out by Reaction Biology Corp. (Malvern, PA). Enzyme activity (in %), calculated as above, was used to determine IC50 values with non-linear regression in GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
Inhibition of Cellular Cathepsins D and E by Grassystatin A (1)
To measure the in vitro inhibition of cellular cathepsins by 1 and pepstatin A, kinetic assays were carried out using lysate prepared from untreated MCF7 cells. MCF7 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) containing 10% fetal bovine serum (FBS, HyClone, Logan, UT), in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were lysed with NP-40 lysis buffer (1% NP-40, 50 mM NaOAc, pH 4.0), and a portion of the lysate (50 μL) was incubated at 37 °C in the presence of varying concentrations of the test compounds and 10 μM Mca–Gly–Lys–Pro–Ile–Leu–Phe–Phe–Arg–Leu–Lys(Dnp)–d-Arg-NH2 in 50 mM NaOAc pH 4.0 (the same substrate used for cathepsins D and E in other assays). The total volume of the reactions was 100 μL, and the reaction was monitored by measuring the increase in fluorescence (λex = 320 nm, λem = 405 nm).
To provide some information of cell permeability, intact live MCF7 cells were treated directly with test compounds, using a procedure previously used for this purpose.24 Cells were cultured as described above, and were seeded into 24-well plates. When cells reached 80–100% confluency, varying concentrations of compound 1 or pepstatin A were added. After 1 h incubation, the medium was removed and the cells were washed then trypsinized for 10 min, in order to remove surface-bound inhibitor.25 Cells were collected by centrifugation and lysed with NP-40 lysis buffer. The lysate (50 μL) from each well was incubated at 37 °C with 10 μM Mca–Gly–Lys–Pro–Ile–Leu–Phe–Phe–Arg–Leu–Lys(Dnp)–d-Arg-NH2 in 50 mM NaOAc (pH 4.0, total volume 100 μL). The reaction was monitored as above.
Isolation of PBMCs and Culture of Monocytes and DCs
PBMCs were isolated from buffy coats (leukopac, PBL) obtained from Lifesouth Community Blood Center (Gainesville, FL, USA) by Ficoll-Hypaque density gradient centrifugation using Lymphoprep (Axis-Shield, Norway). Briefly, the contents of the buffy coat were diluted to three times its volume in sterile 1× PBS pH 7.4 (Gibco, California, USA). The dilution was layered onto the Lymphoprep in a 2:1 ratio. The sample was then centrifuged for 25 min at room temperature and 250 g. The PBMCs were collected at the interface, washed twice with PBS and centrifuged each time for 10 min at 4 °C and 250 g. Cell viability was assessed by trypan blue exclusion. All cultures of human PBMC and derived cells were maintained in RPMI 1640 medium (Sigma, Missouri, USA) supplemented with 2 mM l-glutamine (Life Technologies, Paisley, Scotland), 5000 U/mL penicillin (Sigma, Missouri, USA), 5000 U/mL streptomycin sulfate (Sigma, Missouri, USA), and 10% fetal bovine serum (Gibco, California, USA). PBMCs were either used in an experiment with 1 at various concentrations or separated further.
Monocytes were obtained by adhering 5 × 106 PBMCs/mL to a flask for 2 h at 37 °C. After removing the supernatant containing non-adherent cells, adherent monocytes were washed with 1× PBS pH 7.4. Complete media was added to the remaining cells. To induce differentiation into DCs 50 ng/mL GM-CSF (Leukine, Berlex, Washington, USA) and 20 ng/mL IL-4 (BD Biosciences) were added to the culture for 7 days.
Isolation of CD4+ T Helper Cells from PBMCs
CD4+ T cells were purified from PBMCs by negative-selection using the “Human CD4+ T Cell Enrichment” kit (EasySep, StemCell Technologies, Vancouver, BC, Canada) by following the manufacturer's protocol. Briefly, cells were resuspended in magnesium-free 1× PBS with 2% fetal bovine serum in 12 × 75 mm polystyrene tubes at 5 × 107 cells/mL. 50 μL/mL of enrichment cocktail was added, incubated for 10 min at room temperature and then followed by 100 μL/mL nanoparticle cocktail and extra incubation. Afterwards the total volume was brought to 2.5 mL and the tube was put into an EasySep magnet. After 5 min incubation to allow for magnetic beads to attach to the side of the tube, the contents were decanted into a clean tube. The beads were washed once more to increase cell purity. Cells were subsequently used in MLR reactions as described below.
