

Abstract
Delayed excitotoxic neuronal death after insult from exposure to high glutamate concentrations appears important in several CNS disorders. Although delayed excitotoxicity is known to depend on NMDA receptor (NMDAR) activity and Ca2+ elevation, the electrophysiological mechanisms underlying postinsult persistence of NMDAR activation are not well understood. Membrane depolarization and nonspecific cationic current in the postinsult period were reported previously, but were not sensitive to NMDAR antagonists. Here, we analyzed mechanisms of the postinsult period using parallel current- and voltage-clamp recording and Ca2+ imaging in primary hippocampal cultured neurons. We also compared more vulnerable older neurons [about 22 days in vitro (DIV)] to more resistant younger (about 15 DIV) neurons, to identify processes selectively associated with cell death in older neurons. During exposure to a modest glutamate insult (20 μM, 5 min), similar degrees of Ca2+ elevation, membrane depolarization, action potential block, and increased inward current occurred in younger and older neurons. However, after glutamate withdrawal, these processes recovered rapidly in younger but not in older neurons. The latter also exhibited a concurrent postinsult increase in spontaneous miniature excitatory postsynaptic currents, reflecting glutamate release. Importantly, postinsult NMDAR antagonist administration reversed all of these persisting responses in older cells. Conversely, repolarization of the membrane by voltage clamp immediately after glutamate exposure reversed the NMDAR-dependent Ca2+ elevation. Together, these data suggest that, in vulnerable neurons, excitotoxic insult induces a sustained positive feedback loop between NMDAR-dependent current and depolarization-mediated glutamate release, which persists after withdrawal of exogenous glutamate and drives Ca2+ elevation and delayed excitotoxicity.
INTRODUCTION
Excitotoxic neuronal death (Olney 1986) can be induced by excessive activation of glutamate receptors during exposure to high glutamate concentrations. The induction of such excitotoxicity depends on Ca2+ influx through N-methyl-d-aspartate–type glutamate receptors (NMDARs) (Choi 1992; Hartley et al. 1993; Limbrick et al. 2001; Nicotera and Orrenius 1998; Randall and Thayer 1992; Tymianski et al. 1993) and is widely thought to play an important role in several neuropathological conditions, including ischemia, stroke, trauma, HIV-related neurotoxicity, and, possibly, neurodegenerative disease (Choi 1995; Lipton and Rosenberg 1994; Olney 1986).
However, excitotoxic neuronal death is often delayed for some hours after the termination of a glutamate insult. Several lines of evidence indicate that continuing activity of NMDARs during the postinsult phase is essential for the expression of this delayed excitotoxicity. Perhaps most importantly, it has been found that NMDAR antagonists administered up to an hour or longer after glutamate exposure can be fully neuroprotective (Choi et al. 1988; Levy and Lipton 1990; Munir et al. 1995; Okiyama et al. 1998; Rothman et al. 1987; Shalaby et al. 1992; Smith et al. 1993). Delayed excitotoxicity also appears to be Ca2+ dependent and has been linked to a persistent Ca2+ elevation (PCE) that follows termination of glutamate insult. This PCE has been sensitive to block by NMDAR antagonists in some studies, but not in others (Attucci et al. 2002; Cheng et al. 1999; Dubinsky 1993; Limbrick et al. 2001; Rajdev and Reynolds 1994; Randall and Thayer 1992; Vergun et al. 1999), suggesting there may be multiple phases or sources of Ca2+ elevation.
Despite the critical importance of the postinsult period, however, little is known about the mechanisms that sustain NMDAR activity or the PCE after glutamate withdrawal, in part because of a paucity of electrophysiological studies on delayed excitotoxicity and the sequence of events unfolding during the postexcitotoxic insult period. The few studies on this phase have identified several membrane responses as potential mechanistic mediators of delayed excitotoxicity, including extended neuronal depolarization (Coulter et al. 1992; Limbrick et al. 2001) and an inward nonspecific cationic current (postexposure current, IPE) (Chen et al. 1997, 1998). However, once induced, neither of these membrane responses could be interrupted by NMDAR antagonists, indicating that they do not mediate the toxic actions of persistent NMDAR activity.
Here, we combined parallel current-clamp, voltage-clamp, and Ca2+-imaging techniques, to analyze events during and after excitotoxic exposure. Further, to elucidate which postin-sult responses are most closely associated with cell death, we studied primary hippocampal cultured neurons at different ages because their vulnerability to excitotoxicity increases substantially with developmental age (Adamec et al. 1998; Attucci et al. 2002; Cheng et al. 1999; Choi 1992; Clodfelter et al. 2002; Frandsen and Schousboe 1991; Toescu and Verkhratsky 2000; Xia et al. 1995). Together, the results appear to identify a novel positive feedback loop that sustains NMDAR current and Ca2+ elevation after excitotoxic insult.
METHODS
Cell culture
Primary hippocampal mixed neuronal–glial cell cultures were prepared from fetal pup tissue (embryonic day 18) obtained from pregnant Sprague–Dawley rats using slight modifications of the Banker method (Banker and Cowan 1977) as previously described (Porter et al. 1997). All protocols were approved by the institutional animal care and use committee. Fetal hippocampal tissue was isolated and neurons were dissociated and plated on 35-mm poly-l-lysine–coated dishes and Thermanox plastic coverlips (Nunc, Naperville, IL) as previously described (Blalock et al. 1999). In most studies, age comparisons were performed in cultures of younger neurons [11–17 days in vitro (DIV)] against sister cultures of older neurons (21–24 DIV), although in some experiments (see Fig. 1), somewhat different age ranges (younger, 7–14 DIV; older, 28–35 DIV) were used. All experiments were conducted at room temperature.
FIG. 1.
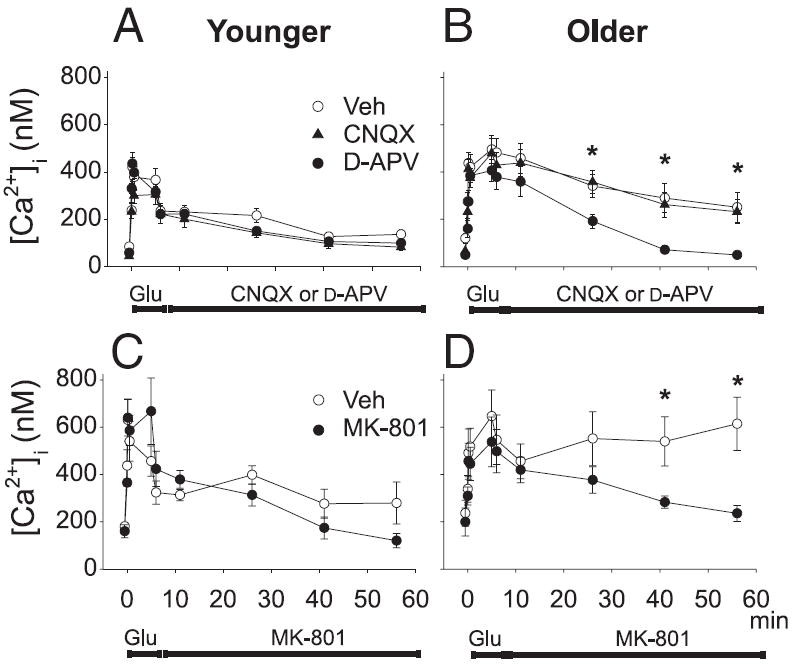
Inhibition of N-methyl-d-aspartate receptors (NMDARs) in the postinsult period reduced the prolonged Ca2+ elevation (PCE). A and B: mean ± SE [Ca2+]i levels obtained using Indo-1 confocal laser scanning microscopy are plotted before, during, and after a 5-min glutamate insult (100 μM) in 7–14 days in vitro (DIV, A) and 10 μM in 28–35 DIV (B) neurons. Immediately after glutamate exposure, medium containing vehicle (open circles), 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, filled triangles) or 100 μM d-(−)-2-amino-5-phosphonovaleric acid (d-APV, filled circles) was perfused through the culture dish. C and D: similar experiment with a noncompetitive NMDAR antagonist showing [Ca2+]i before, during, and after a 5-min glutamate insult in younger (C) and older (D) neurons. Immediately after glutamate exposure, medium containing vehicle (open circles) or 10 μM NMDAR antagonist MK-801 (filled circles) was perfused through the culture dish. For A–D, bars indicate the periods in which glutamate and glutamate receptor antagonists were applied. n = 5–10 cells per group [two-way ANOVA on repeated measures; post hoc Tukey’s pairwise comparisons; B: Veh and CNQX both significantly different from d-APV at indicated time points (*P < 0.05); D: Veh significantly different from MK-801 at indicated time points (*P < 0.05)].
Solutions
For all experiments the external recording/imaging solution contained (in mM): 145 NaCl, 2.5 KCl, 10 HEPES, 10 d-glucose, 2 CaCl2, 1 MgCl2, and 0.01 glycine; pH was adjusted to 7.35 using NaOH. Osmolality was adjusted to 310 mOsm with sucrose. The recording pipette solution contained (in mM): 150 KCH3SO4, 5 HEPES, 4 Tris-ATP, 0.3 Tris-GTP, 1.4 Tris-phospho-creatine, and 0.1 leupeptin; pH was adjusted to 7.35 using KOH and osmolality was adjusted to 290 mOsm by dilution with dH2O. All solutions were sterile-filtered.
