

Abstract
Lanthanide complexes of tetraamide derivatives of DOTA are of interest today because of their application as chemical exchange saturation transfer (CEST) agents for magnetic resonance imaging (MRI). The protonation constants of some simple tetraamide derivatives of DOTA and the stability constants of the complexes formed with some endogenous metal ions, namely Mg2+, Ca2+, Cu2+, Zn2+, and lanthanide(III) ions, have been studied. These complexes were found to be considerably less stable than the corresponding [M(DOTA)]2− complexes, largely due to the lower basicity of the tetraamide ligands. The Mg2+ and Ca2+ complexes are well described by formation of only ML species at equilibrium while the Zn2+ and Cu2+ complexes exhibit one and two additional deprotonation steps above a pH of around 6, respectively. The extra deprotonation that occurs at high pH for the [Zn{DOTA-(amide)4}]2+ complexes has been assigned to an amide deprotonation by 1H NMR spectroscopy. The first deprotonation step for the Cu2+ complexes was traced to formation of the ternary hydroxo complexes ML(OH) (by UV/Vis spectrophotometry) while the second step corresponds to deprotonation of an amide group to form ML(OH)H−1-type complexes. The trends in the stability constants of the [Ln{DOTA-(amide)4}]3+ complexes follow similar trends with respect to ion size as those reported previously for the corresponding [Ln(DOTA)]− complexes, but again, the stability constants are about 10–11 orders of magnitude lower. A kinetic analysis of complex formation has shown that complexes are directly formed between a Ln3+ cation and fully deprotonated L without formation of a protonated intermediate. [Ln{DOTA-(MeAm)4}]3+ complex formation occurs at a rate that is two to three orders of magnitude slower than those of the corresponding [Ln(DOTA)]− complexes, while the variation in complex formation rates with Ln3+ ion size is opposite to that observed for the corresponding [Ln(DOTA)]− complexes. The Ce3+ and Eu3+ complexes of DOTA-(MeAm)4 are kinetically inert with respect to acid-catalyzed dissociation, which suggests that these complexes may potentially be safe for use in vivo.
Keywords: Magnetic resonance imaging, Contrast agents, Protonation constants, Stability constants, Kinetics
Introduction
Magnetic resonance imaging (MRI) is one of the most powerful and versatile techniques in modern medical diagnostics. Contrast agents (CA) are used to enhance MR contrast in 40–50% of all clinical MRI applications. The most common CAs are Gd3+ complexes due to the high spin state (seven unpaired electrons) and favorable relaxation properties of the Gd3+ ion.[1] The first contrast agent used in medical diagnosis was the Gd3+ complex of diethylenetriamine-N,N,N′,N″,N″-pentaacetic acid (H5DTPA), followed shortly thereafter by the Gd3+ complex of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (H4DOTA). These complexes are negatively charged and therefore must be formulated as salts. The next two approved agents, [Gd(DTPA-BMA)] and [Gd(HP-DO3A)], derived from an open-chain DTPA ligand and a macrocyclic DOTA ligand, respectively, are both neutral complexes.[2–4] All four of these systems are nonadentate, with one Gd3+ coordination site occupied by a water molecule. Rapid relaxation of this inner-sphere water molecule by the paramagnetic Gd3+ results in a shortening of the bulk water proton relaxation time (T1) through the rapid exchange of an inner-sphere bound water molecule with tissue water. Lanthanide(III) complexes of some tetraamide derivatives of DOTA have been known for many years but were neglected as possible contrast agents due to the slow water-exchange rates exhibited by the LnIII complexes of these ligands and their less than favorable thermodynamic stablility.[5] However, a recent report by Balaban et al. describing the chemical exchange saturation transfer (CEST) mechanism for generating a negative image contrast has renewed interest in these complexes.[6] CEST contrast begins with application of a presaturation RF pulse applied at the resonance freqnency of an exchangeable OH or NH proton on a CEST agent and this partially saturated spin then exchanges with the protons of bulk water. Given proper exchange kinetics and relaxation times, this results in a reduction in the bulk water signal intensity and negative contrast in an image. More recently, Sherry and coworkers discovered the CEST effect in [Eu{DOTA-(gly-OEt)4}]3+ (Scheme 1) and showed that a particularly useful exchange site for CEST activation is provided by the highly shifted (approx. 50 ppm), slow-exchanging water protons bound to a paramagnetic Eu3+ ion in this complex.[7] Paramagnetic CEST (PARACEST) agents that contain a highly shifted CEST activation site offer some advantages over the diamagnetic agents proposed originally; most importantly, the large chemical-shift difference between the exchanging protons on the agent and the bulk water reduces the possibility of off-resonance saturation of the bulk water signal. This observation has catalyzed interest in the synthesis and design of new tetraamide derivatives of DOTA as potential CEST systems, and this potential new application makes it especially important to evaluate the thermodynamic and kinetic stability of these complexes in solution and in the presence of adventitious ions.
Scheme 1.

Ligands investigated in this work.
Lanthanide(III) complexes of DOTA are among the most thermodynamically stable and kinetically inert complexes known to date.[8–10] A recently published report on the thermodynamic stability and kinetics of [Ln{DOTA-(gly)4}]− complex formation demonstrated that both the basicity of the amide ligands and the stability of the complexes are several orders of magnitude lower than for their [Ln(DOTA)]− analogues.[11] It was also shown that spontaneous dissociation of [Eu{DOTA-(gly)4}]− occurs somewhat faster than [Eu(DOTA)]−, but that acid-catalyzed dissociation of the protonated intermediate [Eu{H4DOTA-(gly)4}]3+ seems to occur more slowly than for its protonated [Eu(H2DOTA)]+ analog.[9,11] This suggests that these systems may in fact be suitable for in vivo imaging applications.
The stability constants of the Ca2+, Sr2+, Ba2+, Cu2+, Zn2+, Hg2+, La3+, and Gd3+ complexes of DOTAM have been reported by Maumela et al.[12] The authors also provided the lower limit for the stability constants of the Cd2+ and Pb2+ complexes (log Kmin = 19.0), because these stabilities are too high to be measured experimentally.[12] A more detailed study involving eleven LnIII ions was published recently by Voss et al., who used competition reactions between various LnIII ions and EuIII and followed the competition by the measurement of the luminescence intensity to calculate the ratio of the stability constants KLnIII/KEuIII.[13] No information about possible hydroxo complex formation (or amide dissociation) for Cu2+, Zn2+, and LnIII complexes are available in these papers. The protonation constants of the ligand DOTA-(MeAm)4 and the stability of the Eu3+ and Gd3+ complexes have also been reported by Virtuani and co-workers,[14] and the formation of MHL and ML [M(OH)L and M(OH)2 for Cu2+ and Zn2+ ions] species in solution was subsequently reported.[15] The stability constants of metal ion complexes of DOTA-(EtAm)4 and DOTA-(gly-OEt)4 have not been reported previously.
These earlier reports leave the impression that these structurally similar ligand systems behave quite differently in solution, which led to the present investigation of detailed thermodynamic studies of the divalent ions Mg2+, Ca2+, Cu2+, and Zn2+ and the trivalent lanthanide(III) ions with the ligands shown in Scheme 1. The rates of complex formation and acid-catalyzed dissociation of some [Ln{DOTA-(MeAm)4}]3+ complexes (where Ln3+ = Ce3+, Eu3+, and Lu3+ in the complex formation studies and Ln3+ = Ce3+ and Eu3+ in the acid-catalyzed dissociation studies) have also been studied.