Mixed Lymphocyte Reactions (MLRs)
Autologous or allogeneic enriched CD4+ T cells were labeled for 15 min with 200 nM carboxyfluorescein diacetate, succinimidyl ester (CFSE, Molecular Probes, Eugene OR) according to manufacturer's protocol and cultured at 1 × 106 cells/mL. Monocyte-derived DCs (MoDC), pulsed with 5 μg/mL Tetanus Toxin C-fragment (TTc, Roche Diagnostics, Mannheim, Germany) and phorbol 12-myristate 13-acetate (PMA, Promega, Madison, WI), were added to the same culture at a ratio of 1:2 in combination with increasing concentrations of 1. The cell mixture was allowed to incubate for 5 days in a water-jacketed incubator at 37 °C. Afterwards, culture cells were collected for flow-cytometric analysis.
Flow Cytometry
Experimental pelleted cells were incubated for 30 min at 4 °C with antibodies and washed with staining buffer (PBS + 2% BSA + 0.1% Na azide). Subsequently intracellular staining was carried out by first adding Cytofix/Cytoperm to increase the permeability of cells followed by another round of staining before fixing with 2% paraformaldehyde. Quantitation was carried out using FCS Express (version 3, De Novo Software, Los Angeles, CA) by gating for lymphocytes based on forward and side scatter properties followed by analysis of the percentages of positively stained quadrants. 50,000 cells were analyzed for each sample and isotype-specific immunoglobulin controls were run for each fluorochrome. Stains used in PBMC experiments were CD3-APC, CFSE and 7-AAD (eBioscience); those used in CD4+ T cells for MLRs were CFSE, 7-AAD, AlexaFluo 647-conjugated anti-IL-17 and PE-conjugated anti-IFN-γ (BD Biosciences).
Molecular Docking
Compounds 1 and 3 were docked into cathepsin D using the crystal structure of pepstatin A in cathepsin D as a starting point (PDB code 1LYB).31 AutoDock Vina 1.030 was used for all docking runs. This program is two orders of magnitude faster than AutoDock 4, and thus renders docking of flexible peptides with ∼25-50 rotatable bonds possible on normal workstations in a reasonable timeframe. The program was able to reproduce the docked conformation of pepstatin A in cathepsin D, with an RMSD of 0.977 Å (Figure S2A).54 In structures of grassystatins A (1) and C (3), all bonds were treated as rotatable, except ring and amide bonds, and the protein was treated as rigid. For 1, the terminal amine was protonated to reflect its likely state at physiological pH. Apart from the Pro amide bond, all amides in the ligand were set to trans configuration. For each compound, separate structures were made with the Pro amide bond either cis or trans. Docking was carried out with an exhaustiveness value of 25, and a maximum output of 100 structures. It was observed that AutoDock was always able to propose docked structures with similar calculated affinities (∼ −9 to −7 kcal/mol), and so the output structures were examined qualitatively. The primary criterion used in choosing the best docked structures was the position of the statine unit relative to the active site aspartates (Asp-33 and Asp-231), with reference to the bound conformation of pepstatin A. The rationale for this is found in the numerous crystal structures of pepstatin A55 and analogs56 bound to many different aspartic proteases.
