

Abstract
A pathologic hallmark of Huntington disease (HD) is the presence of intraneuronal aggregates of polyglutamine-containing huntingtin protein (Htt) fragments. Monoclonal antibody 1C2 is a commercial antibody to normal human TATA-binding protein that detects long stretches of glutamine residues. Using 1C2 as a surrogate marker for mutant Htt, we immunostained 19 HD cases, 10 normal controls, and 10 cases of frontotemporal degeneration with ubiquitinated inclusions as disease controls. In the HD cases there was consistent 1C2 immunoreactivity in the neocortex, striatum, hippocampus, lateral geniculate body, basis pontis, medullary reticular formation, and cerebellar dentate nucleus. The normal and disease controls demonstrated 1C2 immunoreactivity only in the substantia nigra, locus coeruleus, and pituitary gland. Staining of 5 HD cases and 5 normal controls revealed a less consistent and less diagnostically useful morphologic immunoreactivity profile. These results indicate that widespread 1C2 immunoreactivity is present in diverse central nervous system areas in HD and that in the appropriate setting 1C2 staining can be a useful tool in the postmortem diagnosis of HD when neuromelanin-containing neuronal populations are avoided.
Keywords: 1C2, Frontotemporal degeneration with ubiquitinated inclusions, inclusions, Huntington disease, Immunohistochemistry, Neuromelanin, Polyglutamine
INTRODUCTION
Huntington disease (HD) is caused by an abnormal expansion of CAG nucleotide repeats within the IT15 gene located at 4p16.3 (1). The IT15 protein product, huntingtin (Htt), is believed to play a role in vesicle-trafficking, endocytosis, and transcription regulation (2). In wild type form, Htt consists of approximately 3,144 amino acids with a stretch of 10–35 glutamine residues at the N-terminus encoded by a stretch of CAG repeats (1, 3). Incomplete penetrance of the disease is said to occur when the CAG repeat number reaches 36 (4). Due to a CAACAG sequence following the uninterrupted CAG stretch, the glutamine number for a patient is 2 more than the CAG repeat number. Patients with HD develop a characteristic progressive motor, psychiatric, and cognitive dysfunction that is ultimately fatal. The pathophysiology of the disease is believed to involve the cleavage and subsequent aggregation of N-terminal mutant Htt (mHtt) fragments. A toxic gain of function occurs and the death of medium spiny neurons in brain regions including the striatum and cortex ensues (5).
A variety of antibodies has been generated against portions of wild type Htt and mHtt (Fig. 1, Fig. 2). Multiple studies summarized in Table 1 have employed these antibodies to detect protein aggregates in the brains of HD patients (6–17); they have also been used in HD animal model studies. Unfortunately, however, most commercially available antibodies to huntingtin are not very sensitive. This low sensitivity with resultant low intensity staining (low signal-to-noise ratio) complicates the discrimination of anti-huntingtin staining of pathologic aggregates of huntingtin from normally occurring huntingtin. 1C2 (Chemicon, Billerica, MA) is a monoclonal antibody raised against the N-terminus of normal human TATA-binding protein (TBP), a protein integral to transcription and one that contains a long (38 or more) glutamine stretch (LGS) in its most common polymorphism (3). 1C2 was first used to study the binding properties of TBP but was later demonstrated on Western blots by Trottier et al to bind preferentially to LGS-containing mHtt (3). Many investigators have used it since as a surrogate marker for mHtt to detect abnormal protein aggregates in patients and animal models of HD and other CAG repeat diseases; these studies are summarized in Table 2 (9, 12, 18, 19).
Figure 1.

Map of full-length wild type huntingtin and corresponding anti-huntingtin antibodies listed in Table 1 and Table 2.
Figure 2.

Map of N-terminal wild type huntingtin and corresponding anti-huntingtin antibodies listed in Table 1 and Table 2.
Table 1.
Review of Immunohistochemical Studies on Postmortem Huntington Disease Central Nervous System Using Anti-Huntingtin Antibodies
| Reference | Antibody | Location of epitope in Htt amino acid sequence |
Dx | Locations studied | Patterns of IR |
|---|---|---|---|---|---|
| (6) | Ab1 | 1 – 17 aa | HD (n=9) | Cortex | I, N |
| Striatum | I | ||||
| HD (n=9), Nl (n=5) | Globus pallidus, cerebellum | - | |||
| Nl (n=5) | Cortex, striatum | - | |||
| Ab585 | 585 – 725 aa | HD (n=9), Nl (n=5) | Cortex, striatum, globus pallidus, cerebellum | C | |
| (7) | Ab585 | 585 – 725 aa | HD (n=12) | Striatum, globus pallidus | I, C |
| HD (n=12), Nl (n=8) | Cortex | I, C, N in white matter | |||
| Basal forebrain, hippocampus, cerebellum, substantia nigra | C | ||||
| Nl (n=8) | Striatum, globus pallidus | C | |||
| (8) | AP78, AP194 | 1 – 17 aa | HD (n=20) | Neocortex, striatum, dentate nucleus, amygdala, hippocampus, red nucleus | I |
| Thalamus, substantia nigra, olivary complex of medulla, Purkinje cells | - | ||||
| Nl (n=4), DC (n=10) | Neocortex, striatum, cerebellum | - | |||
| AP81 | 650 – 663 aa | HD (n=20), Nl (n=4), DC (N=10) | Neocortex, striatum, cerebellum | C | |
| (9) | 4C8 | 414 – 503 aa | HD (n=5), Nl (n=5) | Cortex, striatum, cerebellum | C, N |
| (10) | EM48 | 1 – 256 aa with 2 glutamines and deletion of polyproline stretch | HD (n=12) | Cortex | I, C, N |