Electrophysiology
Recording pipettes made from glass capillary tubes (Drummond Scientific, Broomall, PA) were pulled on a horizontal micropipette puller (model P-87; Sutter Instruments, Novato, CA). All pipettes were coated with polystyrene Q-dope and were fire-polished immediately before recording (Corey and Stevens 1983). Tip resistances were similar across the two age groups and ranged between 2.4 and 4.5 MΩ (mean = 3.3 ± 0.09 MΩ). Plastic coverslips layered with hippocampal neurons were cut into fragments and transferred to a perfusion chamber (Warner Instruments, Hamdem, CT). Spontaneous synaptic activity was monitored in individual neurons with current-clamp or voltage-clamp techniques using Axopatch 200 amplifiers and pClamp 8.0 software (Axon Instruments, Foster City, CA). Cells unable to maintain a resting potential (Vm) of at least −50 mV for 5 min without current injection through the recording electrode were discarded. Spontaneous excitatory postsynaptic potentials (EPSPs) and action potentials (APs) were filtered at 10 kHz and digitized at 1 kHz. In voltage-clamp experiments, neurons were clamped at −80 mV throughout the experiment. Spontaneous excitatory postsynaptic currents (EPSCs) were filtered at 1 kHz and digitized at 2 kHz.
At the beginning of each experiment, junction potentials were nulled in the bath and pipette capacitance was compensated. Whole cell membrane capacitance was determined off-line by integrating the area of the capacitive transient generated during a 10-ms, 5-mV hyperpolarizing step from −70 mV. Membrane capacitance was 33.8 ± 1.9 pF for younger neurons (n = 47) and 40.1 ± 3.5 pF for older neurons (n = 61) (P < 0.05). Access resistances were 6.9 ± 0.46 and 5.59 ± 0.3 MΩ (P < 0.05), respectively. These values and the differences observed between younger and older cells were similar to those reported earlier (Blalock et al. 1999; Porter et al. 1997).
Indo-1 imaging
For some Ca2+-imaging studies, Indo-1 was used as described previously (Attucci et al. 2002; Clodfelter et al. 2002). Briefly, cultures were placed at room temperature in the dark for a 20-min incubation period in 2 μM of the acetoxymethyl ester of Indo-1, AM (Molecular Probes, Eugene, OR). Cells were then washed three times with indicator-free recording solution and incubated for an additional 15 min to allow deesterification of the intracellular indicator.
Ca2+ transients were acquired on an RCM 8000 UV-compatible confocal laser-scanning microscope (Nikon, New York, NY) equipped with a Nikon Diaphot 300 and a ×40 water-immersion objective. Two emitted wavelengths (<400 and >500 nm) were acquired simultaneously through a dichroic mirror centered at 445 nm. Excitation was provided by an argon laser (351–364 nm). All signals were background-subtracted from a cell-free area adjacent to the cells of interest. The area of interest was outlined by hand using Metamorph imaging software (Universal Imaging, West Chester, PA) and consisted of the somatic region. A ratio of the two wavelength images was constructed in Metamorph and analyzed based on an in situ calibration (see following text). Ratios were acquired immediately before glutamate (Glu) application (rest), and at 2, 10, and 30 s and 5 min during glutamate application, as well as at 1, 5, 15, 30, and 45 min postglutamate.
In situ Indo-1 calibration
Ca2+ concentrations were calibrated in situ using hippocampal neurons prepared and maintained as above. Neurons were dialyzed with Indo-1 (pentapotassium salt; Molecular Probes, Eugene, OR) at two concentrations (50 or 100 μM) using patch pipettes, to determine the extent of intracellular Indo-1 loading. The bathing solution consisted of (in mM): 130 LiCl2, 5 CsCl, 2 MgCl2, 10 glucose, 10 HEPES, 0.001 tetrodotoxin (TTX), and 0.1 d-(−)-2-amino-5-phosphonovaleric acid (d-APV). Ratios were converted to [Ca2+] by the equation [Ca2+]i = KDβ(R − Rmin)/(Rmax − R), where R is the 400/500-nm fluorescence emission ratio of a particular cell and Rmax, Rmin, and KDβ were determined from the in situ calibration (Grynkiewicz et al. 1985; Poenie 1990). Separate calibration curves were developed for younger and older cells and no differences in the parameters were found. The in situ calibration yielded values of 1.3 for Rmin, 4.3 for Rmax, and 0.8 for KDβ (in μM).
Fura-2 imaging and concomitant electrophysiology
Axon Imaging Workbench 2.2 (AIW, Axon Instruments) and pClamp 7 were used for concomitant acquisition of Ca2+ imaging and electrophysiology data. Briefly, glass pieces (broken glass-bottom 35-mm culture dishes) containing primary cultures of hippocampal cells were transferred to an RC-22 perfusion chamber (Warner Instruments) and individual cells were loaded with 10 μM Fura-2 pentapotassium salt (Molecular Probes) by the recording patch pipette. Although this relatively low Fura-2 concentration minimized the intracellular buffering of Ca2+ (Helmchen et al. 1996), it also made it difficult to reliably image Ca2+ dynamics in neuronal processes. As such, all measurements reported here refer to somatic Ca2+ levels.
A CCD camera (MicroMax 5 MHz; Princeton Instruments) in combination with an illumination controller (Lambda DG4; Sutter Instruments) was used to acquire emitted light (510 nm) and excite the fluorophore (340 and 380 nm) through a dichroic mirror. A Nikon E600FN microscope hosted the recording and imaging setup. Exposure times were kept to a minimum (0.5–1 s) and a ratio was acquired every 20 s, starting 2 min before glutamate application. Background-subtracted 340- and 380-nm wavelengths were ratioed and analyzed using AIW. Calibration of Fura-2 ratiometric values was accomplished using a series of increasing free Ca2+ concentrations (1 mM Mg2+ kit; Molecular Probes). Values for Rmin, Rmax, and KDβ were determined from the calibration curve and used to calculate free [Ca2+] using the formula described above for Indo-1. For this calibration set, Rmin, Rmax, and KDβ were 0.6, 7.9, and 1.3 μM, respectively.
In experiments in which a hybrid current-clamp/voltage-clamp protocol was used, the Axopatch 1D (Axon Instruments) was switched from current-clamp to voltage-clamp mode and a command potential of −30 mV was immediately imposed on the cell membrane. Then, over the course of 1 min, Vm was ramped linearly to a final value of −80 mV.
Drug delivery
All drugs, including l-glutamate, d-APV, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor blocker 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), the NMDA receptor antagonist MK-801, and the Na+ channel blocker TTX were dissolved in HPLC-grade H2O and stored as stock solutions (+4°C). When used, these compounds were diluted 1,000 × in recording solution. QX-314 (1 mM; Tocris Cookson, Ellisville, MO) was present in the recording pipette in a subset of voltage-clamp experiments to prevent the possible generation of Na+-driven “action potential” currents in the recorded cell. In each experiment, the control condition consisted of adding H2O vehicle to the recording medium at a 1:1,000 dilution. All external solutions were perfused through the recording chamber at 1 to 1.5 mL/min.
Data analysis
Effects of age and/or treatment were assessed by repeated-measures ANOVA. Maximal [Ca2+]i, Vm, and I values during the glutamate application were substantially larger than pre- and post-glutamate values and therefore analyzed separately by one-way ANOVA.
Properties of spontaneous membrane events (i.e., currents and potentials) including amplitude and total number of events were analyzed using Minianalysis software (Synaptosoft) and pClamp 7. The number of EPSCs was counted over a 2-min period immediately before and 15 min after glutamate exposure and the EPSCs were summed and binned according to amplitude. Grouping the data in this manner revealed that the majority of events in each treatment group fell between 15 and 20 pA (i.e., the peak of the distribution). After the peak, the number of events fell exponentially as the event amplitude increased to about 300 pA. This decay was fit well with a single exponential (black line) and the resulting tau values were compared between groups using a Z-test. z-scores > ∣ 2 ∣ were considered significant. Events >300 pA (i.e., compound events) were excluded from these histograms and analyzed separately, primarily because their amplitudes were narrowly distributed and easily discriminated from small events. The mean number of these large compound events in a 2-min window before and 15 min after glutamate exposure was compared across groups using repeated-measures ANOVA. The Scheff é F-test was used for all post hoc analyses. All statistical tests were conducted with SigmaStat (2.0.3) or Statview (5.0.1). For all statistical tests, significance was set at P ≤ 0.05.
To compare membrane repolarization and [Ca2+]i recovery kinetics, Vm and [Ca2+]i values recorded during the postinsult period were normalized to their respective peak values obtained immediately before glutamate removal. These normalized values were then fit with a Boltzmann equation and the time required for Vm and [Ca2+]i to recover to half-maximal values was compared using a z-test.
RESULTS
NMDAR activity was required for persistent Ca2+ elevation
Figure 1 illustrates experiments in which cultures were exposed for 5 min to glutamate and then imaged for the remainder of 1 h. As cells age in culture they exhibit a greater amplitude and more prolonged duration of Ca2+ elevation after a glutamate insult. The prolonged Ca2+ elevation (PCE) in older neurons after glutamate exposure is not attributable to a larger initial peak in Ca2+ because younger neurons do not reliably show a substantial PCE, even when glutamate concentrations are adjusted to generate similar maximal Ca2+ elevations (Attucci et al. 2002).