Results and Discussion
The simple tetraamide ligands DOTAM (1), its N-methyl analog DOTA-(MeAm)4 (2), the N-ethyl analog DOTA-(EtAm)4 (3), and the glycinate ethyl ester analog DOTA- (gly-OEt)4 (4) were prepared following established procedures by alkylation of 1,4,7,10-tetraazacyclododecane with the appropriate α-halo amide derivative.[12,15–18] The characteristics of these ligands were in a good agreement with those reported previously.
Equilibrium Studies
The protonation constants of the ligands and stability constants of the complexes formed with the DOTA-(amide)4 ligands were determined by pH-potentiometry. The protonation constants (log KiH) of these tetraamide ligands are listed and compared to the literature values for DOTA and DOTA-(gly)4 in Table 1 (the values shown in parentheses are the standard deviations).[11,19] Our data agree well with the published values except for the variation in the first pKa of the ligand DOTAM, where the difference is larger than one order of magnitude. This variation can be attributed to the different ionic strength used in the present study. The protonation constants of DOTAM have been determined in 0.1 M NaCl at 25 °C by Maumela et al.[12] The Na+ ion is known to form more stable complexes with most of the ligands than the K+ ion. For instance, the stability of [Na(DOTA)]3− is log KNaL = 4.2 in contrast to [K(DOTA)]3−, which is log KKL = 1.6.[20] The stability of the Na+ and K+ complexes with DOTAM differ considerably from those reported for DOTA, so one can argue that measurements in a KCl background give more realistic pKa values than those measured in an NaCl background. Only two protonations were found for these ligands above a pH of about 1.8. The highest protonation constant was typically in the range 9.0–9.6, while the second protonation constant was in the range 5.4–6.5. The sum of these protonation constants (reflecting the total ligand basicity) is in the range 15.5–16.5 log K units (Table 1). These values are around 50 % of the total ligand basicity of DOTA, thus demonstrating the importance of the negatively charged acetate side-chains in DOTA in stabilizing the protonated ring nitrogen atoms of DOTA through hydrogen-bonding interactions. Similar variations in pKa values of DOTA-(MeAm)4 to those reported by Virtuani and co-workers were found.[14,15]
Table 1.
Protonation constants of several DOTA-(amide)4 ligands (I = 1.0 M KCl, T = 25 °C, values given in parentheses are standard deviations).
| DOTAM | DOTA-(MeAm)4 | DOTA-(EtAm)4 | DOTA-(gly-OEt)4 | DOTA-(gly)4a | DOTAb | |
|---|---|---|---|---|---|---|
| log K1H | 9.08(1) | 9.56(1) | 9.33(2) | 9.18(3) | 9.19 | 12.60 |
| 7.70c | 9.27d | |||||
| log K2H | 6.44(1) | 5.95(1) | 6.39(3) | 5.47(4) | 6.25 | 9.70 |
| 6.21c | 5.55d | |||||
| log K3H | – | 1.56d | – | 1.82(6) | 4.08 | 4.50 |
| log K4H | – | – | – | – | 3.45 | 4.14 |
| log K5H | – | – | – | – | 3.20 | 2.32 |
| log K6H | – | – | – | – | 1.40 | – |
| Σlog Ki H | 15.52 | 15.51 | 15.72 | 16.47 | 27.57 | 33.26 |
The reaction between these ligands and different divalent endogenous metal ions was rapid enough to allow direct potentiometric titrations for stability determinations. However, under acidic conditions, where complex formation is slow, the interval between base additions was maintained at 30 minutes to ensure complete equilibrium complexation at each titration step. The time needed for the Cu2+ complexes to reach equilibrium was verified by UV/Vis spectrophotometry. These stability constants are summarized and compared with previous literature values in Table 2.
Table 2.
Stability constants (log K) of DOTA-(amide)4 complexes with selected divalent metal ions (I = 1.0 M KCl, T = 25 °C).
| Ligand | Species | Mg2+ | Ca2+ | Cu2+ | Zn2+ |
|---|---|---|---|---|---|
| DOTAM | ML | 4.65(4) | 10.32(1) | 14.50(1) | 13.77(1) |
| 7.54a | 16.31a | 10.47a | |||
| MLH−1 | – | – | 8.41(4) | 10.57(6) | |
| MLH−2 | – | – | 8.82(2) | – | |
| DOTA-(MeAm)4 | ML | 4.33(2) | 10.11(1) | 14.61(4) | 13.66(2) |
| 9.47b | 14.12b | 13.00b | |||
| MLH−1 | – | – | 8.78(7) | 10.71(3) | |
| MLH−2 | – | – | 10.31(6) | – | |
| DOTA-(EtAm)4 | ML | 4.49(1) | 10.74(2) | 16.40(8) | 16.55(10) |
| MLH−1 | – | – | 8.96(9) | 10.86(10) | |
| MLH−2 | – | – | 11.27(9) | – | |
| DOTA-(gly-OEt)4c | ML | 3.97(7) | 9.09(5) | 17.18(9) | 11.44(5) |
| DOTAd | ML | 11.15 | 16.37 | 22.72 | 18.17 |
| MHL | 4.09 | 3.54 | 3.78 | 4.18 | |
| MLH−1 | – | – | 10.54 | 10.62 | |
| DOTA-(gly)4e | ML | 4.34 | 10.39 | 13.39 | – |
| MHL | – | 4.23f | 4.38f | – |
The equilibria involving these ligands with Mg2+ and Ca2+ ions can be described by formation of ML-type complexes only; no protonated complexes or terniary hydroxo complexes [ML(OH)] were detected. The stability of the MgL complexes is so low that competition by hydrolysis of Mg2+ at higher pH values was significant. Hydrolysis of Ca2+, however, occurs at much higher pH values [log Ka(Ca2+) = 12.8] so formation of ML(OH)-type complexes was neither anticipated nor observed.[21] The titration curves measured for the CuL complexes show two extra deprotonation steps above the pH at which the complexes were fully formed. The absorption spectra of [Cu-(DOTAM)]2+ and [Cu{DOTA-(MeAm)4}]2+ are identical and addition of the first equivalent of base to form a CuL-(OH) species resulted in no apparent change in the visible spectra of these complexes. However, addition of a second equivalent of base resulted in a significant blue shift in their absorption maxima from 715 nm to about 650 nm (Figure 1). Such spectral changes are known to occur for Cu2+-oligopeptide complexes, where deprotonation and coordination of an amide group to the Cu2+ ion is known to occur. However, the terminal NH2 group (anchor nitrogen atom) plays a critical role in the deprotonation and coordination of the adjacent amide moiety in the Cu2+-polypeptide systems.[20]
Figure 1.