| Striatum, hypothalamus, thalamus, substantia nigra, cuneate nucleus | Infrequent I, C, N | ||||
| Globus pallidus, hippocampus, cerebellum | Rare I, C, N | ||||
| Nl (n=4) | Not specified | Faint C | |||
| DC (AD n=3) | Cortex, hippocampus | Plaques | |||
| mHD549 | 549 – 679 aa | HD (n=1) | Insular cortex, striatum | C, N | |
| (11) | Antiserum 7 | 1 – 19 aa | HD (n=7) | Cortex, striatum, hippocampus | I, C, N |
| Dentate gyrus, globus pallidus, cerebellum, substantia nigra | C | ||||
| DC (n=2) | Cortex | C | |||
| Antiserum 93 | 1929 – 2421 aa | HD (n=7), DC (n=5) | Cortex, striatum, hippocampus, globus pallidus, cerebellum, substantia nigra | - | |
| (12) | CAG53b | 1 – 118 aa with 51 glutamines | HD (n=6) | Frontal cortex | I, C, N |
| Nl (n=2), DC (AD n =5) | Frontal cortex | - | |||
| HD1 | 1 – 173 aa with 23 glutamines | HD (n=6) | Frontal cortex | C, N | |
| Nl (n=2), DC (AD n =5) | Frontal cortex | - | |||
| HP1 | 80 – 113 aa | HD (n=6) | Frontal cortex | I, C, N | |
| Nl (n=2), DC (AD n =5) | Frontal cortex | - | |||
| P2 | 1187 – 1207 aa | HD (n=6), Nl (n=2), DC (AD n=5) | Frontal cortex | - | |
| HF1 | 1981 – 2580 aa | HD (n=6) | Frontal cortex | “Faint synaptic staining” | |
| Nl (n=2), DC (AD n =5) | Frontal cortex | - | |||
| anti-Hunt3 | 2110 – 2121 aa | HD (n=6), Nl (n=2), DC (AD n =5) | Frontal cortex | C | |
| (13) | 1259, 2B4 | 1 – 82 aa | HD (n=6) | Frontal cortex | I, C |
| Nl (n=3) | Frontal cortex | C | |||
| 1H6 | 115 – 129 aa | HD (n=6), Nl (n=3) | Frontal cortex | C | |
| 214 | 214 – 229 aa | HD (n=6), Nl (n=3) | Frontal cortex | C | |
| 4C8 | 414 – 503 aa | HD (n=6), Nl (n=3) | Frontal cortex | C | |
| (14) | not named | 1 – 17 aa | HD (n=8) | Middle frontal gyrus | I, C |
| Nl (n=8) | Middle frontal gyrus | - | |||
| (15) | mAb5374 (Chemicon) | 1 – 256 aa with deletion of glutamine stretch | HD (n=7) | Motor and prefrontal association cortex | I, C, N |
| (16) | mAb5374 (Chemicon) | 1 – 256 aa with deletion of glutamine stretch | HD (81 CAG# confirmed cases) | Cortex | I |
| DC (4 cases with confirmed normal CAG#) | Cortex | - | |||
| (17) | Htt1-17 | 1 – 17 aa | HD (n=6) | Parietal cortex | N |
| Nl (n=5) | Parietal cortex | - | |||
| N-Del | Portions of N-terminal 1– 90 aa excluding most of polyglutamine domain and all of first polyproline repeat | HD (n=6) | Parietal cortex | N | |
| Nl (n=5) | Parietal cortex | - | |||
| Htt55-65 | 55 – 65 aa | HD (n=6) | Parietal cortex | N | |
| Nl (n=5) | Parietal cortex | - | |||
| Htt81-90 | 81 – 90 aa | HD (n=6) | Parietal cortex | N | |
| Nl (n=5) | Parietal cortex | - | |||
| 1H6 | 115 – 129 aa | HD (n=6), Nl (n=5) | Parietal cortex | C |
HD, Huntington disease; Nl, normal control cases; DC, diseased control cases; IR, immunoreactivity; I, intranuclear; C, cytoplasmic; N, neuropil.
Table 2.
Review of Immunohistochemical Studies Using 1C2 on Postmortem Huntington Disease Central Nervous System
| Reference | Antibody | Epitope | Dx | Locations studied | Patterns of IR |
|---|---|---|---|---|---|
| (9) | IC2 (0.2 µg/ml) | 38 glutamine stretch | HD (n = 5) | Cortex, striatum, globus pallidus, thalamus | C |
| Cerebellum | - | ||||
| Nl (n = 5) | Cortex, striatum, globus pallidus, thalamus | - | |||
| (12) | 1C2 (at 1:4000 to 1:400) | 38 glutamine stretch | HD (n = 6) | Frontal cortex | C |
| Nl (n = 2), DC (AD n = 5) | Frontal cortex | - | |||
| (18) | 1C2 (dilution or conc not given) | 38 glutamine stretch | HD (n = 1) | Cortex, dentate nucleus | I, C |
| Striatum, thalamus, subthalamic nucleus, claustrum, hippocampus, dentate nucleus, midbrain, brainstem reticular formation, basis pontis, hypoglossal nucleus, vestibular nucleus, Anterior horn cells | I | ||||
| (19) | 1C2 (1:10000) | 38 glutamine stretch | HD (n = 6), HDL2 (n = 5) | Motor cortex, amygdala (in all cases of each group), entorhinal cortex, striatum, thalamus (in some cases of each group) | I |
| HD (n = 6), HDL2 (n = 5) | Subthalamic nucleus | - | |||
| HD (n = 6) | Pons, medulla | I |
HDL2, Huntington disease-like 2; HD, Huntington disease; Nl, normal control cases; DC, diseased control cases; IR, immunoreactivity; I, intranuclear; C, cytoplasmic; N, neuropil.
While many have studied the cortex and striatum, a thorough immunohistochemical survey of the central nervous system (CNS) in postmortem brains of HD patients with 1C2 is lacking. Thus, our main objective was to conduct a neuroanatomical and morphologic study of 1C2 immunoreactivity in postmortem CNS tissue of 19 individuals with HD. We performed 1C2 immunostaining on 7 neuroanatomic locations in 9 postmortem brains of individuals who died of non-neurologic causes and in 2 cases of frontotemporal dementia with ubiquitinated inclusions (FTD-Ub) for all locations except substantia nigra and pons for which an additional 10 cases were stained. FTD-Ub, also a neurodegenerative disease, is characterized by nuclear and cytoplasmic inclusions in neurons of hippocampus and cortex; like mHtt aggregates, these inclusions are ubiquitin-immunoreactive. In 6 patients with familial FTD-Ub, Mackenzie et al found no significant CAA/CAG repeat expansions within 63 candidate genes using polymerase chain reaction (PCR) confirmed with high-resolution agarose gel and/or capillary electrophoresis (20). They also demonstrated an absence of 1C2 immunoreactivity in the cortex and striatum of these patients. Thus, cases of this disorder are appropriate negative disease controls for immunohistochemical studies of CAG repeat diseases.