Age-dependent changes and NMDAR sensitivity of the PCE are shown in Fig. 1. As reported previously (Attucci et al. 2002), a 5-min glutamate application (100 μM) to younger (7–14 DIV) neurons produced a rise in [Ca2+]i that endured for the duration of glutamate application, but then began to return gradually to basal levels soon after removal of glutamate (Fig. 1, A and C). In older cells (28–35 DIV), [Ca2+]i also increased during glutamate (10 μM) application (Fig. 1B). However, in contrast to younger neurons, [Ca2+]i remained elevated after glutamate removal and did not recover to basal levels.
In parallel experiments, we confirmed the regulation of this age-dependent PCE by postinsult NMDAR activity, exposing another group of cells to the competitive NMDAR antagonist, d-APV (100 μM) immediately after the termination of the 5-min glutamate insult (Fig. 1, A and B). Other cells were exposed postinsult to the AMPA receptor antagonist CNQX (10 μM). Neither antagonist had a significant effect on younger cultures, in which the PCE was small. However, in older cells, the postinsult application of d-APV significantly facilitated the recovery of [Ca2+]i. In contrast, CNQX had no effect on the PCE in older cells.
Because there appear to be some differences in neuroprotective actions of competitive and noncompetitive NMDAR antagonists (Levy and Lipton 1990), we also tested the effects of the noncompetitive NMDAR antagonist MK-801 in a similar experiment. When given immediately in the postinsult period, MK-801 also reversed the PCE in older cells (Fig. 1, C and D). These results indicate that the age-dependent PCE is maintained by postinsult activity of NMDARs.
Membrane depolarization was age related and NMDAR dependent
To determine whether postinsult membrane depolarization and spontaneous action potential firing also exhibit age-and/or NMDAR dependency, younger (11–17 DIV) and older (21–24 DIV) neurons were examined under current-clamp conditions with whole cell patch pipettes before, during, and after a 5-min glutamate application (20 μM) (Fig. 2). To reduce synaptic connectivity differences between age-in-culture groups during the electrophysiological studies, the younger neurons in this experiment and all subsequent experiments were slightly older (11–17 DIV), and the older neurons were somewhat younger (21–24 DIV) than those used in the preceding experiment (see Fig. 1). Cells <11 DIV exhibit sparse synaptic connectivity (Porter et al. 1997) and little spontaneous action potential activity (data not shown), whereas 11–17 DIV neurons exhibit more extensive synaptic connectivity and increasing spontaneous network activity (see Figs. 2 and 3). Moreover, the degree of Ca2+ elevation in younger (11–17 DIV) and older (≥21 DIV) neurons during a 5-min application of 20 μM glutamate is more nearly comparable (see Fig. 6), although the PCE and probability of cell death are still substantially greater in the older neurons (Attucci et al. 2002).
FIG. 2.
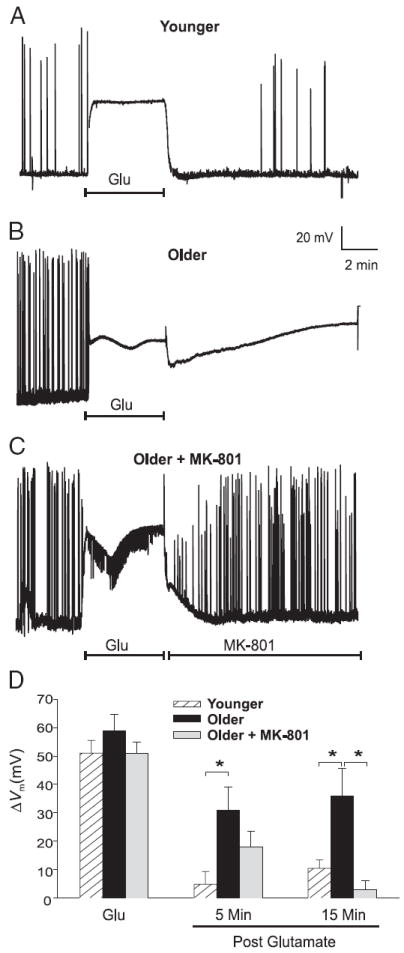
Glutamate exposure induces an age-dependent depolarization that requires postinsult NMDAR activity. A–C: representative experiments illustrating the effects of a 5-min glutamate exposure (Glu, 20 μM) on action potentials and Vm in Younger cultures (n = 17) (A), Older cultures (n = 8) (B), and Older cultures perfused postglutamate with 10 μM MK-801 (n = 8) (C). D: extent of depolarization (means ± SE) measured in Younger (hatched columns), Older (filled columns), and Older MK-801–treated neurons (gray columns) during (Glu) and at 5 and 15 min postglutamate exposure. *P < 0.05. Note that the Younger neurons and Older MK-801–treated neurons were generally able to repolarize and resume action potential activity.
FIG. 3.
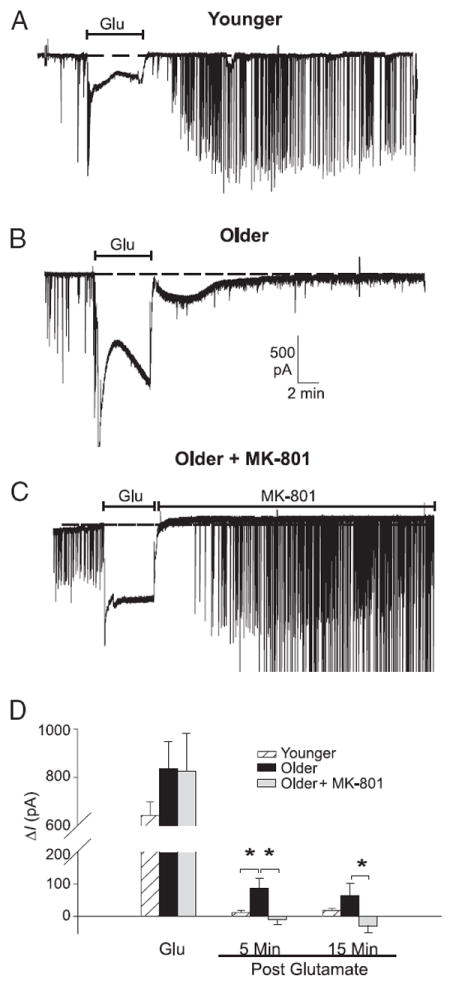
Glutamate exposure induces persistent inward current that is dependent on age and NMDA receptor. A–C: representative examples of spontaneous excitatory postsynaptic current (EPSC) activity and inward current in neurons voltage-clamped at −80 mV, recorded before, during, and after a 5-min application of 20 μM glutamate (Glu). Dashed line shows baseline holding current (Ih). In Younger neurons (n = 25) (A), glutamate application resulted in a large increase in inward current followed by rapid restoration of baseline Ih after glutamate termination. B: Older neurons (n = 14) exhibited a somewhat larger (but variable and nonsignificant) increase in inward current during glutamate exposure. After glutamate exposure, holding current remained elevated above preinsult levels for >5 min before eventually recovering. Postinsult period in Older neurons was also characterized by an increase in the appearance of smaller EPSCs and an almost complete loss of larger (>300 pA) EPSCs. C: postinsult increase in inward holding current and changes in EPSC activity were completely reversed in Older neurons by applying MK-801 (10 μM) immediately after the glutamate insult (n = 9). D: mean ± SE change in holding current (ΔI) during glutamate exposure and at 5 and 15 min after glutamate application (relative to preinsult levels) in Younger (white columns), Older (filled columns), and Older MK-801–treated (gray columns) neurons. *P < 0.05.
FIG. 6.
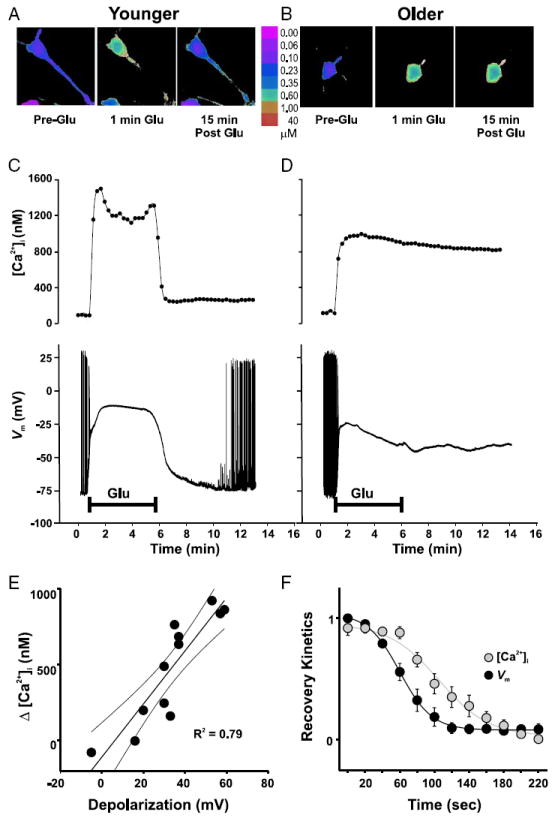
PCE and extended neuronal depolarization occur within the same neurons and exhibit strong quantitative and temporal relationships. Simultaneous [Ca2+]i imaging (with Fura-2) and electrophysiological recording in an individual younger (n = 5, A and C) and an older (n = 7, B and D) neuron before, during, and after 5-min exposure to 20 μM glutamate (Glu). Note that, for illustration purposes, recording electrodes have been subtracted from images. A and B: representative pseudocolor Fura-2 ratiometric images taken immediately before (Pre-Glu), during the first minute of glutamate exposure (1 min Glu), and 15 min after glutamate application (15 min Post-Glu). C and D: time-course plots of [Ca2+]i measurements (top) and continuous Vm measurements in current clamp (bottom) within the same neurons. Similar to the results in Figs. 1 and 2, the younger neuron was able to recover its [Ca2+]i and Vm to near basal levels, whereas the older neuron was not. E: correlation analysis across all younger and older cells between the postinsult change in [Ca2+]i (ΔCa2+i) and the simultaneously recorded postinsult depolarization, indicated a close quantitative relationship between Vm and [Ca2+]i in the postinsult period (measured at 15 min Post-Glu). F: recovery kinetics for [Ca2+]i and Vm immediately after washout of glutamate. Only data from cells in which either the Vm or the [Ca2+]i returned to near preglutamate levels were included (all younger neurons and one older neuron). In all cases, Ca2+ recovery followed the repolarization of the Vm.