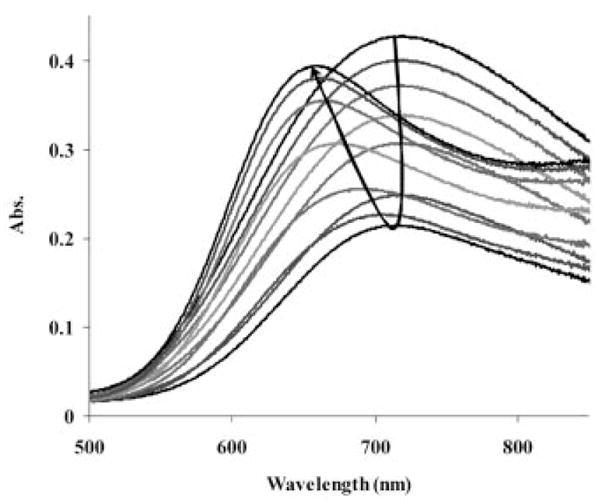
Visible spectra of [Cu{DOTA-(MeAm)4}]2+ (CCu = CL = 2.5 ×10−3 M) as a function of sample pH. The pH of the samples following the arrow are: 6.43, 7.39, 7.76, 8.00, 8.26, 8.72, 9.33, 9.90 10.26, 10.67, 11.05, 11.34, and 11.89.
The peptide hydrogen deprotonation in Zn2+-oligopeptide systems is known to occur only above a pH of 10.[22] The titration curve of [Zn{DOTA-(amide)4}]2+ systems also reflects an extra deprotonation step after the protons associated with the ligand are neutralized (above pH > 10). Furthermore, the X-ray structure of [Zn(DOTAM)]2+ shows a Zn2+ ion coordinated by four nitrogen atoms of the cyclen ring and two oxygen atoms of the amide pendant arms, with no inner-sphere water molecule, so the extra deprotonation likely reflects an amide proton.[12] 1H NMR titrations of [Zn(DOTAM)]2+ and [Zn{DOTA-(MeAm)4}]2+ complexes (Figure S2 in the Supporting Information and Figure 2) over the pH range 7–12 were consistent with this assignment.
Figure 2.
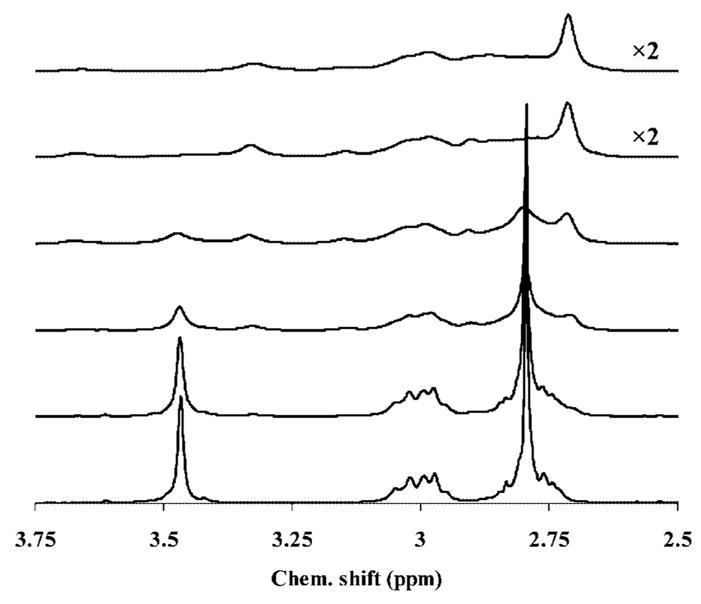
1H NMR titration of the [Zn{DOTA-(MeAm)4}]2+ complex (CZn = CL = 3.0 × 10−2 M). The pD of the samples are as follows (from top to bottom): 12.04 (the intensity of the spectra is doubled), 11.44 (the intensity of the spectra is doubled), 10.94, 10.49, 9.84 and 6.95.
Aqueous solutions of DOTA-(gly-OEt)4 are unstable toward hydrolysis of the ester above a pH of around 5. When dissolved in 0.01 M acid, the ligand is stable enough to allow estimates of the stabilities of its Mg2+, Ca2+, Cu2+, and Zn2+ complexes (Table 2) using titration data up to pH 5. With further increases in pH, hydrolysis of the ester bonds was evident so this precluded an examination of whether MLH−1 type complexes are formed with this ligand. Similarly, it was not possible to calculate the stabilities of the LnL complexes with this ligand because slow kinetics of complex formation dictated that the “out-of-cell” technique be used and this requires sample heating for an extended period to reach equilibrium; this favors ester hydrolysis which, in turn, distorts the pH-potentiometric data.
The stability constants of the complexes formed between several DOTA-(amide)4 ligands and numerous Ln3+ ions are summarized in Table 3. The values increase with a decrease in Ln3+ ion size near the beginning of the lanthanide series, then remain relatively constant for the heavier ions until the very end of the series, where a decrease is noted. A similar trend has been noted by Voss et al. for the [Ln(DOTAM)]3+ complexes, although the changes with lanthanide size differed somewhat.[13] The stability constants of the [Ln{DOTA-(EtAm)4}]3+ and [Ln{DOTA-(gly)4}]− complexes are nearly identical for most lanthanides, as one would expect for systems with similar structures and ligand basicities.[11] The data also show that the stabilities are not influenced by the overall charge on these complexes and that the extended glycinate groups do not coordinate to the central metal ions in ML complexes.
Table 3.
Stability constants (log K) of DOTA-(amide)4 ML-type complexes formed with several trivalent lanthanide ions Ln3+ (I = 1.0 M KCl, T = 25 °C).