A second objective grew from the need within our laboratory to identify an antibody that would be useful for making or confirming the diagnosis of HD in postmortem brain tissue. We have found that multiple factors can prevent a definitive antemortem diagnosis of HD in an individual, including an atypical clinical presentation, lack of family history, a decision by the patient not to seek medical care or testing, or lack of access to such testing. Moreover, in some cases (e.g. forensic cases), definitive clinical information may be unavailable at the time of autopsy. A characteristic immunohistochemical staining pattern with such an antibody in combination with typical gross and microscopic neuropathologic findings of HD would provide a convincing method for diagnosing HD. If this tool were commercially available and relatively straightforward to implement it would be widely useful. Therefore, we compared the 1C2 immunoreactivity profile of HD and control cases to that of another commercially available monoclonal antibody, 2B4 (Chemicon). The 2B4 antibody recognizes the first 82 amino acid residues of wild type Htt assuming a 21 glutamine repeat and therefore detects N-terminal mHtt fragments (13, 17).
MATERIALS AND METHODS
Case Selection
The University of Texas Southwestern Division of Neuropathology database was used to select cases with the autopsy diagnosis of HD, neuropathologically normal controls, or FTD-Ub. This database includes information on postmortem brains submitted to the division for neuropathological consultation since 1980 from a variety of institutions including the Dallas County Medical Examiners Office, Parkland Memorial Hospital, Children's Medical Center of Dallas, the UT Southwestern Alzheimer Disease Center, and several outside referral sources. Patient history was extracted from the database and other available medical records.
Archival pathology paraffin blocks from 19 cases of HD and 9 normal control cases were studied. Initially, 2 FTD-Ub cases were chosen as disease controls. An additional 10 cases were subsequently identified for studying neuromelanin-containing neurons. The clinical information for these cases is detailed in Table 3 and Table 4.
Table 3.
Clinical Profile of Huntington Disease Cases
| Case No. |
Age (yrs) /Sex |
Age of Onset (years) |
Race | No. CAG repeats |
Vonsattel Grade |
|---|---|---|---|---|---|
| 1 | 12/F | 7 | W | - | IV |
| 2 | 26/M | 18 | B | 72 | II |
| 3 | 37/M | 32 | H | - | IV |
| 4 | 38/M | - | W | - | II |
| 5 | 42/M | 37 | - | - | III |
| 6 | 47/F | - | W | - | IV |
| 7 | 48/M | 33 | W | - | III |
| 8 | 51/F | - | W | - | III |
| 9 | 53/F | 42 | W | - | III |
| 10 | 54/F | - | W | 44 | II |
| 11 | 55/M | - | W | - | IV |
| 12 | 61/M | 57 | W | - | III |
| 13 | 62/M | - | - | - | II |
| 14 | 62/M | 53 | W | 42 | IV |
| 15 | 63/M | - | W | - | III |
| 16 | 67/M | - | W | - | III |
| 17 | 68/F | - | W | - | III |
| 18 | 74/M | 59 | W | - | III |
| 19 | 76/F | 59 | W | - | III |
F, female; M, male; W, white; B, black; H, Hispanic.
Table 4.
Clinical Profile of Control Cases
| Age/Sex | Diagnosis |
|---|---|
| 8mo/F | Normal control |
| 3y/M | Normal control |
| 19y/F | Normal control |
| 24y/F | Normal control |
| 33y/M | Normal control |
| 46y/M | Normal control |
| 59y/F | Normal control |
| 64y/M | Normal control |
| 73y/F | Normal control |
| 55y/M | FTD-Ub |
| 59y/F | FTD-Ub |
| 60y/M | FTD-Ub |
| 61y/M | FTD-Ub |
| 64y/F | FTD-Ub |
| 65y/F | FTD-Ub |
| 65y/M | FTD-Ub |
| 68y/M | FTD-Ub |
| 75y/M | FTD-Ub |
| 79y/M | FTD-Ub |
| 80y/M | FTD-Ub |
| 81y/M | FTD-Ub |
Mo, month; y, year; F, female; M, male; FTD-Ub, frontotemporal dementia with ubiquitinated inclusions.
Brain tissue samples from the 3 groups were stained with 1C2. 5 HD cases and 5 age-matched normal controls were selected for staining with 2B4. The locations selected for staining in the anti-Htt group were those found to be 1C2-immunoreactive in some or all of the HD cases studied (Table 5, Table 6).
Table 5.
Summary of 1C2 Immunoreactivity in Huntington Disease Cases
| Location | Immunoreactive cases / Total cases |
Cytoplasm | Nucleus | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| D | A | DA | No | D | A | DA | No | |||
| Immunoreactive in all HD cases studied | Neocortex | 15/15 | 1 | 12 | 2 | - | 6 | 1 | 8 | - |
| Striatum | 9/9 | 1 | 8 | - | - | 4 | - | 5 | - | |
| Hippocampal pyramidal neurons/Subiculum/Entorhinal cortex | 13/13 | - | 7 | 5 | 1 | 6 | - | 7 | - | |
| Lateral geniculate body | 6/6 | - | 6 | - | - | 2 | - | - | 4 | |
| Substantia nigra* | 7/7 | - | 7 | - | - | - | - | 2 | 5 | |
| Basal pontine nuclei | 10/10 | - | 10 | - | - | 4 | 1 | 3 | 2 | |
| Medullary reticular formation | 10/10 | - | 10 | - | - | 8 | - | - | 2 | |
| Dentate nucleus | 9/9 | - | 9 | - | - | 5 | - | - | 4 | |
| Immunoreactive in some of HD cases studied | Dentate gyrus | 3/13 | - | 1 | - | 2 | 1 | - | 2 | - |
| Thalamus | 5/6 | 1 | 2 | 1 | 1 | 3 | - | - | 2 | |
| Locus coeruleus* | 7/8 | 1 | 6 | - | - | - | - | - | 7 | |
| Spinal cord | 3/4 | 1 | 2 | - | - | - | 1 | 1 | 1 | |
| Immunoreactive in none of HD cases studied | Pituitary gland | 0/2 | - | - | - | - | - | - | - | - |
| Cerebellar cortex | 0/10 | - | - | - | - | - | - | - | - | |
D, diffuse; A, aggregated; DA, diffuse and aggregated.
Locations that were immunoreactive in control cases: substantia nigra-immunoreactive in 18/21 control cases and locus coeruleus-immunoreactive in 3/21 control cases.
Table 6.