Before glutamate treatment, resting Vm was slightly but significantly more depolarized in younger versus older neurons (−55.23 ± 2.08 mV, n = 17, vs. −62.19 ± 2.44 mV, n = 16; P < 0.05). In both age groups, neuronal Vm was permitted to fluctuate freely and no holding current was applied during the course of the experiment. Representative examples of these experiments are illustrated in Fig. 2, A–C and group mean data are shown in Fig. 2D. Bath perfusion of glutamate induced substantial membrane depolarization (denoted as ΔVm, or difference from resting Vm), which was maintained for the 5-min duration of glutamate exposure and did not differ between age groups (younger, 51 ± 4.5 mV, n = 17; older, 55 ± 3.5 mV, n = 16; P < 0.55) (Fig. 2, A, B, and D, Glu).
On depolarization, all spontaneous action potential (AP) activity, which tended to be greater in older cultures, was blocked (Fig. 2, A and B). Similar to the pattern of [Ca2+]i changes that occurred after glutamate exposure (Fig. 1, A and B), younger neurons repolarized quickly after washout of glutamate (Fig. 2, A and D, 5 min post). In older neurons, repolarization generally failed, resulting in persistent depolarization (Fig. 2B). Repolarization in the younger group, which generally occurred within 5 min of glutamate removal (Fig. 2D, 5 min, P < 0.005), was typically accompanied by reemergence of spontaneous APs and EPSPs at preinsult or greater frequency (Fig. 2A). AP activity generally did not resume in depolarized older neurons (Fig. 2B).
To test whether the age-related depolarization after glutamate exposure was dependent on postinsult NMDAR activity, a subset of older neurons (n = 8) was subjected to the same 5-min, 20-μM glutamate insult, followed immediately by perfusion with normal recording medium containing 10 μM MK-801. Similar to its effects on the PCE (Fig. 1D), postinsult application of MK-801 reduced or prevented the development of sustained depolarization (Fig. 2, C and D, P < 0.005, 15 min post). MK-801–treated older neurons behaved similarly to younger neurons and were able to restore membrane potential and spontaneous APs within minutes of the removal of glutamate, indicating that this depolarization, like the PCE, was both age- and NMDAR activity dependent.
Persistent postinsult current was age- and NMDAR dependent
In a series of voltage-clamp experiments, paralleling current-clamp studies in Fig. 2, younger (about 15 DIV, n = 25) and older (about 22 DIV, n = 23) neurons were voltage clamped at −80 mV before, during, and after 5 min glutamate exposure (20 μM). A subset (n = 9) of older cells was also treated in the postinsult period with MK-801 (see following text). Representative examples of these experiments are illustrated in Fig. 3.
No age-related differences were found in holding current (I) during the preglutamate period (younger, −86.5 ± 69.6 pA; older, −123.11 ± 91.9 pA). Bath perfusion of glutamate (5 min) induced a substantial increase in current (ΔI) for both age groups (P < 0.0001), which tended nonsignificantly to be greater in the older group (−939 ± 110 pA) relative to the younger group (−728 ± 61 pA, P > 0.05) (Fig. 3, A, B, and D, Glu). During the postinsult period, ΔI was significantly larger in older neurons (P < 0.01) (Fig. 3, B and D, 5 min post). However, under these hyperpolarized conditions, the postinsult current in older neurons declined considerably during the next 15 min.
MK-801 treatment blocked or reversed the postinsult current in older neurons (5 min, P < 0.01; 15 min, P < 0.05), indicating dependency on persistent NMDAR activity. Because NMDAR current is appreciably attenuated at negative holding potentials (−80 mV here), it seems highly likely that the postinsult NMDAR-mediated current would have been substantially larger and lasted longer if the membrane had been clamped to a more positive potential. Nonetheless, even at these hyperpolarized levels, there was substantially greater NMDAR-dependent current in older neurons during the post-insult period.
Changes in network activity after glutamate exposure
The voltage-clamp mode provides an advantageous configuration for monitoring synaptic activity and transmitter release of neighboring connected cells and for gauging network-wide behavior during and after glutamate exposure. That is, neurons clamped at a constant hyperpolarized potential are relatively protected against the subsequent postinsult [Ca2+]i elevation experienced by neighboring unclamped cells (see following text, Fig. 4). Moreover, because the clamped neuronal membrane does not fluctuate, driving force on excitatory postsynaptic currents (EPSCs) is constant and the clamped neuron can serve as a sensitive monitor of alterations in presynaptic glutamate release by assessing spontaneous network EPSCs before, during, or after toxic glutamate exposure.
FIG. 4.
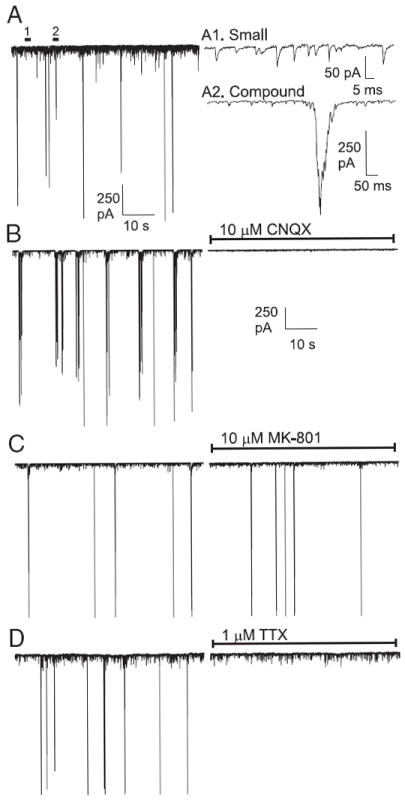
Pharmacological characterization of spontaneous EPSCs in hippocampal neuronal cultures. A: typical record of spontaneous, ongoing synaptic activity (EPSCs) in a voltage-clamped cultured hippocampal neuron (left). Activity recorded at time points 1 and 2 are magnified (right) to illustrate small (A1) and compound (A2) EPSCs. B: nearly all synaptic activity was abolished by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist CNQX (10 μM, n = 12). C: NMDAR antagonist MK-801 (10 μM, n = 10) had little effect on EPSC frequency. D: voltage-sensitive Na+ channel blocker tetrodotoxin (TTX, 1 μM, n = 5) did not affect spontaneous small EPSCs, but eliminated compound EPSCs, suggesting that these large events were the result of presynaptic action potentials.
Pharmacological approaches were used to define and identify the sources of EPSCs observed in the postsynaptic, voltage-clamped neuron (Fig. 4). Spontaneous EPSCs could be divided roughly into two main categories (Fig. 4A), based on their amplitudes: Smaller (<40 pA) EPSCs, most of which are likely unitary or mini-EPSCs, and larger compound inward currents (>300 pA) that often lasted for >100 ms (“compound EPSCs”) (Fig. 4A, right). Both small and compound EPSCs were blocked by CNQX, a specific AMPA receptor (AMPAR) blocker, showing that they were generated by presynaptic glutamate release and mediated by AMPARs (Fig. 4B). NMDARs appeared to make little contribution to spontaneous EPSC activity under these hyperpolarized clamped conditions because MK-801 application did not alter the total number of small or large EPSCs (Fig. 4C). The Na+ channel blocker TTX blocked only the larger burstlike compound EPSCs, whereas small unitary EPSCs were scarcely affected by blockade of Na+ channels (Fig. 4D). Large compound currents were also not inhibited by intracellular dialysis with the Na+ channel blocker QX-314 (1 mM) (data not shown). Thus the large compound EPSCs result from synchronous release of transmitter quanta evoked by (unseen) presynaptic action potentials.
To quantify the age-related changes in synaptic activity after glutamate (Fig. 3), histograms of EPSC amplitudes were constructed before and after exposure to glutamate (Fig. 5). The numbers of small and compound EPSCs were greater in older than younger neurons in the preglutamate period (number of small EPSCs, P < 0.001; number of compound EPSCs, P < 0.05). In the postglutamate period, the clearest change for younger neurons was a general increase in the number of EPSCs in the 15- to 60-pA range (Fig. 5A). Exponential regression analysis revealed a postinsult increase in the histogram τ (from pre-Glu, 10.03 pA to post-Glu, 14.45 pA; P < 0.05), reflecting a shift to slightly larger EPSCs and a nonsignificant increase in the peak of the distribution (pre-Glu, 126 observations vs. post-Glu, 190 observations; P > 0.05). The number of compound EPSCs (i.e., ≥300 pA), which reflected impinging action potentials, also tended to increase in the postglutamate period, although this effect was not significant (Fig. 5A, inset). Nevertheless, for many younger neurons (e.g., the one illustrated in Fig. 3A), the number of compound EPSCs increased dramatically after glutamate exposure.