| Ln3+ | DOTAM | DOTA-(MeAm)4 | DOTA-(AmEt)4 | DOTA-(gly)4a | DOTAb |
|---|---|---|---|---|---|
| Ce3+ | 11.93(7) | 12.68(1) | 13.00(8) | 13.02 | 23.4 |
| Nd3+ | 12.40(5) | 13.08(6) | 14.37(2) | 14.45 | 23.0 |
| Eu3+ | 13.80(2) | 13.67(8)c | 15.09(6) | 14.84 | 23.5 |
| Gd3+ | 13.12(4)d | 13.54(4)e | 14.83(25) | 14.54 | 24.7 |
| Dy3+ | 13.57(14) | 13.84(5) | 15.09(2) | 14.37 | 24.8 |
| Tm3+ | 13.46(2) | 14.08(5) | 14.74(3) | – | 24.4 |
| Lu3+ | 13.53(5) | 13.91(3) | 14.62(3) | 14.25f | 25.4 |
The stabilities of [Eu{DOTA-(MeAm)4}]3+ and [Gd{DOTA-(MeAm)4}]3+ previously reported by Virtuani et al. are in good agreement with the current results; the deviation of about 0.5 log K units in these two studies can be traced to differences in the log KiH values of the ligands themselves.[14,15] A noticeable difference in the equilibrium model is that we could find no evidence for formation of protonated (LnHL) complexes (either from “out-of-cell” data or from the titration data obtained for fully formed LnL complexes with HCl), while Virtuani et al. included protonated species in their model.[14,15]
An extra deprotonation was evident for all three [Ln{DOTA-(amide)4}]3+ complexes at pH values above 10.5. Virtuani, et al. have also reported formation of ternary hydroxo complexes [Ln(OH)L2+], although the pH range for this deprotonation was much lower in their study (in the range pH 7–9).[14,15] These differences may be due to the presence of excess free metal ions in their study since our titration curves resemble the titration curve of a strong acid with a strong base (no buffering region is observed below pH 10.5, Figure S3). So, if one accepts that a deprotonated species does form in these complexes, does the site of deprotonation correspond to an amide deprotonation or does deprotonation of the coordinated water molecule occur in these complexes? To answer this question, several additional experiments were performed. First, high resolution 1H NMR spectra of the diamagnetic Lu3+ complexes of DOTAM and DOTA-(MeAm)4 showed no change in the chemical shift of the CH2 protons of the amide side-arms (AB coupling pattern) upon addition of up to 2.5 equivalents of base (Figures S4 and S5 show the 1H NMR titrations of the [Lu(DOTAM)]3+ and [Lu(DOTA-(MeAm)4}]3+ complexes, respectively). This result is inconsistent with dissociation of an amide proton. The most convincing evidence that deprotonation occurs at the coordinated water molecule was obtained by luminescence studies with the Eu3+ complexes. It is known from the literature that both [Eu(DOTAM)]3+ and [Eu{DOTA-(MeAm)4}]3+ contain one inner-sphere coordinated water molecule (q = 1).[23] This was verified by measuring the luminescence rate constants for depopulation of the Eu3+ excited states in H2O vs. D2O. At pH 7 and 25 °C, these lifetimes are 1.79 (H2O)/0.44 ms−1 (D2O) and 1.96 (H2O)/0.46 ms−1 (D2O) for [Eu(DOTAM)]3+ and [Eu{DOTA-(MeAm)4}]3+, respectively. These values give an estimated q of 1.02 and 1.20 for the two complexes.[23] With an increase in pH (pD) to 11.8, the measured q values decrease to 0.47 and 0.52 for [Eu-(DOTAM)]3+ and [Eu{DOTA-(MeAm)4}]3+, respectively, consistent with deprotonation of the coordinated water molecule and a decrease in OH (OD) oscillators of about 50 %. These data do not, however, rule out the possibility of amide deprotonation in these complexes at even higher pH values (> 11.8) but it is not likely that amide deprotonation would lead to a change in the coordination environment around the chelated Ln3+ ion. This is in good agreement with the results published previously by Aime et al.[24]
Formation Kinetics of Some [Ln{DOTA-(MeAm)4}]3+ Complexes
The complexation reaction involving macrocyclic ligands is usually several orders of magnitude slower than that with linear polyamino-polycarboxylate type ligands.[25] It is now generally accepted that complex formation in LnDOTA-type systems takes place in several discrete steps.[26] First, an unusually stable intermediate where the Ln3+ ion is positioned “out of cage” and one or two of the macrocyclic amino groups remain protonated, which has been detected by several spectroscopic techniques, is formed quickly. Subseqnent slow base-catalyzed deprotonation of those amines and rearrangement leads to the formation of a final [Ln(DOTA)]− complex where the Ln3+ ion is trapped “in cage”.[8–10,27] Evidence for a similar stable intermediate has been reported during formation of the [Ln{DOTA-(gly)4}]− complexes as well.[11] Furthermore, an interesting structure that is thought to be the reactive intermediate in the formation of [Ln(DOTAM)]3+ complexes was characterized recently by X-ray diffraction.[28]
The visible spectra of solutions recorded after mixing DOTA-(MeAm)4 with CeCl3 (both reagents were present at 5 ×10−4 M) at pH 5.26 are shown in Figure 3. At this pH, the ligand is distributed between the forms H2L (80 %) and HL (20 %), both of which are favorable for formation of such an intermediate. With time, two new absorption bands, with λmax = 269 and 323 nm, respectively, appear in the spectra, the intensities of which increase with time as the “in cage” complex forms. The rate constants calculated from each absorption band agree with each other to within 3 %, thereby indicating that both bands characterize the same reaction process. These changes are similar to those described for formation of [Ce(DOTA)]− except that the λmax of the [Ce(H2DOTA)]+ kinetic intermediate occurs at 297 nm. Another major difference is that the [Ce{DOTA-(MeAm)4}]3+ reaction requires about 8 hours to reach equilibrium at pH 5.26, which is considerably more than the time required for [Ce(DOTA)]− complexation to reach equilibrium at pH 4.45.[8] The presence of one isosbestic point at λ = 260 nm indicates that only two absorbing components are present – the reactant (Ce3+) and the product [Ce{DOTA-(MeAm)4}]3+.
Figure 3.
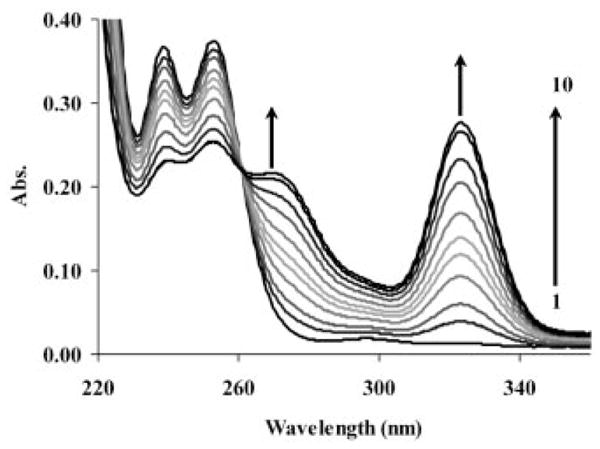
Formation of [Ce{DOTA-(MeAm)4}]3+ as a function of time. Absorption spectra were recorded (150 nm/min) at 1 (1), 9 (2), 17 (3), 33 (4), 49 (5), 65 (6), 97 (7), 161 (8), 241 (9), and 401 (10) minutes after the reaction was started and at equilibrium [CCe = CL = 5 ×10− 4 M in NMP buffer (CNMP = 2.5 ×10− 2 M) with pH 5.26].
It is also possible to follow the rate of [Eu{DOTA-(MeAm)4}]3+ complex formation by following changes in the Eu3+ charge-transfer (CT) bands (broad “shoulder” between 240 and 305 nm, Figure S6). A wavelength of 255 nm was selected for these studies since the absorbance of Eu3+ and ligand are not significant at this wavelength and is the only absorbing species [Eu{DOTA-(MeAm)4}]3+ (Figure S6).
Kasprzyk et al. have reported that the reaction between metal and ligand can proceed by three possible mechanisms,[29] one of which is a simple second-order reaction where saturation kinetics is not observed and the rate constant is a linear function of the metal ion concentration. The linear variation of pseudo-first-order rate constants (kobs) with analytical concentration of Ln3+ at different pH values is displayed graphically in Figure 4 for Ce3+ (similar data were also observed for Eu3+ and Lu3+, see Figures S7 and S8, respectively, in the Supporting Information). kobs was found to vary linearly with Ln3+ concentration for all systems, and since [L] ≤ 10 [M], these data may be described by Equation (1).
Figure 4.