Central Nervous System Locations and Number of Cases Stained
| IC2 | 2B4 | ||||
|---|---|---|---|---|---|
| Location | HD cases | Normal controls |
FTD-Uh cases |
HD cases | Normal controls |
| Neocortex | 15 | 7 | 2 | 4 | 5 |
| Striatum | 9 | 4 | 2 | 4 | 2 |
| Hippocampus | 13 | 7 | 2 | 3 | 4 |
| Lateral geniculate body | 7 | 4 | 1 | 1 | 3 |
| Thalamus | 6 | 7 | 2 | 5 | 4 |
| Cerebellum | 10 | 8 | 2 | 4 | 5 |
| Pituitary gland | 2 | - | 1 | - | - |
| Midbrain | 7 | 9 | 12 | 5 | 5 |
| Pons | 10 | 9 | 12 | 5 | 5 |
| Medulla | 10 | 8 | 2 | 5 | 5 |
| Spinal cord | 4 | 1 | 2 | - | - |
HD, Huntington disease; 1C2, polyglutamine antibody; 2B4, N-terminal huntingtin antibody.
Immunohistochemistry
Four-µm-thick sections were prepared from paraffin blocks of formalin-fixed CNS tissue obtained at the time of autopsy.
Polyglutamine (1C2)
Following deparaffinization, sections were pretreated with concentrated (98%) formic acid for 5 minutes at room temperature. Heat-induced epitope retrieval was carried out using citrate buffer and a modified pressure cooker. All cases were stained with the monoclonal antibody 1C2, MAB1574 clone 5T1-1C2-17.2 (Chemicon), at 1:500 – 1:1000 using a Benchmark XT automatic immunostainer (Ventana Medical Systems, Tucson, AZ). For most sections, streptavidin-horseradish peroxidase/diaminobenzidine (DAB) detection was employed. For selected sections the XT Ultraview red chromogen (Ventana) was used either in addition to or instead of the DAB technique to distinguish between nonspecific brown signal and true polyglutamine immunoreactivity. Sections from the medulla oblongata underwent Nissl staining to aid in the identification of medullary nuclei. Selected sections containing substantia nigra and locus coeruleus were stained with a monoclonal antibody to desmin (Dako, Glostrup, Denmark) and detected with red chromogen as negative tissue controls. Appropriate positive and negative reagent controls were performed.
Huntingtin (2B4)
Deparaffinization and staining with the monoclonal N-terminal huntingtin antibody, MAB5492 clone 2B4, was performed on the Ventana Benchmark XT automatic immunostainer with an EDTA-based retrieval solution. Locations expected to contain neurons with neuromelanin were detected with the Ultraview detection system using a red chromogen at an antibody dilution of 1:500. The remaining sections were detected with Ultraview horseradish peroxidase/diaminobezaidine (DAB) with an antibody dilution of 1:250. Appropriate positive and negative reagent controls were performed.
Assessment
The original hematoxylin and eosin slides of the HD cases were reviewed and graded according to the scale described by Vonsattel et al (21). The cases were graded as: 4 grade II, 10 grade III, and 5 grade IV cases. 1C2 and 2B4 stained sections were evaluated for topographical and intracellular location of immunoreactivity.
RESULTS
1C2 Staining in HD Cases
1C2 immunoreactivity within the cytoplasm or nucleus fell into 4 general categories: diffuse, aggregated, diffuse and aggregated, or no staining (Fig. 3). The diffuse pattern consisted of homogeneous immunoreactivity, whereas the aggregated pattern encompassed a range of finely granular to coarsely punctate immunoreactivity. Within the nucleus, the aggregated designation was used for what are typically termed ‘inclusions,’ as well as for smaller concentrated foci of staining (Table 5).
Figure 3.

Representative nuclear patterns of 1C2 immunoreactivity: (A) diffuse; (B) aggregated; (C)diffuse and aggregated. Representative cytoplasmic patterns of 1C2 immunoreactivity: (D) diffuse; (E) aggregated; (F) diffuse and aggregated. Scale bar = 20 µm.
Cortex
All neocortical sections contained immunoreactive neurons. There were no significant differences in topographical or cellular pattern of neuronal staining among the 4 lobes (data not shown). Immunoreactive neurons were most common in the deeper cortical layers. Combinations of various nuclear and cytoplasmic staining patterns were found within each case. Staining of the neuronal nucleus typically consisted of 1 to 2 round or oval inclusions, approximately 2 to 3 µm in diameter, with a background of diffuse nuclear positivity (Fig. 4A). Frequently, however, there was only diffuse nuclear immunoreactivity without inclusions. Cytoplasmic staining often consisted of scattered flecks of immunoreactivity with little or no diffuse staining. Occasional pinpoint or thread-like foci of immunoreactivity could be identified within neuropil. Rare oligodendrocyte nuclei in the grey or white matter were also immunoreactive (Fig. 4B).
Figure 4.