Docking to cathepsin E was carried out in the same manner, but with some differences. There is only one crystal structure of cathepsin E available (PDB code 1TZS),32 where the inhibitory prodomain is still resident in the active site. This structure probably corresponds to an early intermediate in the maturation of the enzyme. In addition to the prodomain in the active site, the N-terminal region (Lys-14 to Asp-22) is blocking the active site tunnel so that the enzyme is in the closed conformation. A structure more consistent with the mature enzyme had to be produced in order to carry out effective docking. For this purpose, homology modeling was carried out using the SWISS-MODEL web server.57 Amongst the protein structures in the PDB, human cathepsin E has the highest sequence homology with porcine pepsinogen (PDB code 2PSG)58 and its mature form, pepsin (PDB code 4PEP).33 The activation intermediate structure for cathepsin E (1TZS) agrees very well with that for pepsinogen (2PSG, RMSD 0.784 Å), therefore the structure of the corresponding mature enzyme (4PEP) is most likely a good template for homology modeling. Indeed, the structure obtained was in excellent agreement with 1LYB (RMSD 0.833 Å). Docking of pepstatin A into the homology model was successful (see Figure S2B, Supporting Information). The conformation obtained was close to that of pepstatin A bound to cathepsin D (RMSD 1.893 Å). Grassystatin A (1) was docked using the same protocol as above. For grassystatin C (3), a larger value of exhaustiveness was used (50).
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health, NIGMS grant P41GM086210 and the University of Florida College of Pharmacy. We acknowledge NSF for funding through the External User Program of the National High Magnetic Field Laboratory (NHMFL), which supported initial NMR studies at the Advanced Magnetic Resonance Imaging and Spectroscopy (AMRIS) facility in the McKnight Brain Institute of the University of Florida (UF). The 600 MHz 1-mm triple-resonance HTS cryogenic probe used for NMR of compound 1 in DMSO-d6 and 3 in CDCl3 was developed through collaboration between UF, NHMFL, and Bruker Biospin. We thank J. R. Rocca (UF) and K. Horiuchi (RBC) for technical assistance.
Abbreviations used
- 7-AAD
7-AminoActinomycin D
- ACE
Angiotensin Converting Enzyme
- APC
Antigen Presentation Cell
- APT
Attached Proton Test
- ADAM
protein with A Disintegrin And a Metalloprotease domain
- BSA
Bovine Serum Albumin
- CAD
Collisionally Activated Decomposition
- CE
Collission Energy
- CFSE
CarboxyFluorescein diacetate, Succinimidyl Ester
- COSY
Correlation SpectroscopY
- CUR
CURtain gas
- CXP
Collision-cell eXit Potential
- DC
Dendritic Cell
- DMEM
Dulbecco's Modified Eagle Medium
- DP
Declustering Potential
- EP
Entrance Potential
- ESIMS
ElectroSpray Ionization Mass Spectrometry
- FDLA
1-Fluoro-2,4-Dinitro-5-LeucinAmide
- GS1
Gas 1
- GS2
Gas 2
- GM-CSF
Granulocyte Macrophage Colony-Stimulating Factor
- HETLOC
HEtero half-filtered TOCSY for measurement of LOng-range Coupling constants
- HMBC
Heteronuclear Multiple-Bond Correlation spectroscopy
- HMQC
Heteronuclear Multiple-Quantum Correlation spectroscopy
- HRESI/APCIMS
High Resolution Electrospray Ionization/Atmospheric Pressure Chemical Ionization Mass Spectrometry (dual probe)
- HSQC
Heteronuclear Single-Quantum Correlation spectroscopy
- IFN-γ
Interferon-γ
- IL-4
InterLeukin-4
- Ii
Invariant Chain
- IL-17
Interleukin-17
- IS
IonSpray voltage
- MHC
Major Histocompatability Complex
- MLR
Mixed Lymphocyte Reaction
- MMP
Matrix MetalloProtease
- MoDC
Monocyte-derived Dendritic Cell
- MRM
Multiple Reaction Monitoring
- NRPS
Non-Ribosomal Peptide Synthetase
- PBMC
Peripheral Blood Mononuclear Cells
- PBS
Phosphate-Buffered Saline
- PDB
Protein Data Bank
- PKS
PolyKetide Synthetase
- PE
Phycoerythrin
- PMA
Phorbol 12-Myristate 13-Acetate
- RMSD
Root Mean Square Deviation
- ROESY
Rotating frame nuclear Overhauser Effect SpectroscopY
- TACE
Tumor necrosis factor α-Converting Enzyme
- TEM
TEMperature
- TH
T Helper cell
- TOCSY
TOtal Correlation SpectroscopY
- TTc
Tetanus Toxin C-fragment
Footnotes