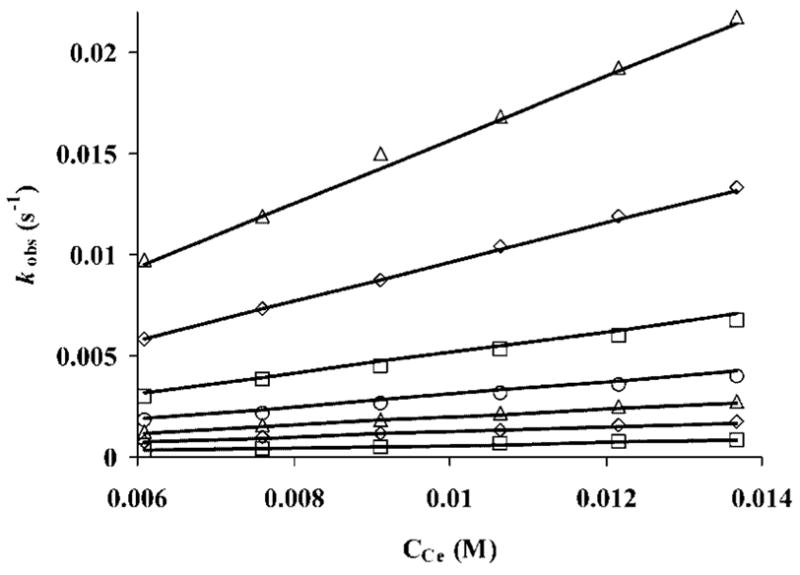
Dependence of kobs on metal concentration for formation of [Ce{DOTA-(MeAm)4}]3+ at different pH values (5.71, 5.59, 5.43, 5.31, 5.20, 5.09, and 4.92 from top to bottom).
| (1) |
The rates of complex formation are pH-dependent, slowing considerably with increasing the acid concentrations. Based on this, one can assume that some protonated species may be involved in the formation reaction. Taking into account all possible pathways, the rate of complex formation may be described by Equation (2), where k0, k1, and k2 are rate constants characterizing the reaction of the nonprotonated (L), monoprotonated (HL+), and diprotonated (H2L2+) forms of the ligand, respectively.
| (2) |
Given that the analytical concentration of the ligand is [L]t = [L] + [HL+] + [H2L2+], and substituting the appropriate concentrations from the definition of the protonation constants of the ligand (K1H = [HL+]/[H+][L] and K2H = [H2L2+]/[H+][HL+]), rearranging Equation (2) yields Equation (3).
| (3) |
The kobs values obtained at different metal concentrations for each pH were fitted to Equation (3) using the ligand protonation constants (K1H and K2H, Table 1) to obtain k0, k1, and k2 (Figure 4). The k2 values were either negligible or negative for all metal ions examined here, and hence were ultimately omitted during final data refinement. This indicates that a diprotonated form of the ligand is not involved in this reaction. The fitting yielded positive k1 values, although the errors in these values were about an order of magnitude higher than the rate constants themselves (for [Ce{DOTA-(MeAm)4}]3+ k1= 0.0026 ± 0.0146 M− 1 S− 1; for[Eu{DOTA-(MeAm)4}]3+ k1 = 0.0025 ± 0.0227 M− 1 S− 1; for [Lu{DOTA-(MeAm)4}]3+ k1 = 0.081 ± 0.184 M− 1 S− 1;). This result suggests that although the monoprotonated ligand may contribute somewhat to the reaction, the fully deprotonated ligand is clearly the most reactive species. This is a surprising conclusion because, over the pH range investigated, the concentration of HL is about four to five orders of magnitude higher than the concentration of L (the ratio [HL]/[L] decreases from 9 ×105 to 7 ×104). These combined data suggest that the pathway involving the monoprotonated ligand can also be ignored in the overall reaction and that formation of the [Ln{DOTA-(MeAm)4}]3+ complexes occurs due to the direct encounter between the Ln3+ ions and the fully deprotonated ligand. Thus, Equation (3) can be simplified to Equation (4).
| (4) |
The rate constants k0 determined here for three [Ln{DOTA-(MeAm)4}]3+ complexes and the rate constants characterizing the formation of the corresponding [Ln(DOTA)]− and [Ln(DOTAM)]3+ complexes are compared in Table 4. These data show that formation of [Ln{DOTA-(MeAm)4}]3+ occurs about two to three orders of magnitude slower than for the corresponding [Ln(DOTA)]− complexes. This may be partially attributed to differences in ligand charge since DOTA, in its active from, is negatively charged ([H2DOTA]2− is the primary species in most cases) while the tetraamide ligand is neutral. Interestingly, the rates of [Ln{DOTA-(MeAm)4}]3+ complex formation are somewhat slower than for formation of the [Ln(DOTAM)]3+ complexes, likely due to the presence of the methyl group on the amide pendant arm.[30] The rates of complex formation can be altered further by increasing the bulkiness of the substituent on the amide pendant arms (formation of [Ln{DOTA-(pipAm)4}]3+ complexes).[31] The trends in formation rates across the lanthanide series follow a similar trend to that observed for [Ln(DOTAM)]3+ complexes recently and opposite to that observed for [Ln(DOTA)]− complexes.[9,30] The water-exchange rates of [Ln{DOTA-(amide)4}]3+ complexes have been found to behave identically when they are plotted against the size of the Ln3+ ions (maximum at Eu3+).[32,33] This indicates that the water-exchange rates may influence the rates of complex formation of [Ln{DOTA-(amide)4}]3+ complexes and can be used to interpret the trends in the rates of complex formation. Similar phenomena have often been observed for the formation of lanthanide complexes of with mono- and multidentate ligands.[34]
Table 4.
Comparison of the formation rate constants for [Ln{DOTA-(MeAm)4}]3+ (k0, M−1 s−1), [Ln(DOTAM)]3+, and [Ln(DOTA)]− complexes.
Acid-Catalyzed Dissociation of [Ln{DOTA-(MeAm)4}]3+Complexes
Lanthanide complexes used for medical diagnosis must be kinetically stable because neither product of dissociation (the Ln3+ aqua ions or the ligands) is well tolerated in vivo. Several groups have demonstrated that dissociation of [Ln(DOTA)]− and Ln(DOTA) derivatives takes place by a proton-assisted pathway and that endogenous metal ions such as Cu2+ or Zn2+ have little affect on the dissociation rates.[8–10,35] As the structures of the complexes examined here do not differ substantially from those of the [Ln(DOTA)]− complexes, one could anticipate a similar kinetic dissociation behavior for the LnDOTA-tetraamides. Acid-catalyzed dissociation of [Ce{DOTA-(MeAm)4}]3+ and [Eu{DOTA-(MeAm)4}]3+ was examined at a pH below 2, where one would predict complete dissociation of the complexes based upon the thermodynamic measurements. At pH 2, the complexes dissociate so slowly that it was difficult to collect kinetic data over a reasonable period of time. Ultimately, it was found that the kinetic measurements could be performed over the range 0.5 < [HClO4] < 3.0 M and by maintaining a constant ionic strength with addition of NaClO4 when necessary [(H+ + Na+)ClO4− constant at 3.0 M]. As the total acid concentration was more than two orders of magnitude greater than the complex concentration in all cases, the dissociation rates follow pseudo-first-order kinetics (kobs = kd) and can be described by Equation (5), where [LnL]t is the total complex concentration.