FIG. 5.
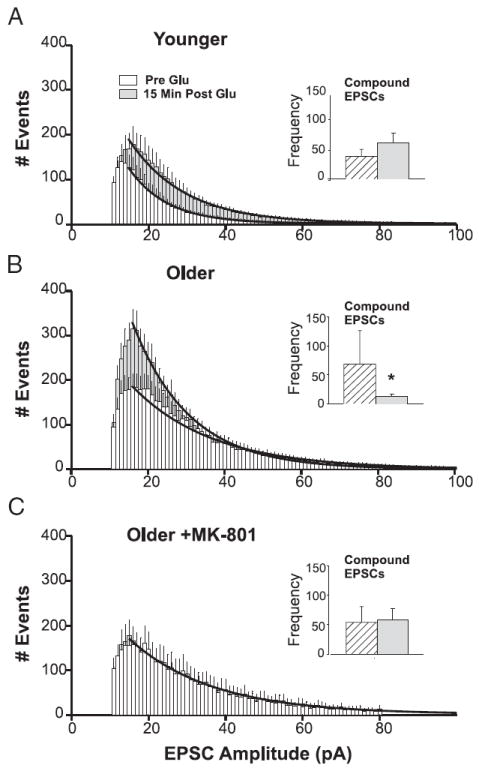
Glutamate exposure alters the distribution of EPSC amplitudes in an age- and NMDAR-dependent manner. EPSC activity exhibited a bimodal distribution. A–C: first mode consisted of TTX-insensitive, small EPSCs (<100 pA). Number of small EPSCs observed during two 2-min windows, one before and one 15 min after, glutamate perfusion were binned as a function of amplitude (in pA) and averaged across cells. Changes in distribution were assessed by exponential regression (bold lines). Insets: second mode consisted of TTX-sensitive (see Fig. 4) compound EPSCs (>300 pA). Mean number of compound EPSCs before (hatched columns) and after (gray columns) glutamate exposure are illustrated in the inset of each panel. A: in younger neurons (n = 25), the number and amplitude of small EPSCs increased after glutamate application, and there was a nonsignificant trend for compound EPSCs to increase (inset). B: in older neurons (n = 14), the number of events in the lowest-amplitude bins (<40 pA) increased substantially at 15 min after glutamate exposure, whereas compound EPSCs were nearly eliminated (inset). C: postinsult application of MK-801 to older neurons (n = 9), produced a remarkable stabilizing effect. Both the amplitude/frequency histogram and the number of compound EPSCs (inset) were unchanged after glutamate exposure (see also Figs. 2C and 3C). *Difference from preglutamate baseline, P < 0.05. See text for analyses of number of events.
In contrast, the postinsult period for older neurons (Fig. 5B) was characterized by a dramatic and selective increase in the number of smaller EPSCs, primarily in the range of 10–25 pA, with little change in the number of EPSCs >30 pA. Regression analysis revealed a postglutamate increase in distribution peak height (pre-Glu, 187 observations vs. post-Glu, 331 observations; P < 0.05) along with a reduction in τ (from pre-Glu, 21.05 pA to post-Glu, 14.86 pA; P < 0.05). Moreover, compound EPSCs, which were prominent in older neurons during the preglutamate period (see Fig. 3, B and C), essentially disappeared (P < 0.01) in the postglutamate period (Figs. 3B and 5B, inset), reflecting the loss of action potential activity in depolarized, unclamped neighboring neurons (Fig. 2B) and the resulting lack of coordinated, synchronous glutamate release from multiple terminals in axonal ramifications within the neuronal network. These changes in the EPSC amplitude distributions for both age groups were maintained for the duration of recording (i.e., ≤30–45 min after glutamate wash-out). Together these alterations indicate that postglutamate EPSCs and action potentials (and synchronous multiple EPSC bursts) are unchanged or increased somewhat in younger neurons, whereas in older neurons, there is a complete loss of action potential–related compound EPSC events, along with a substantial increase in small EPSCs.
In older neurons treated with MK-801 postglutamate, however, synaptic activity was qualitatively and quantitatively similar across the pre- (peak of 170 observations, τ = 21.55) and post- (peak of 168 observations, τ = 21.46) glutamate periods (Fig. 5C), much as in younger neurons. Also, compound EPSCs remained constant in the postinsult period for older cells treated with MK-801 (Fig. 5C, inset). Thus changes in network activity in the postinsult period were age related and critically dependent on the ongoing activity of NMDARs, similar to the changes observed in intracellular Ca2+, membrane depolarization, and inward current.
Repolarization of neuronal Vm in the postinsult period reverses the PCE
To test whether correlative and/or causative relationships exist between the PCE and depolarization, we assessed [Ca2+]i and Vm simultaneously in a subset of neurons before, during, and after a glutamate insult. In these neurons, glutamate induced an elevation in Ca2+ and a depolarization of the Vm that usually recovered soon after removal of glutamate in younger (n = 5) (Fig. 6, A and C) but not older (n = 7) (Fig. 6, B and D) neurons. Across all cells (younger and older), the magnitude of the depolarization measured 15 min after glutamate washout exhibited a strong positive correlation with the magnitude of the PCE at 15 min (P < 0.01) (Fig. 6E). Moreover, for six neurons (all five younger neurons and one older neuron) in which the [Ca2+]i and the Vm spontaneously recovered to near baseline values, the repolarization of Vm preceded the recovery of [Ca2+]i (Fig. 6F). ΔVm declined to half of its maximal value in 78.6 ± 6.67 s, whereas Δ[Ca2+]i fell to its half-maximal value in 123.8 ± 6.92 s (z = 4.71, P < 0.05).
The more rapid recovery of Vm compared with [Ca2+]i after glutamate withdrawal suggests that restoration of Vm may be a requisite for reestablishing [Ca2+]i homeostasis during the postinsult period. Thus to test the hypothesis that the PCE depends on Vm, the Vm of another subset of older neurons (n = 7) in the experiment above was directly controlled in a hybrid current-clamp/voltage-clamp experiment in which Vm was monitored in current clamp before and during glutamate exposure, but was then ramped to −80 mV by switching to voltage clamp immediately after glutamate washout (Fig. 7B). On imposition of voltage clamp, I showed a steep reduction within 2 min, which was generally followed by the restoration of [Ca2+]i to near preglutamate levels. There were no differences in [Ca2+]i between younger and voltage-clamped older neurons in the postinsult period, and at 15 min after glutamate removal, [Ca2+]i in voltage-clamped older neurons was significantly lower than that in older neurons recorded in current clamp throughout (Fig. 7C). Although some space-clamp problems may occur in these older neurons, voltage clamp of slow potentials even in ramified neurons is generally effective after a delay. Therefore in the present study, the hybrid clamp likely established relatively good control of membrane voltage, at least in the soma (Brown and Johnston 1983).
FIG. 7.
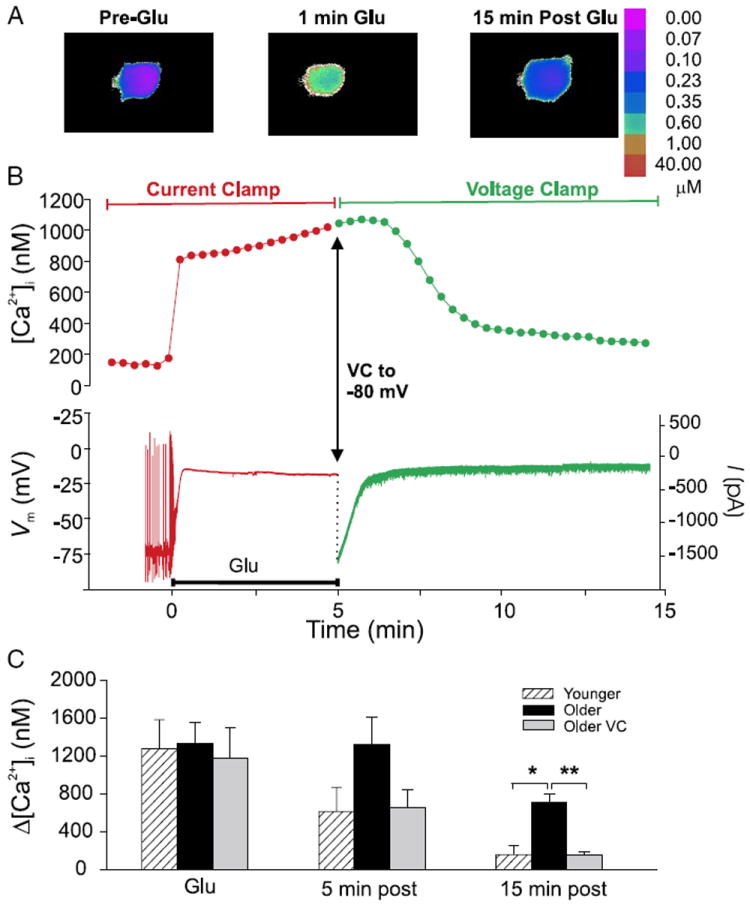
Active repolarization of the membrane immediately after glutamate exposure restores [Ca2+]i to preinsult levels. A and B: [Ca2+]i imaging (with Fura-2) and electrophysiological recording in an individual older neuron studied by hybrid current clamp/voltage clamp. Note that, for illustration purposes, the recording electrode has been subtracted from images. Vm was recorded in current-clamp mode before and during glutamate application (20 μM, 5 min), then voltage-clamped to −80 mV immediately after glutamate application. A: representative pseudocolor Fura-2 ratiometric images taken immediately before (Pre-Glu), during the first minute of glutamate exposure (1 min Glu), and 15 min after glutamate application (15 min Post-Glu). B: time-course plots of [Ca2+]i measurements (top) and continuous Vm and current measurements (bottom) in the neuron illustrated in A. Plot symbols and traces in B that correspond to current-clamp conditions are in red, whereas those after the switch to voltage clamp are shown in green. Point at the end of the 5-min glutamate exposure (Glu) at which the Vm was ramped to −80 mV is shown by the arrow. D: mean ± SE change in [Ca2+]i (Δ[Ca2+]i) at 5- and 15-min postglutamate application in younger (n = 5, hatched columns) and older (n = 7, filled columns) neurons held under current clamp only, and older neurons switched from current clamp (CC) to voltage clamp (VC) in the postglutamate period (n = 7, gray columns). Similarly to younger neurons, older neurons hyperpolarized in the postinsult period were able to restore [Ca2+]i to near basal levels. Older neurons held under current clamp throughout the experiment were not. *P ≤ 0.05; **P ≤ 0.01.