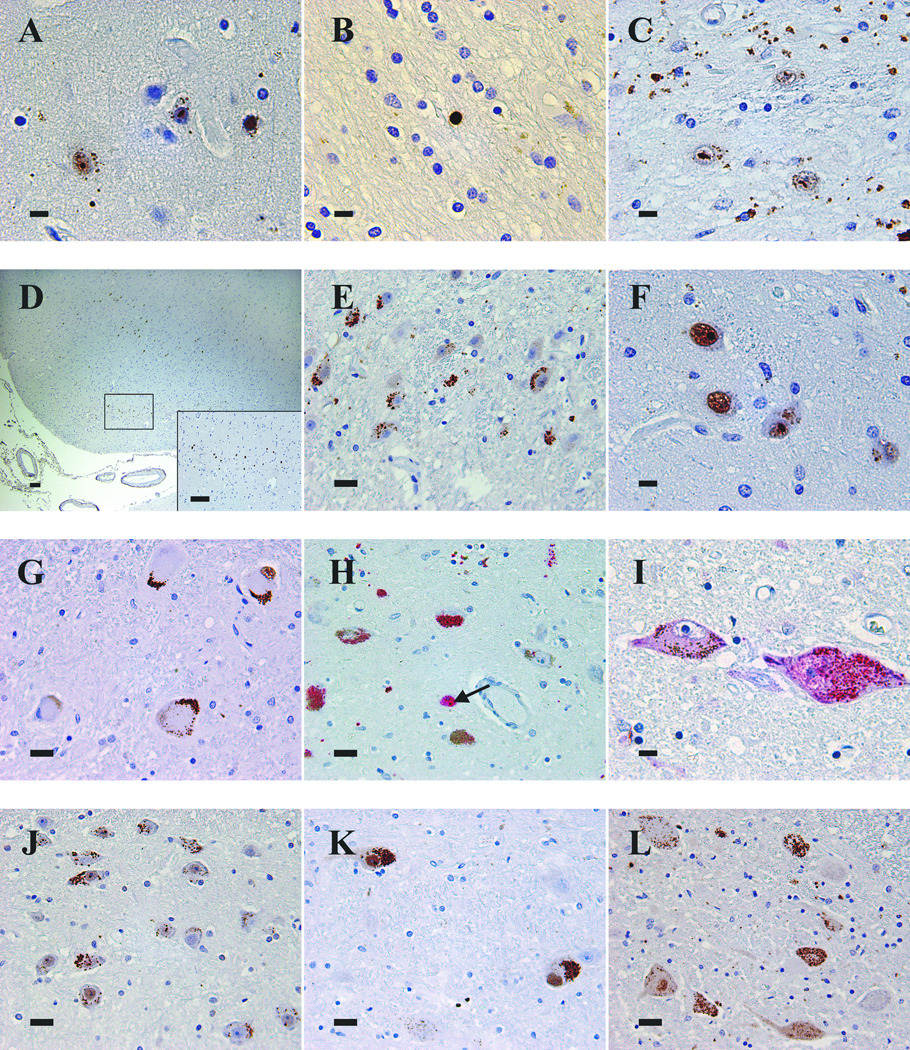
Patterns of 1C2 immunoreactivity in the CNS of HD cases. (A) Frontal cortex of Case 2 demonstrating types of immunoreactivity: coarse granules in neuropil, finely granular aggregated staining in neuronal cytoplasm, diffuse nuclear staining, and round and oval aggregated nuclear immunoreactivity. (B) Subcortical white matter with a diffusely immunoreactive glial nucleus in Case 11. (C) Caudate nucleus of Case 1 with intranuclear immunoreactivity and abundant granular neuropil staining. (D) Entorhinal cortex of Case 5 showing immunoreactive layer 2 pre-alpha clusters. (E) Lateral geniculate body of Case 3 displaying multiple neurons with aggregated cytoplasmic immunoreactivity. (F) Thalamus of Case 2 demonstrating nuclear and cytoplasmic immunoreactivity. (G) Aggregated cytoplasmic immunoreactivity of dentate nucleus neurons of Case 11. (H) Substantia nigra neurons containing aggregated cytoplasmic immunoreactivity (red) intermixed with brown neuromelanin in Case 2. Some neurons have neuromelanin alone. Aggregated nuclear immunoreactivity (arrow) is present in one neuron. (I) Two neurons of the locus coeruleus of Case 15 demonstrating diffuse and aggregated cytoplasmic immunoreactivity, one also with faint, diffuse nuclear immunoreactivity. (J) Basis pontis of Case 2 with frequent immunoreactive neurons. (K) Reticular formation neurons in medulla of Case 5 with intense aggregated cytoplasmic staining and diffuse and aggregated nuclear immunoreactivity. (L) Nucleus dorsalis of thoracic spinal cord of Case 5 showing diffuse and aggregated cytoplasmic immunoreactivity. Several neurons have diffusely immunoreactive nuclei. Scale bars: A, B, C, F, I = 10 µm; E, G, H, J, K, L = 25 µm; D and inset = 100 µm.
Striatum
The caudate and putamen of all 9 cases demonstrated 1C2-immunoreactive neurons. There was a variety of staining patterns but overall cytoplasmic staining tended to be aggregated. In case 1, a juvenile-onset case, there were frequent immunoreactive neurons with striking punctate, granular staining in the neuropil. We speculate that in this case the 1C2 antibody binds aggregated mHtt in the neuropil that was released from neurons during the process of cell death (Fig. 4C).
Hippocampus
Moving from the hilus of the dentate gyrus through the hippocampal formation to the subiculum and entorhinal cortex, increasing numbers of immunoreactive neurons were identified in each of the 13 cases studied (one case contained only minimal entorhinal cortex). The predominant cellular staining pattern was a combination of diffuse or diffuse and aggregated nuclear staining and aggregated cytoplasmic immunoreactivity. In contrast, neurons of the dentate gyrus were immunonegative in 10 out of 13 cases. In the remaining 3 cases there were very rare immunoreactive cells with various morphologic staining patterns (Fig. 4D).
Lateral Geniculate Body
All 6 cases containing the lateral geniculate body demonstrated 1C2-immunoreactive neurons. In 4 cases aggregated cytoplasmic staining with no nuclear staining was the predominant immunoreactivity pattern (Fig. 4E). The remaining cases displayed diffuse nuclear staining in addition to aggregated cytoplasmic immunoreactivity.
Thalamus
Thalamic neurons demonstrated 1C2 immunoreactivity in 5 out of 6 cases studied. A variety of nuclear and cytoplasmic staining patterns was present and the number of immunoreactive neurons also varied from case to case (Fig. 4F).
Cerebellum
The cerebellar cortex of all 10 cases studied was immunonegative. The dentate nucleus was present in 9 of these cases, and it consistently contained immunoreactive neurons usually with aggregated cytoplasmic staining with either diffuse or no nuclear staining (Fig. 4G).
Pituitary
Both of the pituitary glands studied were immunonegative.
Substantia nigra
A red chromogen was used to distinguish positive signal from neuromelanin. The substantia nigra of all 7 cases studied contained frequent neurons with aggregated cytoplasmic immunoreactivity. The location of the signal within the cytoplasm usually but not exclusively corresponded to that of the neuromelanin (Fig. 4H). There was diffuse and aggregated nuclear immunoreactivity in 2 cases but in the other 5 cases no nuclear staining was identified.
Locus coeruleus
As with the midbrain sections, a red chromogen was used to distinguish positive signal from neuromelanin. Neurons of the locus coeruleus were immunoreactive in 7 of 8 cases studied. Finely granular aggregated staining intermixed with neuromelanin was the predominant cytoplasmic pattern, similar to the nigra (Fig. 4I). Nuclei were typically immunonegative.
Pontine nuclei
1C2-immunoreactive neurons were present in the basis pontis of all 10 cases studied. Occasional to frequent neurons were immunoreactive, most often with finely granular aggregated cytoplasmic positivity and a variety of nuclear patterns (Fig. 4J).