| (5) |
Plots of first-order dissociation rates vs. acid concentration were found to be similar for both complexes (Figure 5). Similar results have been obtained in a study of the dissociation rates of [Gd(DOTA)]− and [Eu{DOTA-(gly)4}]−.[10,11] It has been shown previously that dissociation of [Ce(DOTA)] − is second order with respect to [H+] while dissociation of [Eu(DOTA)]− shows saturation behaviour.[8,9] Like other complexes, dissociation of [Ln{DOTA-(MeAm)4}]3+ is initiated by protonation, probably at an amide pendant arm. This is in agreement with the observation that there is no change in the absorption spectrum of either complex immediately after mixing the complex with a strong acid. The proton must therefore be transferred to the ring nitrogen atom, after which the metal ion moves out from the “coordination cage” (rearrangement), since the “in cage” complexes cannot dissociate directly. A protonation constant of KH[Eu(DOTA)]− = 14, which corresponds to the protonation of the acetate arm coordinated to the Eu3+ in the complex, was found for [Eu(DOTA)]−.[9] Complexes formed with extended phosphonate or glycinate pendant arms have even higher log KiH values. For example, three to four protonation steps can be measured for the LnIII complexes of DOTA-(gly)4 (log KiH = 2–4) and four steps for the [Ln(DOTP)]5− complexes (log KiH = 4–8).[11,36] The protonation constants of the [Ln{DOTA-(MeAm)4}]3+ complexes are very low and no protonation constant can be obtained from the titration data of 0.01 M complex solutions with 0.1 M HCl. This can be explained by the large difference in basicity of the donor atoms in the pendant arms of the amides compared to acetates or phosphonates. This is easily understood if one compares log KH values for aminomethanephosphonic acid, glycine, and glycine amide. The protonation constant of aminomethanephosphonic acid falls in the range 5.32–5.40 and the protonation constant of glycine is in the range 2.33–2.79, while the protonation of the amide nitrogen of glycine amide occurs only at high acid concentrations and log KH can not be determined by pH-potentiometry.[20,37]
Figure 5.
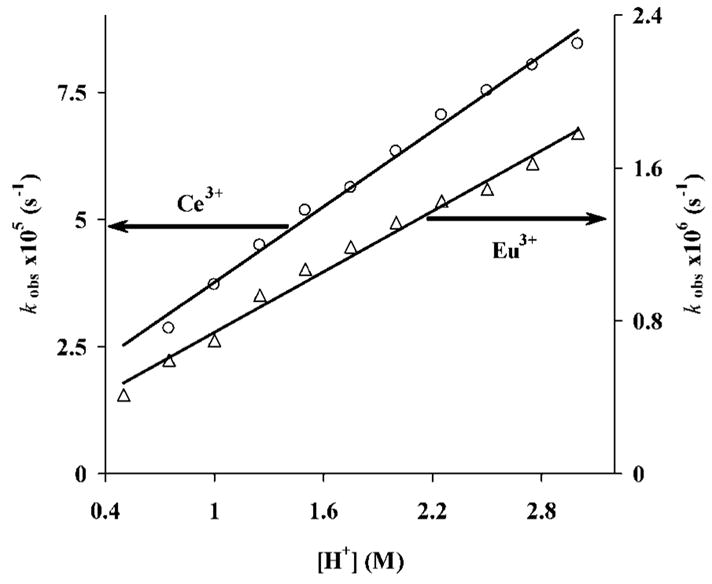
Rates of dissociation of [Ln{DOTA-(MeAm)4}]3+ {Ln3+ = Ce3+ (circles) and Eu3+ (triangles)}. The solid line represents the fitted curves while the points are the measured values.
The data in Figure 5 demonstrate that the rates of dissociation of the complexes are linearly proportional to the acid concentration and are hence described by Equation (6), where k0 corresponds to dissociation of the complex (most likely spontaneous) while k1 corresponds to acid-catalyzed dissociation of the monoprotonated LnLH complex.
| (6) |
The rate constants calculated from these data are listed in Table 5 along with comparative values for [Ce(DOTA)]− and [Eu(DOTA)]−.
Table 5.
Proton-assisted dissociation rates for two [Ln{DOTA-(MeAm)4}]3+ complexes.
Spontaneous dissociation of [Ln{DOTA-(MeAm)4}]3+ (k0) is somewhat faster than [Gd(DOTA)] − but the corresponding data (k0) for [Ce(DOTA)] − and [Eu(DOTA)] − are not available. However, the acid-catalyzed rates of dissociation (k1) are about 25–30-fold slower than the corresponding rates for the [Ln(DOTA)] − complexes, perhaps reflecting the ease of protonation of a charged carboxylate side-arm in [Ln(DOTA)] − compared to the difficulty of protonating a neutral amide side-arm in [Ln{DOTA-(MeAm)4}]3+. There is also a noticeable difference in the rates of acid-catalyzed dissociation of [Eu{DOTA-(MeAm)4}]3+vs. [Eu{DOTA-(gly)4}]− (both tetraamide complexes) – the latter complex dissociates about 15-fold faster than the former.[11] This likely reflects the greater overall positive charge on [Eu{DOTA-(MeAm)4}]3+ compared to [Eu{DOTA-(gly)4}]−. Thus, even though the LnDOTA-tetraamide complexes are thermodynamically considerably less stable than the corresponding [Ln(DOTA)]− complexes, it appears to be more difficult to protonate the neutral amide side-chains in these complexes and this ultimately limits their rates of acid-catalyzed dissociation. Similar conclusions have been reported previously for some phosphinate and phosphonate ester derivatives of DOTA.[38,39] This suggests that the kinetic inertness of the LnDOTA-tetraamide complexes may ultimately allow for their use as imaging agents in vivo.
Conclusions
The protonation constants of several tetraamide derivatives of DOTA show similar log KiH, values with the highest protonation constant falling in the range 9.0–9.6 and the lowest in the range 5.5–6.5. In comparison to DOTA, the basicity (Σlog KiH) of the tetraamide ligands is about 15–16 orders of magnitude less than that of DOTA. This indicates that the DOTA-tetraamide ligands have a greatly reduced ability to bind H+ and metal ions. Only ML type complexes are formed between these ligands and Mg2+ or Ca2+ ions while complexes formed with Zn2+ and Cu2+ undergo additional deprotonation steps either at a bound water molecule (first deprotonation step) or an amide side-chain (first deprotonation step for Zn2+ and second deprotonation step for Cu2+ complexes). The thermodynamic stability constants of several LnL complexes determined using the “out-of-cell” technique show an unusual trend in that they increase between Sm3+ and Eu3+ then remain relatively constant with further decreases in Ln3+ size until the very end of the lanthanide series. The thermodynamic constants are about 10–11 orders of magnitude lower than the corresponding [Ln(DOTA)]− complexes. The [Ln{DOTA-(amide)4}]3+ complexes also undergo an additional deprotonation above pH 10.5 which is accompanied by formation of the ternary Ln(OH)L complex, with the deprotonation likely occurring at the coordinated water molecule in these systems.
The rates of [Ln{DOTA-(MeAm)4}]3+ complex formation are about two to three orders of magnitude slower than those of the corresponding [Ln(DOTA)]− complexes. The variation of the formation rates with Ln3+ ion size is the opposite of that observed for the [Ln(DOTA)]− complexes. This is most likely due to the higher positive charge density of the heavier Ln3+ ions resulting in greater repulsion between the positively charged Ln3+ ions and the ligand. [Ce{DOTA-(MeAm)4}]3+ and [Eu{DOTA-(MeAm)4}]3+ are kinetically inert toward acid-catalyzed dissociation due to the unfavorable acid-base properties of the uncharged amide pendant arms in these complexes. Based on the slow rates of acid-catalyzed dissociation, LnDOTA-(amide)4 systems may prove safe for in vivo imaging applications.