Thus [Ca2+]i in younger neurons and older voltage-clamped neurons was indistinguishable during the postinsult period. These effects of clamping the membrane at −80 mV after the insult on [Ca2+]i were similar to those of NMDAR blockade in reversing the PCE in older neurons (Fig. 1D), although the PCE reversal occurred more rapidly with voltage clamp (Fig. 7). However, the time-course differences were likely the result of several methodological differences. The cells shown in Fig. 1 were considerably older than those in Fig. 7, making it more difficult to reverse the PCE. Moreover, the cells in Fig. 7 were studied under whole cell patch recording conditions and were also loaded with a lower concentration of Ca2+ buffering indicator, factors that may have accelerated cytosolic decay kinetics.
DISCUSSION
The results here show that multiple electrophysiological and [Ca2+]i responses persist during the postglutamate exposure period in older, vulnerable neurons. These persisting responses include [Ca2+]i elevation, inward current, membrane depolarization, block of action potentials, and increased frequency of miniature glutamate EPSCs (presumably driving NMDAR activity in depolarized neurons). Importantly, these processes appear to be interrelated because each could be attenuated by MK-801 blockade of NMDAR current in the postinsult period. Conversely, repolarization of the membrane by voltage clamp was followed by termination of inward current and a return to near baseline of the elevated [Ca2+]i (Fig. 7). These reciprocal dependencies suggest the operation of a positive feedback loop, as discussed further below.
Conflicting results on the NMDAR dependency of postinsult responses
As noted, prior studies of the postinsult phase also saw extended depolarization (Coulter et al. 1992; Limbrick et al. 2001) and persistent inward current (IPE; Chen et al. 1998). However, once induced, those responses were resistant to NMDAR antagonists. Conflicting results on NMDAR dependency could arise from several factors, including, of course, activation of different membrane currents in those studies than in the present study. Alternatively, the results could reflect differences in insult intensity. Substantially more intense excitotoxic insults were used in most prior studies [e.g., 500 μM glutamate for 10 min by Limbrick et al. (2001)] than here. Conceivably, more intense insults could trigger irreversible sequelae or processes that are independent of NMDARs, such as mitochondrial depolarization and disruption of electron transport (Vergun et al. 1999). In addition, irreversible NMDAR-independent changes might accumulate with time after the insult, although the present study examined the effects of NMDAR antagonists delivered only immediately on glutamate washout. Nonetheless, although additional dosage and time-course studies will be needed to resolve these discrepancies, the present data clearly indicate that major postinsult electrophysiological and [Ca2+]i responses are, at least initially, NMDAR dependent.
Altered network activity and sources of postinsult glutamate
An important and long-standing question regarding delayed excitotoxicity has been how NMDAR activity is maintained in the postinsult, glutamate washout period. One prominent hypothesis addressing this question proposes that high glutamate insult induces persistent neuronal hyperexcitability, resulting in amplified network firing frequencies and increased release/ accumulation of endogenous glutamate (Choi 1992; Olney 1986). However, our findings from voltage- and current-clamped neurons showed that instead, insulted older neurons were depolarized and quiescent, exhibiting marked reductions in action potential activity and related synchronous compound EPSCs (Figs. 2B, 3B, and 5B, inset). Nonetheless, despite the blockade of synchronized compound release, overall network glutamate release may well have been maintained or even increased because, concomitantly, there was a substantial increase in the frequency of smaller (<40 pA), TTX-insensitive AMPAR-mediated mini-EPSCs (Fig. 5). Interestingly, this conclusion is consistent with studies using direct measurement of glutamate that found greater release in older neurons (Fogal et al. 2005).
However, it should be emphasized that the critical issue is not whether total release is elevated after insult, but whether NMDAR activation is increased. NMDAR-mediated EPSCs are not readily observed in neurons voltage clamped at these hyperpolarized levels (Fig. 4), and therefore the voltage-clamped neuron serves as a monitor for glutamate release but not for NMDAR activation. In the absence of voltage clamp, however, the Mg2+ block would be relieved by the extended depolarization in depolarized older neurons after insult (Fig. 2), allowing the glutamate release reflected in the mini-EPSCs to strongly activate NMDARs. Therefore regardless of whether total release is increased, its impact on NMDAR activation should be substantially greater postinsult, during extended depolarization, than before insult, when depolarization occurs primarily during brief 1- to 3-ms action potentials. Moreover, spontaneous release from numerous depolarized cells in a network presumably activates NMDARs over a larger extent of the dendritic arborization and soma than does the compound release from the ramifications of a single axon.
One caveat to the above interpretation is that NMDARs are modulated by both desensitization and Ca2+-dependent inactivation (Erreger and Traynelis 2005; Krupp et al. 1999; Vicini et al. 1998). These inhibitory processes could reduce NMDAR sensitivity in depolarized neurons. However, the effectiveness seen here of an NMDAR antagonist in reversing postinsult changes in depolarized neurons (Figs. 1, 2, 3, and 5) indicates that NMDARs retain activity under these conditions.
In the absence of action potentials, it seems likely that the high rate of EPSCs in the older neurons resulted from sustained depolarization of the neuronal network and asynchronous glutamate release from presynaptic terminals. As noted, slow shifts in membrane potential, such as the prolonged depolarization seen here, can spread for considerable electrotonic distances and should reach axon terminals from initial sources in dendrites and soma (Alle et al. 2006; Johnston and Brown 1983; Shu et al. 2006).
An alternative explanation for increased EPSCs is that glutamate treatment might have altered dendritic electrotonic structure. If dendritic electrotonic distance were reduced in the older group, small EPSCs might emerge from the noise more often after glutamate insult (Brown and Johnston 1983; Johnston and Brown 1983). In the present studies, an increase in smaller-amplitude EPSCs after glutamate was observed. Nevertheless, the effects of glutamate insult did not shift the overall amplitude distribution (Fig. 5B), suggesting that altered electrotonic structure was not a major factor in the results.
In addition, a possible alternative to EPSCs as a primary source of depolarizing postinsult glutamate might be reverse glutamate transport from neurons or glia, a mechanism previously implicated in some forms of excitotoxicity (Billups and Atwell 1996). Reverse transport can occur by several distinct high-affinity transporters when extracellular K+ levels are exceedingly high (e.g., after a glutamate insult), resulting in higher levels of extracellular glutamate and greater toxicity (Billups and Attwell 1996; Szatkowski and Attwell 1994). In this view, transporters in neurons or glia that take up glutamate during the insult could reverse their glutamate transport during the postinsult period, providing a source of glutamate release for postinsult NMDAR activation. However, it is unlikely that reverse transport can account for the increased EPSC activity because the size, shape, and duration of mini-EPSCs after glutamate exposure appear to reflect vesicular or quantal release from axon terminals (or surrounding astrocytes; e.g., Fiacco and McCarthy 2004) rather than extrusion by a membrane pump. In addition, glutamate release was recently found elevated in older cortical cultures compared with younger cultures, even though a major source of reverse glutamate transport was blocked by dl-threo-β-benzyloxyaspartate (TBOA). The enhanced release was nearly restored to basal levels by application of MK-801 (Fogal et al. 2005). It is also important to note that several postinsult changes, highly similar to those described in the present study, including the PCE (Hartley et al. 1993), extended depolarization (Sombati et al. 1991), and increased inward current (Chen et al. 1997, 1998), can also be induced by application of NMDA that, unlike glutamate, is nontransportable. Consequently, although reverse transport of glutamate may contribute, it seems likely that the major source of postinsult glutamate driving the NMDAR-dependent changes is persistent release from depolarized pre-synaptic terminals.