Medulla
The medulla oblongata from 10 cases was studied. Rare neurons of the reticular formation stained consistently in all 10 cases with aggregated cytoplasmic immunoreactivity. In all but 2 cases diffuse nuclear immunoreactivity was the predominant pattern although rare neuronal nuclei with diffuse and aggregated staining could also be found (Fig. 4K). We noted immunoreactivity in rare neurons of other medullary nuclei. These nuclei demonstrated a variety of nuclear and cytoplasmic staining patterns.
Spinal Cord
Three of the 4 cases studied showed rare 1C2-immunoreactive neurons within the spinal cord grey matter with a variety of cytoplasmic and nuclear immunoreactivity patterns. Punctate neuropil staining in the grey and white matter suggestive of cross-sectional axonal immunoreactivity was also present (Fig. 4L).
1C2 Staining in Control Cases
1C2 immunoreactivity was identified in substantia nigra, locus coeruleus, and pituitary gland of control cases. 18 of 21 normal and diseased control cases demonstrated 1C2 immunoreactivity in substantia nigra neurons. The staining pattern consisted of aggregated cytoplasmic immunoreactivity that often, but not exclusively, localized with neuromelanin (Fig. 5A). In many cases, clusters of granular immunoreactivity were present in the neuropil around nigral neurons, presumably representing the cytoplasmic immunoreactivity of macrophages. No control cases demonstrated immunoreactivity in the nuclei of nigral neurons.
Figure 5.

1C2 immunoreactivity in diseased control cases. (A)Substantia nigra with aggregated cytoplasmic immunoreactivity in neurons and adjacent macrophages. (B) Neurons of locus coeruleus with cytoplasmic immunoreactivity. (C) Neurohypophysis with cytoplasmic immunoreactivity.
The locus coeruleus of 3 of 21 normal and diseased control cases demonstrated finely granular aggregated cytoplasmic 1C2 immunoreactivity that mostly accompanied neuromelanin (Fig. 5B). As in the substantia nigra of the 18 control cases described above, no nuclear immunoreactivity was identified.
The pituitary gland in one diseased control demonstrated foci of thread-like 1C2 immunoreactivity within the neurohypophysis often accompanied by granular golden-brown deposits of hemosiderin (Fig. 5C). The adenohypophysis was immunonegative for 1C2.
To investigate whether the 1C2 immunoreactivity in the substantia nigra and locus coeruleus was the result of nonspecific binding of excess antibody, a dilution study of 1C2 was performed on the substantia nigra from a 59 year-old normal control patient and cortex of a 55-year-old HD patient. 1C2 immunoreactivity was extinguished at the same dilution, 1:4000, in both cases (Fig. 6).
Figure 6.

1C2 antibody dilution study. Immunoreactivity in the substantia nigra of a 59-year-old normal control patient (A–D) and the cortex of a 55-year-old patient with HD (E–H) disappears at a 1C2 dilution of 1:4000. Scale bars = 50 µm.
To eliminate the possibility of non-specific detection by the Ultraview red chromogen (Ventana), we performed the identical staining protocol on the substantia nigra and locus coeruleus from 1 HD case and 2 diseased control cases using the red chromogen and an antibody to desmin rather than the 1C2 antibody. Signal was only present within rare blood vessel walls (i.e. normal native staining), making non-specificity of the detection system a less likely cause for this immunoreactivity.
2B4 Staining
2B4, an antibody to amino acids 1 through 82 of normal Htt, had a neuroanatomic and intracellular immunoreactivity pattern unlike that of 1C2 (Table 7, Fig. 7). In each major neuroanatomic area, neurons in normal control cases demonstrated some degree of either cytoplasmic or nuclear immunoreactivity or both. Aggregated 2B4 staining within neuronal cytoplasm or nucleus was often composed of smaller caliber and less distinct foci than with 1C2. Aggregated nuclear immunoreactivity was generally but not exclusively limited to neurons of HD cases. Although no distinct immunoreactive puncta such as that frequently seen in 1C2 staining of HD cases were identified, the neocortex of a normal control demonstrated a finely granular aggregated nuclear pattern. Finally, glial cells were frequently immunoreactive in both HD and normal control cases.
Table 7.
2B4 Immunoreactivity in Huntington Disease and Normal Control Cases
| Location | Cytoplasm | Nucleus | |||||||
|---|---|---|---|---|---|---|---|---|---|
| D | A | DA | No | D | A | DA | No | ||
| Neocortex | 4 HD cases | 1 | 3 | - | - | - | 2 | - | 2 |
| 5 normal cases | - | 3 | - | 2 | - | 1 | - | 4 | |
| Caudate and/or putamen | 4 HD cases | 1 | 3 | - | - | 1 | - | - | 3 |
| 2 normal cases | - | 1 | - | 1 | 1 | - | - | 1 | |
| Hippocampus - CA1/Subiculum | 3 HD cases | - | - | - | 3 | - | 2 | - | 1 |
| 5 normal cases | - | 2 | - | 3 | - | - | - | 5 | |
| Hippocampal dentate gyrus | 3 HD cases | - | - | - | 3 | - | 2 | - | 1 |
| 5 normal cases | - | 2 | - | 3 | - | - | - | 5 | |
| Entorhinal cortex | 3 HD cases | 1 | - | - | 2 | - | 1 | - | 2 |
| 4 normal cases | - | 3 | - | 1 | 1 | - | - | 3 | |
| Lateral geniculate body | 1 HD case | - | - | - | 1 | - | 1 | - | - |
| 3 normal cases | - | 2 | - | 1 | - | - | - | 3 | |
| Thalamus | 5 HD cases | 1 | 1 | - | 3 | 2 | 2 | - | 1 |
| 4 normal cases | - | 3 | - | 1 | - | - | - | 4 | |
| Substantia nigra | 5 HD cases | 4 | - | - | 1 | - | - | - | 5 |
| 5 normal cases | 4 | - | - | 1 | - | - | - | 5 | |
| Locus coeruleus | 4 HD cases | 1 | - | - | 3 | - | - | - | 4 |
| 5 normal cases | 1 | - | - | 4 | - | - | - | 5 | |
| Basal pontine nuclei | 5 HD cases | 1 | 2 | - | 2 | - | 5 | - | - |
| 5 normal cases | - | 3 | - | 2 | - | - | - | 5 | |
| Inferior olives | 5 HD cases | 3 | - | - | 2 | 3 | - | - | 2 |
| 5 normal cases | 5 | - | - | - | 5 | - | - | - | |
| Cerebellar dentate nucleus | 4 HD cases | 1 | - | - | 3 | 1 | - | - | 3 |
| 5 normal cases | 1 | 1 | - | 3 | - | - | - | 5 | |
| Cerebellar granular neurons | 4 HD cases | - | - | - | 4 | 1 | - | - | 3 |
| 5 normal cases | - | - | - | 5 | 3 | - | - | 2 | |
| Cerebellar Purkinje cells | 4 HD cases | - | 3 | - | 1 | - | - | - | 4 |
| 5 normal cases | - | 4 | - | 1 | - | - | - | 5 | |
HD, Huntington disease; D, diffuse; A, aggregated; DA, diffuse and aggregated.