Supporting Information Available: Table S1, Table S2, Figure S1, Figure S2 and NMR spectra for compounds 1–3. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.(a) López-Otín C, Bond JS. Proteases: Multifunctional enzymes in life and disease. J Biol Chem. 2008;283:30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Turk B. Targeting proteases: successes, failures and future prospects. Nature Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 2.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2378–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 3.(a) Luesch H, Yoshida WY, Moore RE, Paul VJ, Corbett TH. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J Am Chem Soc. 2001;123:5418–5423. doi: 10.1021/ja010453j. [DOI] [PubMed] [Google Scholar]; (b) Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 4.(a) Matthew S, Ross C, Rocca JR, Paul VJ, Luesch H. Lyngbyastatin 4, a dolastatin 13 analogue with elastase and chymotrypsin inhibitory activity from the marine cyanobacterium Lyngbya confervoides. J Nat Prod. 2007;70:124–127. doi: 10.1021/np060471k. [DOI] [PubMed] [Google Scholar]; (b) Taori K, Matthew S, Rocca JR, Paul VJ, Luesch H. Lyngbyastatins 5–7, potent elastase inhibitors from Floridian marine cyanobacteria, Lyngbya spp. J Nat Prod. 2007;70:1593–1600. doi: 10.1021/np0702436. [DOI] [PubMed] [Google Scholar]
- 5.Dittman E, Neilan BA, Börner T. Molecular biology of peptide and polyketide biosynthesis in cyanobacteria. Appl Microbiol Biotechnol. 2001;57:467–473. doi: 10.1007/s002530100810. [DOI] [PubMed] [Google Scholar]
- 6.Kittendorf JD, Sherman DH. The methymycin/pikromycin pathway: A model for metabolic diversity in natural product biosynthesis. Bioorg Med Chem. 2009;17:2137–2146. doi: 10.1016/j.bmc.2008.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Morishima H, Takita T, Aoyagi T, Takeuchi T, Umezawa H. The structure of pepstatin. J Antibiotic. 1970;23:263–265. doi: 10.7164/antibiotics.23.263. [DOI] [PubMed] [Google Scholar]; (b) Umezawa H, Aoyagi T, Morishima H, Matsuzaki M, Hamada M, Takeuchi T. Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J Antibiotic. 1970;23:259–262. doi: 10.7164/antibiotics.23.259. [DOI] [PubMed] [Google Scholar]
- 8.Morishima H, Sawa T, Takita T, Aoyagi T, Takeuchi T, Umezawa H. Biosynthetic studies on pepstatin, biosynthesis of (3S,4S)-4-amino-3-hydroxy-6-methylheptanoic acid moiety. J Antibiotic. 1974;27:267–273. [PubMed] [Google Scholar]
- 9.Williams PG, Yoshida WY, Moore RE, Paul VJ. Tasiamide, a cytotoxic peptide from the marine cyanobacterium Symploca sp. J Nat Prod. 2002;65:1336–1339. doi: 10.1021/np020184q. [DOI] [PubMed] [Google Scholar]
- 10.Williams PG, Yoshida WY, Moore RE, Paul VJ. The isolation and structure elucidation of tasiamide B, a 4-amino-3-hydroxy-5-phenylpentanoic acid containing peptide from the marine cyanobacterium Symploca sp. J Nat Prod. 2003;66:1006–1009. doi: 10.1021/np030114z. [DOI] [PubMed] [Google Scholar]
- 11.(a) Zaidi N, Hermann C, Hermann T, Kalbacher H. Emerging functional roles of cathepsin E. Biochem Biophys Res Commun. 2008;377:327–330. doi: 10.1016/j.bbrc.2008.10.034. [DOI] [PubMed] [Google Scholar]; (b) Zaidi N, Kalbacher H. Cathepsin E: A mini review. Biochem Biophys Res Commun. 2008;367:517–522. doi: 10.1016/j.bbrc.2007.12.163. [DOI] [PubMed] [Google Scholar]
- 12.Kwan JC, Rocca JR, Abboud KA, Paul VJ, Luesch H. Total structure determination of grassypeptolide, a new marine cyanobacterial cytotoxin. Org Lett. 2008;10:789–792. doi: 10.1021/ol702946d. [DOI] [PubMed] [Google Scholar]
- 13.The minor signals present in the CDCl3 NMR spectra are due to conformers and not impurities, as these were not observed in the spectra obtained in DMSO-d6
- 14.The threonine hydroxyl was observed only in the NMR spectra collected in DMSO-d6 - see supporting information
- 15.The presence of L-Asp in the hydrolysate is consistent with the presence of L-Asn in the intact molecule, the primary amide having undergone hydrolysis
- 16.Marfey P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res Commun. 1984;49:591–596. [Google Scholar]
- 17.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. Stereochemical determination of acyclic structures based on carbon-proton spin-couping constants. A method of configuration analysis for natural products. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 18.A modification of the pulse sequence in ref. 19 was used; see, Luesch et al. 2001 (ref 3a).