Ca2+ sources of the persistent Ca2+ elevation
As noted, all PCEs may not reflect the same process. The sources of Ca2+ in excitotoxicity apparently vary under different conditions and have been suggested to include, in addition to NMDARs, release from, or impaired uptake into, the mitochondria (Nicholls and Budd 2000; Vergun et al. 1999), influx through voltage-gated calcium channels (VGCCs) (Blalock et al. 1999; Porter et al. 1997; Sucher et al. 1991), or nonspecific cation channels (Chen et al. 1997), group 1 mGluR activation of intracellular Ca2+ release from IP3-sensitive stores (Attucci et al. 2002; Conn and Pin 1997; Moroni et al. 1998), or impaired Ca2+ extrusion (Crespo et al. 1990; Khodorov et al. 1993; Limbrick et al. 2001; Sanchez-Armass and Blaustein 1987). Moreover, increasing evidence indicates that the PCE or excitotoxicity in older neurons can be blocked by inhibiting Ca2+-induced Ca2+ release (CICR) from ryanodine receptors (Clodfelter et al. 2002; Frandson and Schousboe 1991; Lei et al. 1992), which, together with similar effects of NMDAR and VGCC antagonists, suggests that the PCE seen here may be generated by the interplay of Ca2+ influx through NMDARs/VGCCs and consequent CICR.
Possible relevance to brain aging and Alzheimer’s disease (AD)
There is mounting evidence that Ca2+ dysregulation also plays a role in brain aging and neurodegenerative conditions such as Alzheimer’s disease (Disterhoft et al. 2004; LaFerla 2002; Mattson et al. 2000; Thibault et al. 1998; Toescu et al. 2004). Although aging in cell culture models is clearly very different from aging in animal models, some intriguing similarities in Ca2+ regulation have been found. These include aging-related increases in neuronal L-type VGCC activity (Porter et al. 1997; Thibault and Landfield 1996), alterations in the clearance of Ca2+ (Adamec et al. 1998; Cheng et al. 1999; Korkotian and Segal 1996; Toescu and Verkhratsky 2000), changes in CICR (Clodfelter et al. 2002; Gant et al. 2006; Lei et al. 1992), and increased vulnerability to degeneration (Attucci et al. 2002; Choi et al. 1987; McDonald et al. 1997). Given these parallels, cells aging in mixed neuronal cultures may be a useful tool for studying alterations in Ca2+ homeostasis relevant to aging and AD.
One major difference in Ca2+ regulation between aging-inculture and aging in animals, however, is that NMDAR activity appears to decrease with aging (Barnes et al. 1997; Clayton et al. 2002; Magnusson 1998). Nevertheless, increased Ca2+ levels arising from other sources that do change with aging (e.g., L-type Ca2+ channels and ryanodine sensitive Ca2+ stores; Gant et al. 2006; Thibault and Landfield 1996; Thibault et al. 2001) could amplify the impact of episodes of excessive NMDAR activity, providing a mechanism for excitotoxic contributions to age-related neurodegenerative conditions and for positive results with the low-affinity, noncompetitive NMDAR antagonist memantine, in treating Alzheimer’s disease (Ferris 2003; Le and Lipton 2001).
Positive feedback model of NMDAR current maintenance
The well-established dependency of NMDAR current on both depolarization and the presence of glutamate (Mayer and Westbrook 1987) indicates that postinsult NMDAR current (Fig. 3) likely is maintained by the combination of prolonged depolarization (Fig. 2) and persisting glutamate release (asynchronous mini-EPSC activity) (Figs. 4 and 5). Conversely, the prolonged depolarization and asynchronous presynaptic release appear to depend on NMDAR activity (Fig. 2C). The NMDAR current may modulate the membrane potential directly, or indirectly, by activation of a secondary conductance (e.g., Chen et al. 1997, 1998). Furthermore, the PCE depends both on NMDAR activity (Fig. 1) and on membrane depolarization (Fig. 7). Taken together, these data are consistent with a model of delayed excitotoxicity in which large NMDAR current (and depolarization) is initially triggered by excessive glutamate release, as might occur in a traumatic head injury or ischemic incident. On clearance (or washout) of excessive glutamate, outward currents are not large enough to repolarize the membrane against the persisting inward currents that are sustained by glutamate release from depolarized terminals. NMDAR current is likely a major component of the inward current, particularly given the slow kinetics and long decay constants of NMDARs (Clements et al. 1992). These reciprocal interactions lead to establishment of a positive feedback loop between the depolarizing NMDAR current, on one hand, and the enabling depolarization and resulting glutamate release, on the other. In turn, the sustained NMDAR activity appears to generate persistent [Ca2+]i elevation through a combination of direct Ca2+ influx by NMDARs (and possibly, VGCCs) and CICR, resulting in excitotoxic neuronal death.
Interruption of this putative loop requires activation of outward currents sufficient to repolarize the membrane on termination of excessive glutamate exposure. The high density of K+ channels throughout the soma and, particularly, the dendrites of pyramidal and other neurons (Johnston et al. 2000) apparently can repolarize the membrane in younger but not older neurons, perhaps because of age-dependent increases in NMDAR current (Brewer et al. 2001; Cheng et al. 1999; Priestley et al. 1996; Xia et al. 1995). However, other mechanisms also might account for the inability of older neurons to repolarize, including changes in the relative number of inhibitory synapses, neuronal or glial energy metabolism, or levels of reactive oxygen species, and will require further investigation.
In summary, the present work provides the first comprehensive electrophysiological and Ca2+-imaging overview of the critical postinsult phase of excitotoxicity and suggests a model in which positive feedback interactions between NMDAR activation, on one hand, and membrane depolarization/glutamate release, on the other, maintain NMDAR current after the initial high glutamate exposure is terminated. The sustained NMDAR current, in turn, drives [Ca2+]i elevation, resulting in toxicity. This positive feedback model of excitotoxicity, if supported in future studies, might well have important therapeutic implications. A positive feedback loop can be disrupted by damping any of its elements and thus elucidating reciprocal feedback mechanisms in delayed excitotoxicity may reveal new targets for intervention in a wide range of neuropathologies.
Acknowledgments
GRANTS This work was supported by National Institute on Aging Grants AG-04542, AG-10836, and AG-020251.
References
- Adamec E, Didier M, Nixon RA. Developmental regulation of the recovery process following glutamate-induced calcium rise in rodent primary neuronal cultures. Brain Res Dev Brain Res. 1998;108:101–110. doi: 10.1016/s0165-3806(98)00034-0. [DOI] [PubMed] [Google Scholar]
- Alle H, Geiger JR. Combined analog and action potential coding in hippocampal mossy fibers. Science. 2006;311:1290–1293. doi: 10.1126/science.1119055. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Attucci S, Clodfelter GV, Thibault O, Staton J, Moroni F, Landfield PW, Porter NM. Group I metabotropic glutamate receptor inhibition selectively blocks a prolonged Ca2+ elevation associated with age-dependent excitotoxicity. Neuroscience. 2002;112:183–194. doi: 10.1016/s0306-4522(02)00002-7. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–342. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Rao G, Shen J. Age-related decrease in the N-methyl-d-aspartate receptor-mediated excitatory postsynaptic potential in hippocampal region CA1. Neurobiol Aging. 1997;18:445–452. doi: 10.1016/s0197-4580(97)00044-4. [DOI] [PubMed] [Google Scholar]
- Billups B, Attwell D. Modulation of non-vesicular glutamate release by pH. Nature. 1996;379:171–174. doi: 10.1038/379171a0. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Porter NM, Landfield PW. Decreased G-protein-mediated regulation and shift in calcium channel types with age in hippocampal cultures. J Neurosci. 1999;19:8674–8684. doi: 10.1523/JNEUROSCI.19-19-08674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21:98–108. doi: 10.1523/JNEUROSCI.21-01-00098.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TH, Johnston D. Voltage-clamp analysis of mossy fiber synaptic input to hippocampal neurons. J Neurophysiol. 1983;50:487–507. doi: 10.1152/jn.1983.50.2.487. [DOI] [PubMed] [Google Scholar]
- Chen QX, Perkins KL, Choi DW, Wong RK. Secondary activation of a cation conductance is responsible for NMDA toxicity in acutely isolated hippocampal neurons. J Neurosci. 1997;17:4032–4036. doi: 10.1523/JNEUROSCI.17-11-04032.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QX, Perkins KL, Wong RK. Zn2+ blocks the NMDA- and Ca2+-triggered postexposure current IPE in hippocampal pyramidal cells. J Neurophysiol. 1998;79:1124–1126. doi: 10.1152/jn.1998.79.2.1124. [DOI] [PubMed] [Google Scholar]
- Cheng C, Fass DM, Reynolds IJ. Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca(2+)](i) increases and NMDA receptor mRNA levels. Brain Res. 1999;849:97–108. doi: 10.1016/s0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA, Grosshans DR, Browning MD. Aging and surface expression of hippocampal NMDA receptors. J Biol Chem. 2002;277:14367–14369. doi: 10.1074/jbc.C200074200. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Clodfelter GV, Porter NM, Landfield PW, Thibault O. Sustained Ca2+-induced Ca2+-release underlies the post-glutamate lethal Ca2+ plateau in older cultured hippocampal neurons. Eur J Pharmacol. 2002;447:189–200. doi: 10.1016/s0014-2999(02)01843-5. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corey DP, Stevens CF. Science and technology of patch recording electrodes. In: Sakmann B, Neher E, editors. Single Channel Recording. New York: Plenum; 1983. pp. 53–58. [Google Scholar]