Figure 7.

Comparison of HD case (A, C) and normal control case (B, D) with 1C2 (A, B) and 2B4 (C, D) staining. Arrows indicate aggregated nuclear staining. Scale bar = 20 µm.
DISCUSSION
One objective in this study was to construct a neuroanatomic and morphologic profile for polyglutamine antibody 1C2 immunoreactivity in the HD CNS. 1C2 immunoreactivity was present in diverse areas of the HD CNS, and a variety of cytoplasmic and nuclear staining patterns was identified in neurons, neuropil, and glia. All of the HD cases showed immunoreactivity in the neocortex, striatum, hippocampal pyramidal neurons/subiculum/entorhinal cortex, lateral geniculate body, substantia nigra, basal pontine nuclei, medullary reticular formation, and cerebellar dentate nucleus (Table 5). Some cases showed 1C2 immunoreactivity in the dentate gyrus, thalamus, locus coeruleus, and spinal cord. None of the HD cases demonstrated immunoreactivity in the cerebellar cortex or pituitary gland.
1C2 immunoreactivity was identified in normal and diseased control cases, a finding not reported to date in the HD immunohistochemistry literature. Neurons and occasional adjacent macrophages in the substantia nigra and locus coeruleus of these controls demonstrated aggregated cytoplasmic immunoreactivity. This immunoreactivity was not present when staining with an antibody to desmin was performed under similar conditions.
What is the epitope of 1C2 in these locations? Is it truly the long glutamine stretch? In addition to polyglutamine stretches, 1C2 has been shown to bind polyleucine- and polyalanine-containing proteins (22, 23). We cannot exclude the possibility that the 1C2 immunoreactivity in nigral neurons may represent detection of leucine or alanine residues present in neuromelanin. Neuromelanin is a pigmented material that accumulates in neuronal cytoplasm over time and is believed to arise from the spontaneous autoxidation of dopamine in the substantia nigra and of norepinephrine in the locus coeruleus (24). Neuromelanin extracted from the nigras of 18 normal brains by Double et al (24) contained a variety of amino acids, including leucine and alanine, but not glutamine.
Another possibility is that substantia nigra 1C2 immunoreactivity represents an accumulation of normal TBP. The detection of TBP by 1C2 was proposed as a possible explanation by Rudnicki et al (19) for neuronal 1C2 immunoreactivity in cases of Huntington disease-like 2 (HDL2). HDL2 is an autosomal dominant disease with clinical features and neuropathology similar to that of HD but is not associated with a CAG expansion in the IT15 gene at 4p16.3. Rather, it is caused by a CAG/CTG repeat expansion in a variably spliced exon of the junctophilin-3 gene at 16p24.3. Intraneuronal protein aggregates immunoreactive for 1C2 have been identified in all reported cases to date, but it is not clear whether LGS are present within the aggregates. Besides TBP, polyalanine and polyleucine stretches were also proposed as possible antigens for 1C2 in these HDL2 cases. Fortunately, a difference in distribution of 1C2 immunoreactivity is one feature that distinguishes HD from HDL2. In the 5 cases of HDL2 studied, all lacked 1C2-immunoreactive intranuclear aggregates in the pons and medulla, 2 locations in which we and others have demonstrated reproducible 1C2 immunoreactivity in HD (18).
Despite this 1C2 immunoreactivity in 3 limited areas of control cases, we feel that its consistent, reproducible staining in numerous CNS locations in HD cases provides a supplemental diagnostic tool in the postmortem diagnosis of HD. Our experience with 2B4, on the other hand, leads us to believe that it would be difficult to use in a diagnostic setting. Although our survey was small, the morphologic patterns of 2B4 staining were less reproducible in distinguishing between HD and controls.
Immunohistochemistry has been integral to the advancement of knowledge about HD with the work of Lunkes et al (13) and Schilling et al (17) as prime examples. They narrowed the mHtt cleavage site to an area between amino acids 81 and 90 and 115 and 129 using multiple anti-Htt antibodies. While the identity of the enzyme(s) performing this cleavage in the neuron and the exact location of the cleavage remain unknown, this information puts the results of the many immunohistochemical studies listed in Table 1 into a clearer molecular framework. These anti-Htt antibodies (by no means an exhaustive list), segregate roughly into 2 groups: those that recognize the N-terminal fragment of mHtt and those that recognize the portion of mHtt on the carboxy end of the putative cleavage site. With cleavage between amino acids approximately 90 and 115 in HD, the latter antibodies will not label the aggregates that are characteristic of HD and thus will not differentiate between HD and non-HD CNS tissue. With rare exceptions, this holds true in the studies listed in Table 1 and Table 2. The 1C2 antibody is unique in that its epitope should be present within an HD neuron regardless of where mHtt is cleaved and which fragments of the protein aggregate. But clearly, LGS and therefore 1C2 binding are not specific for HD; 8 other CAG repeat diseases (spinal bulbar muscular atrophy, dentatorubral pallidoluysian atrophy, and spinocerebellar atrophy 1, 2, 3, 7, 8, and 17) are currently known to contain expanded CAG repeats that are translated in LGS (25).
Immunohistochemical technique is also a worthwhile consideration when working with HD antibodies. Technical methods, especially for epitope retrieval, have repeatedly been shown to play an important role in the interpretability and reproducibility of immunohistochemical staining. Recent work by BrainNet Europe centers demonstrates that variability in alpha-synuclein staining between laboratories can significantly influence interrater reliability (26). 1C2-immunoreactive intranuclear inclusions were a prominent finding in many of the CNS locations that we studied, but they have not been a consistent finding in other studies. Yamada et al (18) and Rudnicki et al (19), but not Gourfinkel-An et al (9) or Sieradzan et al (12), identified neuronal intranuclear inclusions in their studies. Technical differences in epitope retrieval and staining methods may explain these dissimilar results although Yamada et al did not describe their methods. We and others have found that formic acid pretreatment of CNS tissue improves antigenicity for 1C2 (19, 27, 29). Our use of chemical and heat pretreatment may have unmasked epitopes within the neuronal nucleus that had been undetectable in other studies.