- 19.Uhrin D, Batta G, Hruby VJ, Barion PN, Kövér KE. Sensitivity- and gradient-enhanced hetero (ω1) half-filtered TOCSY experiment for measuring long-range heteronuclear coupling constants. J Magn Reson. 1998;130:155–161. doi: 10.1006/jmre.1997.1308. [DOI] [PubMed] [Google Scholar]
- 20.Preciado A, Williams PG. A simple microscale method for determining the rlative stereochemistry of statine units. J Org Chem. 2008;73:9228–9234. doi: 10.1021/jo8012429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The presence of N-Me-L-Glu in the hydrolysate is consistent with the presence of N-Me-L-Gln in the intact molecule, the primary amide having undergone hydrolysis
- 22.Berglund L, Björling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CAK, Persson A, Ottosson J, Wernérus H, Nilsson P, Lundberg E, Sivertsson Ǻ, Navani S, Wester K, Kampf C, Hober S, Pontén F, Uhlén M. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.The relative expression of cathepsins D and E are given as 55% and 54%, respectively, in the Human Protein Atlas. However, each of these figures is relative to the tissue/cell line that has the highest expression of that particular protein. Therefore, from this data alone, one cannot compare the expression of cathepsins D and E directly in this cell line.
- 24.Zaidi N, Burster T, Sommandas V, Hermann T, Boehm BO, Driessen C, Voelter W, Kalbacher H. A novel cell penetrating aspartic protease inhibitor blocks processing and presentation of tetanus toxoid more efficiently than pepstatin A. Biochem Biophys Res Commun. 2007;364:243–249. doi: 10.1016/j.bbrc.2007.09.114. [DOI] [PubMed] [Google Scholar]
- 25.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 26.(a) Burster T, Reich M, Zaidi N, Voelter W, Boehm BO, Kalbacher H. Cathepsin E regulates the presentation of tetanus toxin C-fragment in PMA activated primary human B cells. Biochem Biophys Res Commun. 2008;377:1299–1303. doi: 10.1016/j.bbrc.2008.10.162. [DOI] [PubMed] [Google Scholar]; (b) Chain BM, Free P, Medd P, Swetman C, Tabor AB, Terrazzini N. The expression and function of cathepsin E in dendritic cells. J Immunol. 2005;174:1791–1800. doi: 10.4049/jimmunol.174.4.1791. [DOI] [PubMed] [Google Scholar]; (c) Nishioku T, Hashimoto K, Yamashita K, Liou SY, Kagamiishi Y, Maegawa H, Katsube N, Peters C, von Figura K, Saftig P, Katunuma N, Tamamoto K, Nakanishi H. Involvement of cathepsin E in exogenous antigen processing in primary cultured murine microglia. J Biol Chem. 2002;277:4816–4822. doi: 10.1074/jbc.M108382200. [DOI] [PubMed] [Google Scholar]; (d) Zhang T, Maekawa Y, Yasumoto K, Ishikawa H, Nashed BF, Dainichi T, Hisaeda H, Sakai T, Kasai M, Mizuochi T, Asao T, Katunuma N, Himeno K. Pepstatin A-sensitive aspartic proteases in lysosome are involved in degradation of the invariant chain and antigen-processing in antigen presenting cells of mice infected with Leishmania major. Biochem Biophys Res Commun. 2000;276:693–701. doi: 10.1006/bbrc.2000.3538. [DOI] [PubMed] [Google Scholar]