- Coulter DA, Sombati S, DeLorenzo RJ. Electrophysiology of glutamate neurotoxicity in vitro: induction of a calcium-dependent extended neuronal depolarization. J Neurophysiol. 1992;68:362–373. doi: 10.1152/jn.1992.68.2.362. [DOI] [PubMed] [Google Scholar]
- Crespo LM, Grantham CJ, Cannell MB. Kinetics, stoichiometry and role of the Na–Ca exchange mechanism in isolated cardiac myocytes. Nature. 1990;345:618–621. doi: 10.1038/345618a0. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Wu WW, Ohno M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer’s disease. Ageing Res Rev. 2004;3:383–406. doi: 10.1016/j.arr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci. 1993;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Traynelis SF. Allosteric interaction between zinc and glutamate binding domains on NR2A causes desensitization of NMDA receptors. J Physiol. 2005;569:381–393. doi: 10.1113/jphysiol.2005.095497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris SH. Evaluation of memantine for the treatment of Alzheimer’s disease. Expert Opin Pharmacother. 2003;4:2305–2313. doi: 10.1517/14656566.4.12.2305. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogal B, Trettel J, Uliasz TF, Levine ES, Hewett SJ. Changes in secondary glutamate release underlie the developmental regulation of excitotoxic neuronal cell death. Neuroscience. 2005;132:929–942. doi: 10.1016/j.neuroscience.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Frandsen A, Schousboe A. Dantrolene prevents glutamate cytotoxicity and Ca2+ release from intracellular stores in cultured cerebral cortical neurons. J Neurochem. 1991;56:1075–1078. doi: 10.1111/j.1471-4159.1991.tb02031.x. [DOI] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–3490. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Brown TH. Interpretation of voltage-clamp measurements in hippocampal neurons. J Neurophysiol. 1983;50:464–486. doi: 10.1152/jn.1983.50.2.464. [DOI] [PubMed] [Google Scholar]
- Johnston D, Hoffman DA, Magee JC, Poolos NP, Watanabe S, Colbert CM, Migliore M. Dendritic potassium channels in hippocampal pyramidal neurons. J Physiol. 2000;525:75–81. doi: 10.1111/j.1469-7793.2000.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Golovina V, Fajuk D, Andreeva N, Uvarova T, Khaspekov L, Victorov I. On the origin of a sustained increase in cytosolic Ca2+ concentration after a toxic glutamate treatment of the nerve cell culture. FEBS Lett. 1993;324:271–273. doi: 10.1016/0014-5793(93)80132-e. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Lasting effects of glutamate on nuclear calcium concentration in cultured rat hippocampal neurons: regulation by calcium stores. J Physiol. 1996;496:39–48. doi: 10.1113/jphysiol.1996.sp021663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Le DA, Lipton SA. Potential and current use of N-methyl-d-aspartate (NMDA) receptor antagonists in diseases of aging. Drugs Aging. 2001;18:717–724. doi: 10.2165/00002512-200118100-00001. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Zhang D, Abele AE, Lipton SA. Blockade of NMDA receptor-mediated mobilization of intracellular Ca2+ prevents neurotoxicity. Brain Res. 1992;598:196–202. doi: 10.1016/0006-8993(92)90183-a. [DOI] [PubMed] [Google Scholar]
- Levy DI, Lipton SA. Comparison of delayed administration of competitive and uncompetitive antagonists in preventing NMDA receptor-mediated neuronal death. Neurology. 1990;40:852–855. doi: 10.1212/wnl.40.5.852. [DOI] [PubMed] [Google Scholar]
- Limbrick DD, Jr, Churn SB, Sombati S, DeLorenzo RJ. Inability to restore resting intracellular calcium levels as an early indicator of delayed neuronal cell death. Brain Res. 1995;690:145–156. doi: 10.1016/0006-8993(95)00552-2. [DOI] [PubMed] [Google Scholar]
- Limbrick DD, Jr, Pal S, DeLorenzo RJ. Hippocampal neurons exhibit both persistent Ca2+ influx and impairment of Ca2+ sequestration/extrusion mechanisms following excitotoxic glutamate exposure. Brain Res. 2001;894:56–67. doi: 10.1016/s0006-8993(00)03303-5. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Magnusson KR. The aging of the NMDA receptor complex. Front Biosci. 1998;3:e70–e80. doi: 10.2741/a368. [DOI] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000;23:222–229. doi: 10.1016/s0166-2236(00)01548-4. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Behrens MI, Chung C, Bhattacharyya T, Choi DW. Susceptibility to apoptosis is enhanced in immature cortical neurons. Brain Res. 1997;759:228–232. doi: 10.1016/s0006-8993(97)00248-5. [DOI] [PubMed] [Google Scholar]
- Moroni F, Nicoletti F, Pellegrini-Giampietro DE. Metabotropic Glutamate Receptors and Brain Function. London: Portland Press Proceedings; 1998. [Google Scholar]
- Munir M, Lu L, McGonigle P. Excitotoxic cell death and delayed rescue in human neurons derived from NT2 cells. J Neurosci. 1995;15:7847–7860. doi: 10.1523/JNEUROSCI.15-12-07847.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Orrenius S. The role of calcium in apoptosis. Cell Calcium. 1998;23:173–180. doi: 10.1016/s0143-4160(98)90116-6. [DOI] [PubMed] [Google Scholar]
- Okiyama K, Smith DH, White WF, McIntosh TK. Effects of the NMDA antagonist CP-98,113 on regional cerebral edema and cardiovascular, cognitive, and neurobehavioral function following experimental brain injury in the rat. Brain Res. 1998;792:291–298. doi: 10.1016/s0006-8993(98)00158-9. [DOI] [PubMed] [Google Scholar]
- Olney JW. Inciting excitotoxic cytocide among central neurons. Adv Exp Med Biol. 1986;203:631–645. doi: 10.1007/978-1-4684-7971-3_48. [DOI] [PubMed] [Google Scholar]
- Poenie M. Alteration of intracellular Fura-2 fluorescence by viscosity: a simple correction. Cell Calcium. 1990;11:85–91. doi: 10.1016/0143-4160(90)90062-y. [DOI] [PubMed] [Google Scholar]
- Porter NM, Thibault O, Thibault V, Chen KC, Landfield PW. Calcium channel density and hippocampal cell death with age in long-term culture. J Neurosci. 1997;17:5629–5639. doi: 10.1523/JNEUROSCI.17-14-05629.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley T, Ochu E, Macaulay AJ. Time-dependent changes in NMDA receptor expression in neurones cultured from rat brain. Brain Res Mol Brain Res. 1996;40:271–274. doi: 10.1016/0169-328x(96)00056-3. [DOI] [PubMed] [Google Scholar]
- Rajdev S, Reynolds IJ. Glutamate-induced intracellular calcium changes and neurotoxicity in cortical neurons in vitro: effect of chemical ischemia. Neuroscience. 1994;62:667–679. doi: 10.1016/0306-4522(94)90468-5. [DOI] [PubMed] [Google Scholar]
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22:471–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- Sanchez-Armass S, Blaustein MP. Role of sodium–calcium exchange in regulation of intracellular calcium in nerve terminals. Am J Physiol Cell Physiol. 1987;252:C595–C603. doi: 10.1152/ajpcell.1987.252.6.C595. [DOI] [PubMed] [Google Scholar]
- Shalaby IA, Chenard BL, Prochniak MA, Butler TW. Neuroprotective effects of the N-methyl-d-aspartate receptor antagonists ifenprodil and SL-82,0715 on hippocampal cells in culture. J Pharmacol Exp Ther. 1992;260:925–932. [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Smith DH, Okiyama K, Thomas MJ, McIntosh TK. Effects of the excitatory amino acid receptor antagonists kynurenate and indole-2-carboxylic acid on behavioral and neurochemical outcome following experimental brain injury. J Neurosci. 1993;13:5383–5392. doi: 10.1523/JNEUROSCI.13-12-05383.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sombati S, Coulter DA, DeLorenzo RJ. Neurotoxic activation of glutamate receptors induces an extended neuronal depolarization in cultured hippocampal neurons. Brain Res. 1991;566:316–319. doi: 10.1016/0006-8993(91)91716-e. [DOI] [PubMed] [Google Scholar]
- Sucher NJ, Lei SZ, Lipton SA. Calcium channel antagonists attenuate NMDA receptor-mediated neurotoxicity of retinal ganglion cells in culture. Brain Res. 1991;551:297–302. doi: 10.1016/0006-8993(91)90944-q. [DOI] [PubMed] [Google Scholar]
- Szatkowski M, Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci. 1994;17:359–365. doi: 10.1016/0166-2236(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Tenneti L, Lipton SA. Involvement of activated caspase-3-like proteases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. 2000;74:134–142. doi: 10.1046/j.1471-4159.2000.0740134.x. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Thibault O, Porter NM, Chen KC, Blalock EM, Kaminker PG, Clodfelter GV, Brewer LD, Landfield PW. Calcium dysregulation in neuronal aging and Alzheimer’s disease: history and new directions. Cell Calcium. 1998;24:417–433. doi: 10.1016/s0143-4160(98)90064-1. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. Neuronal ageing in long-term cultures: alterations of Ca2+ homeostasis. Neuroreport. 2000;11:3725–3729. doi: 10.1097/00001756-200011270-00027. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A, Landfield PW. Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci. 2004;27:614–620. doi: 10.1016/j.tins.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Xia Y, Ragan RE, Seah EE, Michaelis ML, Michaelis EK. Developmental expression of N-methyl-d-aspartate (NMDA)-induced neurotoxicity, NMDA receptor function, and the NMDAR1 and glutamate-binding protein subunits in cerebellar granule cells in primary cultures. Neurochem Res. 1995;20:617–629. doi: 10.1007/BF01694545. [DOI] [PubMed] [Google Scholar]