Experimental Section
Synthesis of the Ligands
The tetraamide ligands DOTAM (1),[12] DOTA-(MeAm)4 (2)[15,16] DOTA-(EtAm)4 (3),[16,17] and DOTA-(gly-OEt)4 (4)[18] were prepared by established procedures and their purity established by 1H and 13C NMR spectroscopy, EI mass spectrometry, elemental analysis, and melting point determinations for all solids.
Potentiometric Measurements
The pH-potentiometric titrations were carried out with a Thermo Orion expandable ion analyzer EA940 pH meter using Thermo Orion semi-micro combination electrode 8103BN in a mixing vessel thermostatted at 25.0 °C. A Metrohm DOSIMATE 665 autoburette (5 mL capacity) was used for base additions and 1.0 M KCl was used to maintain the ionic strength. All equilibrium measurements (direct titrations) were carried out in 10.00 mL sample volumes with magnetic stirring. Argon was passed over the top of the sample during the titrations to maintain the cell free of CO2. Standard buffers (Borax: 0.01 M, pH 9.180 and KH-phthalate 0.05 M, pH 4.005) were used to calibrate the electrode. The titrant, a carbonate-free KOH solution, was standardized against 0.05 M KH-phthalate solution by pH-potentiometry. A standard solution of hydrochloric acid was prepared using doubly distilled water and the concentration was determined by pH-potentiometry with standard KOH solution. A method proposed by Irving et al. was used to obtain H+ ion concentrations from the measured pH values.[40] The pKw of water was also calculated from these titration data and was found to be 13.845. The protonation constants (log KiH) and stability constants were evaluated from the potentiometric data by using the PSEQUAD computer program.[41]
Stock Solutions
Stock solutions of metal ions (prepared from chloride salts) at about 0.02–0.06 M were calibrated by complexometric titration against standardized [Na2(H2EDTA)] using the appropriate end-point indicator.[42] [Na2(H2EDTA)] was purchased from Alfa-Aesar and standardized by a pH-potentiometric titration using standardized KOH. Stock solutions of the ligands synthesized in this work (0.02–0.05 M) were prepared in deionized water maintaining the pH in the range 3–4 with acid (HCl). It was found important to maintain the stock solution of DOTA-(gly-OEt)4 somewhat acidic (pH ≤ 3) due to slow hydrolysis of the ester bonds. The concentrations of stock solutions of these ligands were determined by pH-potentiometry from the titration data obtained in the absence and in the presence of an approximately 50-fold excess of CaCl2.
Protonation and Metal Binding (Stability) Constants
The protonation constants of the ligands were calculated from the data obtained by titrating 2 and 5 mM samples (180–268 data points) with standardized KOH solution (0.18 M) in the absence of Ca2+ over the pH range 1.8–12.0. The protonation constants of the ligand (log KiH) are defined according to Equation (7), where i = 1, 2, and 3 and [Hi−1L] and [H+] are the equilibrium concentrations of the ligand (i = 1), protonated forms of the ligand (i = 2 or 3) and hydrogen ions, respectively.
| (7) |
Relatively rapid equilibrium was observed in the systems containing Mg2+, Ca2+, Zn2+, or Cu2+. This proved fast enough to be able to use a direct titration method for stability constant determinations. In these measurements, a time period of 900–1200 seconds was allowed between each addition of KOH to ensure equilibrium before the sample pH was read. The next addition of base was made only when four consecutive pH readings collected over 60 seconds were identical within ±0.0006 pH units. The titrations were carried out at 1:1 and 2:1 metal to ligand concentration ratios and the concentration of the ligands was maintained at around 2 mM. These combined titration data were fitted simultaneously and the thermodynamic stability constants (KML) were evaluated and reported based on Equations (8) and (9).
| (8) |
| (9) |
The complexation of lanthanide(III) ions with macrocyclic ligands is very slow (about four to five weeks are required to achieve equilibrium at room temperature), so “out-of-cell” experiments (also known as the batch method) were performed instead of direct titrations. Typically, 25.0 mL of a mixture of Ln3+ and ligand each at 2–2.5 mM was prepared and divided into 16 individual solutions (1.5 mL each). The pH of the samples was adjusted with HCl or KOH to span the range pH 2.5–4.5 (where complex formation is ongoing but not complete). The samples were sealed and placed in a 45 °C incubator for 21 days followed by an additional 28 days at room temperature to re-equilibrate the mixtures. The pH of each individual sample was then measured. Acid/base titrations were also performed under identical conditions before and after measuring the pH of the samples. The average of these measurements was used to correct the pH of the samples for the difference between the measured and calculated pH by following the method proposed by Irving et al.[40] This difference was usually greater than the one obtained in the stirred samples and can be attributed to the difference in the diffusion of the ions in the samples not being stirred.
Formation and Dissociation Kinetics of the [Ln{DOTA-(MeAm)4}]3+Complexes
The formation rates of [Ce{DOTA-(MeAm)4}]3+ (λmax = 323 nm) and [Eu{DOTA-(MeAm)4}]3+ (λmax = 255 nm) were studied at 25 °C and 1.0 M KCl ionic strength by spectrophotometry using a Cary 300 Bio UV/Vis spectrophotometer equipped with thermostatted cell holders and semi-micro quartz cells (Starna, optical path length 1 cm). All the reactions were performed under pseudo-first-order conditions where the metal ions were in 10–25-fold excess. The concentration of the ligand in the formation reactions of Ce3+, Eu3+, and Lu3+ complexes was set to 5.62 ×10− 4, 9.83 ×10− 4, and 1.12 ×10−3 M, respectively. Since Lu3+ ions do not absorb in the UV/Vis region, the indicator method proposed by Kasprzyk et al. using indicators bromocresol green (pH 4.0–5.6) and bromocresol purple (pH 5.2–6.8) was used to record the absorbance change at a wavelength of 615 nm.[29] An appropriate concentration of indicator and buffer at different pH values was established experimentally to allow slight pH change in these systems (ΔpH ≤ 0.1). The pH of each sample was re-measured after the equilibrium was reached (usually the next day) and any samples with a pH change greater than 0.1 pH units were either excluded from the calculations or the process was repeated at a higher buffer concentration. Formation kinetic studies were carried out with the non-coordinating buffers N-methylpiperazine (NMP, log K2H = 4.83) at a concentration of 0.05 M for Ce3+ and Eu3+ and dimethylpiperazine (DMP, log K2H = 4.18) at 0.025–0.05 M for Lu3+ to maintain constant pH.[20]
Absorbance data recorded as a function of time were fitted to Equation (10) to evaluate the first-order rate constants, where A0, Ae, and At are the absorbance values measured at the start of the reaction (t = 0), at equilibrium, and at time t, respectively. These data were fitted to Equation (10) with the Scientist® (Micromath) software using a standard least-squares procedure.[43]
| (10) |
Proton-assisted dissociation of the complexes takes place slowly after addition of excess strong acid. The dissociation kinetics were studied in 0.5–3.0 M HClO4 acidic solutions at 25 °C and 3.0 M ionic strength provided by (Na+ + H+)ClO4 in samples where the concentration of complexes was 1 mM. The decrease in absorbance was measured periodically at 323 nm for [Ce{DOTA-(MeAm)4}]3+ and at 255 nm for [Eu{DOTA-(MeAm)4}]3+. The complete kinetics curve was registered for the Ce3+ complex and used for the fitting procedure, whereas the dissociation curves for the Eu3+ complex were followed until 60–70 % conversion was reached.