- 27.Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology, The Immune System in Health and Disease. 6th. Garland Publishing; New York: 2005. T cell-mediated immunity. [Google Scholar]
- 28.Zaidi N, Herrmann T, Baechle D, Schleicher S, Gogel J, Driessen C, Voelter W, Kalbacher H. A new approach for distinguishing cathepsin E and D activity in antigen-processing organelles. FEBS J. 2007;274:3138–3149. doi: 10.1111/j.1742-4658.2007.05846.x. [DOI] [PubMed] [Google Scholar]
- 29.Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology, The Immune System in Health and Disease. 6th. Garland Publishing; New York: 2005. Autoimmunity and transplantation; pp. 557–612. [Google Scholar]
- 30.Trott O, Olsen AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009 doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baldwin ET, Bhat TN, Gulnik S, Hosur MV, Sowder RC, Cachau RE, Collins J, Silva AM, Erickson JW. Crystal structures of native and inhibited forms of human cathepsin D: implications for lysosomal targeting and drug design. Proc Natl Acad Sci USA. 1993;90:6796–6800. doi: 10.1073/pnas.90.14.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostermann N, Gerhartz B, Worpenberg S, Trappe J, Eder J. Crystal structure of an activation intermediate of cathepsin E. J Mol Biol. 2004;342:889–899. doi: 10.1016/j.jmb.2004.07.073. [DOI] [PubMed] [Google Scholar]
- 33.Sielecki AR, Fedorov AA, Boodhoo A, Andreeva NS, James MNG. Molecular and crystal structure of monoclinic porcine pepsin refined at 1.8 Å resolution. J Mol Biol. 1990;214:143–170. doi: 10.1016/0022-2836(90)90153-D. [DOI] [PubMed] [Google Scholar]
- 34.Rao-Naik C, Guruprasad K, Batley B, Rapundalo S, Hill J, Blundell T, Kay J, Dunn BM. Exploring the binding preferences/specificity in the active site of human cathepsin E. Proteins Struc Func Genet. 1995;22:168–181. doi: 10.1002/prot.340220209. [DOI] [PubMed] [Google Scholar]
- 35.Ghoneim OL, Legere JA, Golbraikh A, Tropsha A, Booth RG. Novel ligands for the human histamine H1 receptor: Synthesis, pharmacology, and comparative molecular field analysis studies of 2-dimethylamino-5-(6)-phenyl-1,2,3,4-tetrahydronaphthalenes. Bioorg Med Chem. 2006;14:6640–6658. doi: 10.1016/j.bmc.2006.05.077. [DOI] [PubMed] [Google Scholar]
- 36.For example, conformations where the N-C direction was reversed, where the ligand folded back on itself, or where a unit other than statine resided at the catalytic center.