Our finding of 1C2 immunoreactivity in neuromelanin-containing neurons and adjacent macrophages in normal and diseased controls raises interesting and important questions about the nature of these neurons and the epitope recognized by 1C2. Aside from these areas, we have identified widespread, reproducible 1C2 immunoreactivity in postmortem CNS tissue of individuals with HD that is absent in controls. Compared to staining with the 2B4 anti-huntingtin antibody, 1C2 may therefore be of diagnostic utility in postmortem CNS evaluation for HD when other CAG repeat diseases are not under consideration and when the substantia nigra, locus coeruleus, and pituitary gland are avoided.
ACKNOWLEDGMENTS
This work was supported in part by NIH ADC grant AG12300, the Winspear Family Center for Research on the Neuropathology of Alzheimer's Disease, and the McCune Foundation.
The authors thank Gwen Beasley, HT(ASCP), for her excellent histologic preparation and Amy Davis, HTL(ASCP)QIHC, for her skillful immunochemistry.
REFERENCES
- 1.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.del Toro D, Canals JM, Gines S, et al. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci. 2006;26:12748–12757. doi: 10.1523/JNEUROSCI.3873-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trottier Y, Lutz Y, Stevanin G, et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 4.Tibben A. Genetic counselling and presymptomatic testing. In: Bates G, Harper P, Jones L, editors. Huntington's Disease. Oxford: Oxford University Press; 2002. pp. 198–248. [Google Scholar]
- 5.Jones L. The cell biology of Huntington's disease. In: Bates G, Harper P, Jones L, editors. Huntington's Disease. Oxford: Oxford University Press; 2002. pp. 348–386. 5. [Google Scholar]
- 6.DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 7.Sapp E, Schwarz C, Chase K, et al. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 8.Becher MW, Kotzuk JA, Sharp AH, et al. Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 9.Gourfinkel-An I, Cancel G, Duyckaerts C, et al. Neuronal distribution of intranuclear inclusions in Huntington's disease with adult onset. Neuroreport. 1998;9:1823–1826. doi: 10.1097/00001756-199806010-00028. [DOI] [PubMed] [Google Scholar]
- 10.Gutekunst CA, Li SH, Yi H, et al. Nuclear and neuropil aggregates in Huntington's disease: Relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maat-Schieman ML, Dorsman JC, Smoor MA, et al. Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J Neuropathol Exp Neurol. 1999;58:129–137. doi: 10.1097/00005072-199902000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Sieradzan KA, Mechan AO, Jones L, et al. Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol. 1999;156:92–99. doi: 10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- 13.Lunkes A, Lindenberg K, Ben-Haiem L, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 14.van Roon-Mom MCW, Reid SJ, Jones AL, et al. Insoluble TATA-binding protein accumulation in Huntington's disease cortex. Mol Brain Res. 2002;109:1–10. doi: 10.1016/s0169-328x(02)00450-3. [DOI] [PubMed] [Google Scholar]
- 15.van Roon-Mom MCW, Hogg VM, Tippett LJ, et al. Aggregate distribution in frontal and motor cortex in Huntington's disease brain. Neuroreport. 2006;17:667–670. doi: 10.1097/00001756-200604240-00022. [DOI] [PubMed] [Google Scholar]
- 16.Maat-Schieman M, Roos R, Losekoot M, et al. Neuronal intranuclear and neuropil inclusions for pathological assessment of Huntington's disease. Brain Pathol. 2007;17:31–37. doi: 10.1111/j.1750-3639.2006.00040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schilling G, Klevytska A, Tebbenkamp A, et al. Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J Neuropathol Exp Neurol. 2007;66:313–320. doi: 10.1097/nen.0b013e318040b2c8. [DOI] [PubMed] [Google Scholar]
- 18.Yamada M, Tsuji S, Takahashi H. Pathology of CAG repeat diseases. Neuropathol. 2000;20:319–325. doi: 10.1046/j.1440-1789.2000.00354.x. [DOI] [PubMed] [Google Scholar]
- 19.Rudnicki D, Pletnikova O, Vonsattel J-P, et al. A comparison of Huntington disease and Huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol. 2008;67:366–374. doi: 10.1097/NEN.0b013e31816b4aee. [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Butland SL, Devon RS, et al. Familial frontotemporal dementia with neuronal intranuclear inclusions is not a polyglutamine expansion disease. BMC Neurol. 2006;6:1471–2377. doi: 10.1186/1471-2377-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vonsattel JP, Myers RH, Stevens TJ, et al. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Dorsman J, Pepers B, Langenberg D, et al. Strong aggregation and increased toxicity of polyleucine over polyglutamine stretches in mammalian cells. Hum Mol Genet. 2002;11:1487–1496. doi: 10.1093/hmg/11.13.1487. [DOI] [PubMed] [Google Scholar]
- 23.Sugaya K, Matsubara S, Miyamoto K, et al. An aggregate-prone conformational epitope in trinucleotide repeat diseases. Neuroreport. 2003;14:2331–2335. doi: 10.1097/00001756-200312190-00009. [DOI] [PubMed] [Google Scholar]
- 24.Double K, Zecca L, Cost P, et al. Structural characteristics of human substantia nigra neuromelanin and synthetic dopamine melanins. J Neurochem. 2000;75:2583–2589. doi: 10.1046/j.1471-4159.2000.0752583.x. [DOI] [PubMed] [Google Scholar]
- 25.Butland S, Devon R, Huang Y, et al. CAG-encoded polyglutamine length polymorphism in the human genome. BMC Genomics. 2007;8:126. doi: 10.1186/1471-2164-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alafuzoff I, Parkkinen L, Al-Sarraj S, et al. Assessment of alpha-synuclein pathology: A study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol. 2008;67:125–143. doi: 10.1097/nen.0b013e3181633526. [DOI] [PubMed] [Google Scholar]
- 27.White CL, III, Hladik C, Stark J, et al. Formic acid pretreatment enhances immunostaining of polyglutamine-containing inclusions in Huntington disease brains. Brain Pathol. 2006;16:S55. [Google Scholar]
- 28.Osmand AP, Berthelier V, Wetzel R. Imaging polyglutamine deposits in brain tissue. Methods Enzymol. 2006;412:106–122. doi: 10.1016/S0076-6879(06)12008-X. [DOI] [PubMed] [Google Scholar]