- 37.Rich DH. Pepstatin-derived inhibitors of aspartic proteinases. A close look at an apparent transition-state analogue inhibitor. J Med Chem. 1985;28:263–273. doi: 10.1021/jm00381a001. [DOI] [PubMed] [Google Scholar]
- 38.Scarborough PE, Dunn BM. Redesign of the substrate specificity of human cathepsin D: the dominant role of position 287 in the S2 subsite. Protein Eng. 1994;7:495–502. doi: 10.1093/protein/7.4.495. [DOI] [PubMed] [Google Scholar]
- 39.Arnold D, Keilholz W, Schild H, Dumrese T, Stevanovic S, Rammensee HG. Substrate specificity of cathepsins D and E determined by N-terminal and C-terminal sequencing of peptide pools. Eur J Biochem. 1997;249:171–179. doi: 10.1111/j.1432-1033.1997.t01-1-00171.x. [DOI] [PubMed] [Google Scholar]
- 40.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: a Guide for Medicinal Chemists and Pharmacologists. Wiley & Sons; Hoboken: 2005. Slow binding inhibitors; pp. 141–177. [PubMed] [Google Scholar]
- 41.Marcinkeviciene J, Luo Y, Graciani NR, Combs AP, Copeland RA. Mechanism of inhibition of β-site amyloid precursor protein-cleaving enzyme (BACE) by a statine-based peptide. J Biol Chem. 2001;276:23790–23794. doi: 10.1074/jbc.M101896200. [DOI] [PubMed] [Google Scholar]
- 42.Bull HG, Thorberry NA, Cordes MHJ, Patchett AA, Cordes EH. Inhibition of rabbit lung angiotensin-converting enzyme by Nα-[(S)-1-carboxy-3-phenylpropyl]L-alanyl-L-proline and Nα-[(S)-1-carboxy-3-phenylpropyl]L-lysyl-L-proline. J Biol Chem. 1985;260:2952–2962. [PubMed] [Google Scholar]
- 43.Bernardo MM, Brown S, Li ZH, Fridman R, Mobashery S. Design, synthesis, and characterization of potent, slow-binding inhibitors that are selective for gelatinases. J Biol Chem. 2002;277:11201–11207. doi: 10.1074/jbc.M111021200. [DOI] [PubMed] [Google Scholar]
- 44.Dunn GP, Ikeda H, Bruce AT, Koebel C, Uppaluri R, Bui J, Chan R, Diamond M, White JM, Sheehan KC, Schreiber RD. Interferon-gamma and cancer immunoediting. Immunol Res. 2005;32:231–245. doi: 10.1385/ir:32:1-3:231. [DOI] [PubMed] [Google Scholar]
- 45.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology, The Immune System in Health and Disease. 6th. Garland Publishing; New York: 2005. Antigen presentation to T lymphocytes; pp. 169–202. [Google Scholar]
- 47.Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, de Sanctis GT, Ploegh HL, Chapman HA. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest. 1998;101:2351–2363. doi: 10.1172/JCI1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marić MA, Taylor MD, Blum JS. Endosomal aspartic proteinases are required for invariant-chain processing. Proc Natl Acad Sci USA. 1994;91:2171–2175. doi: 10.1073/pnas.91.6.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Constantino CM, Hang HC, Kent SC, Hafler DA, Ploegh HL. Lysosomal cystein and aspartic proteases are heterogeneously expressed and act redundantly to initiate human invariant chain degradation. J Immunol. 2008;180:2876–2885. doi: 10.4049/jimmunol.180.5.2876. [DOI] [PubMed] [Google Scholar]
- 50.Keilová H, Tomášek V. Effect of pepsin inhibitor from Ascaris lumbricoides on cathepsin D and E. Biochim Biophys Acta. 1972;284:461–464. doi: 10.1016/0005-2744(72)90143-x. [DOI] [PubMed] [Google Scholar]
- 51.Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H, Köster A, Hess B, Evers M, von Figura K, Peters C. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995;14:3599–3608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brey WW, Edison AS, Nast RE, Rocca JR, Saha S, Withers RS. Design, construction, and validation of a 1-mm triple-resonance high-temperature-superconducting probe for NMR. J Magn Reson. 2006;179:290–293. doi: 10.1016/j.jmr.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Kwan JC, Rocca JR, Abboud KA, Paul VJ, Luesch H. Total structure determination of grassypeptolide, a new marine cyanobacterial cytotoxin. Org Lett. 2008;10:789–792. doi: 10.1021/ol702946d. [DOI] [PubMed] [Google Scholar]
- 54.The default value of exhaustiveness (8) was sufficient to reproduce the bound conformation of pepstatin A, but since compounds 1 and 3 have more rotatable bonds, a higher value (25) was used for all docking studies.
- 55.(a) Bernstein NK, Cherney MM, Yowell CA, Dame JB, James MNG. Structural insights into the activation of P. vivax plasmepsin. J Mol Biol. 2003;329:505–524. doi: 10.1016/s0022-2836(03)00444-3. [DOI] [PubMed] [Google Scholar]; (b) Borelli C, Ruge E, Schaller M, Monod M, Korting HC, Huber R, Maskos K. The crystal structure of the secreted aspartic proteinase 3 from Candida albicans and its complex with pepstatin A. Proteins Struc Func Genet. 2007;68:738–748. doi: 10.1002/prot.21425. [DOI] [PubMed] [Google Scholar]; (c) Fitzgerald PMD, McKeever BM, VanMiddlesworth JF, Springer JP, Heimbach JC, Leu CT, Herber WK, Dixon RAF, Darke PL. Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl-pepstatin at 2.0 Ǻ resolution. J Biol Chem. 1990;265:14209–14219. [PubMed] [Google Scholar]; (d) Fujimoto Z, Fujii Y, Kaneko S, Kobayashi H, Mizuno H. Crystal structure of aspartic proteinase from Irpex lacteus in complex with inhibitor pepstatin. J Mol Biol. 2004;341:1227–1235. doi: 10.1016/j.jmb.2004.06.049. [DOI] [PubMed] [Google Scholar]; (e) Fujinaga M, Chernaia MM, Tarasova NI, Mosimann SC, James MNG. Crystal structure of human pepsin and its complex with pepstatin. Protein Sci. 1995;4:960–972. doi: 10.1002/pro.5560040516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kamitori S, Ohtaki A, Ino H, Takeuchi T. Crystal structures of Aspergillus oryzae aspartic proteinase and its complex with an inhibitor pepstatin at 1.9 Ǻ resolution. J Mol Biol. 2003;326:1501–1511. doi: 10.1016/s0022-2836(03)00078-0. [DOI] [PubMed] [Google Scholar]; (g) Yang J, Quail JW. Structure of the Rhizomucor miehei aspartic proteinase complexed with the inhibitor pepstatin A at 2.7 Ǻ resolution. Acta Cryst D. 1999;D55:625–630. doi: 10.1107/s0907444998013961. [DOI] [PubMed] [Google Scholar]
- 56.(a) Asojo OA, Gulnik SV, Afonina E, Yu B, Ellman JA, Haque TS, Silva AM. Novel uncomplexed and complexed structures of plasmepsin II, as aspartic protease from Plasmodium falciparum. J Mol Biol. 2003;327:173–181. doi: 10.1016/s0022-2836(03)00036-6. [DOI] [PubMed] [Google Scholar]; (b) Bone R, Vacca JP, Anderson PS, Holloway MK. X-ray crystal structure of the HIV protease complex with L-700,417, an inhibitor with pseudo C2 symmetry. J Am Chem Soc. 1991;113:9382–9384. [Google Scholar]; (c) Coates L, Erskine PT, Wood SP, Myles DAA, Cooper JB. A neutron Laue diffraction study of endothiapepsin: Implications for the aspartic proteinase mechanism. Biochem. 2001;40:13149–13157. doi: 10.1021/bi010626h. [DOI] [PubMed] [Google Scholar]; (d) Fraser ME, Strynadka NCJ, Bartlett PA, Hanson JE, James MNG. Crystallographic analysis of transition-state mimics bound to penicillopepsin: Phosphorous-containing peptide analogs. Biochem. 1992;31:5201–5214. doi: 10.1021/bi00137a016. [DOI] [PubMed] [Google Scholar]; (e) James MNG, Sielecki AR, Hayakawa K, Gelb MH. Crystallographic analysis of transition state mimics bounds to penicillopepsin: Difluorostatine- and difluorostatine-containing peptides. Biochem. 1992;31:3872–3886. doi: 10.1021/bi00130a019. [DOI] [PubMed] [Google Scholar]
- 57.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 58.Sielecki AR, Fujinaga M, Read RJ, James MNG. Refined structure of porcine pepsinogen at 1.8 Ǻ resolution. J Mol Biol. 1991;219:671–692. doi: 10.1016/0022-2836(91)90664-r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.