Acknowledgments
The authors thank the National Institutes of Health (NIH) (RR-02584, CA-115531 and EB-004582 A. D. S.), the Robert A. Welch Foundation (AT-584 A. D. S.) and the EMIL Project funded by the EC FP6 Framework Program (LSCH-2004-503569 E. B.) for financial assistance.
Footnotes
Supporting information for this article is available on the WWW under http://www.eurjic.org or from the author.
Supporting Information: Visible spectra of the [Cu(DOTAM)]2+ complex as a function of pH (Figure S1); 1H NMR titration of the [Zn(DO-TAM)]2+ complex (Figure S2); titration curves obtained for some [Ln{DOTA-(MeAm)4}]3+ complexes (Ln3+ = Ce3+, Eu3+, and Lu3+; Figure S3); 1H NMR titration of the [Lu(DOTAM)]3+ complex (Figure S4); 1H NMR titration of the [Lu{DOTA-(MeAm)4}]3+ complex (Figure S5); formation of [Eu{DOTA-(MeAm)4}]3+ with time (Figure S6); dependence of kobs on the Eu3+ concentration for the formation of [Eu{DOTA-(MeAm)4}]3+ at different pH values (Figure S7); dependence of kobs on the Lu3+ concentration for the formation of [Lu{DOTA-(MeAm)4}]3+ at different pH.
References
- 1.Lauffer RB. Chem Rev. 1987;87:901–927. [Google Scholar]
- 2.Carr DH, Brown J, Bydder GM, Weinmann HJ, Speck U, Thomas DJ, Young IR. Lancet. 1984;1:484–486. doi: 10.1016/s0140-6736(84)92852-6. [DOI] [PubMed] [Google Scholar]
- 3.Parizel PM, Degryse HR, Gheuens J, Martin JJ, Vanvyve M, Delaporte C, Selosse P, Vandeheyning P, Deschepper AM. J Comp Assisted Tomography. 1989;13:378–385. doi: 10.1097/00004728-198905000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Meyer D, Schaefer M, Bonnemain B. Invest Radiol. 1988;23:S232–S235. doi: 10.1097/00004424-198809001-00048. [DOI] [PubMed] [Google Scholar]
- 5.Aime S, Barge A, Botta M, Parker D, DeSousa AS. J Am Chem Soc. 1997;119:4767–4768. [Google Scholar]
- 6.Ward KM, Aletras AH, Balaban RS. J Magn Reson. 2000;143:79–87. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- 7.Zhang SR, Winter P, Wu KC, Sherry AD. J Am Chem Soc. 2001;123:1517–1518. doi: 10.1021/ja005820q. [DOI] [PubMed] [Google Scholar]
- 8.Brucher E, Laurenczy G, Makra Z. Inorg Chim Acta. 1987;139:141–142. [Google Scholar]
- 9.Toth E, Brucher E, Lazar I, Toth I. Inorg Chem. 1994;33:4070–4076. [Google Scholar]
- 10.Wang XY, Jin TZ, Comblin V, Lopezmut A, Merciny E, Desreux JF. Inorg Chem. 1992;31:1095–1099. [Google Scholar]
- 11.Baranyai Z, Brucher E, Ivanyi T, Kiraly R, Lazar I, Zekany L. Helv Chim Acta. 2005;88:604–617. [Google Scholar]
- 12.Maumela H, Hancock RD, Carlton L, Reibenspies JH, Wainwright KP. J Am Chem Soc. 1995;117:6698–6707. [Google Scholar]
- 13.Voss DA, Farquhar ER, Horrocks WD, Morrow JR. Inorg Chim Acta. 2004;357:859–863. [Google Scholar]
- 14.Alderighi L, Bianchi A, Calabi L, Dapporto P, Giorgi C, Losi P, Paleari L, Paoli P, Rossi P, Valtancoli B, Virtuani M. Eur J Inorg Chem. 1998:1581–1584. [Google Scholar]
- 15.Bianchi A, Calabi L, Giorgi C, Losi P, Mariani P, Paoli P, Rossi P, Valtancoli B, Virtuani M. J Chem Soc Dalton Trans. 2000:697–705. [Google Scholar]
- 16.Weaver WE, Whaley WM. J Am Chem Soc. 1947;69:515–516. doi: 10.1021/ja01195a012. [DOI] [PubMed] [Google Scholar]
- 17.Shinoda S, Nishimura T, Tadokoro M, Tsukube H. J Org Chem. 2001;66:6104–6108. doi: 10.1021/jo010396c. [DOI] [PubMed] [Google Scholar]
- 18.Zhang SR, Wu KC, Biewer MC, Sherry AD. Inorg Chem. 2001;40:4284–4290. doi: 10.1021/ic0003877. [DOI] [PubMed] [Google Scholar]
- 19.Burai L, Fabian I, Kiraly R, Szilagyi E, Brucher E. J Chem Soc Dalton Trans. 1998:243–248. [Google Scholar]
- 20.Martell AE, Smith RM, Motekaitis RJ. Critically Selected Stability Constants of Metal Complexes. Texas A&M University; 2004. Database Version 8.0. [Google Scholar]
- 21.Baes CFJ, Mesmer RE. The Hydrolysis of Cations. John Wiley & Sons Inc; New York: 1976. [Google Scholar]
- 22.Sigel H, Martin RB. Chem Rev. 1982;82:385–426. [Google Scholar]
- 23.Beeby A, Clarkson IM, Dickins RS, Faulkner S, Parker D, Royle L, de Sousa AS, Williams JAG, Woods M. J Chem Soc Perkin Trans 2. 1999:493–503. [Google Scholar]
- 24.Aime S, Barge A, Bruce JI, Botta M, Howard JAK, Moloney JM, Parker D, de Sousa AS, Woods M. J Am Chem Soc. 1999;121:5762–5771. [Google Scholar]
- 25.Brucher E. Top Curr Chem. 2002;221:103–122. [Google Scholar]
- 26.Csajbok E, Banyai I, Brucher E. Dalton Trans. 2004:2152–2156. doi: 10.1039/B405116E. [DOI] [PubMed] [Google Scholar]
- 27.Wu SL, Horrocks WD. Inorg Chem. 1995;34:3724–3732. [Google Scholar]
- 28.Stenson PA, Thompson AL, Parker D. Dalton Trans. 2006:3291–3293. doi: 10.1039/b605876k. [DOI] [PubMed] [Google Scholar]
- 29.Kasprzyk SP, Wilkins RG. Inorg Chem. 1982;21:3349–3352. [Google Scholar]
- 30.Baranyai Z, Bányai I, Brücher E, Király R, Terreno E. Eur J Inorg Chem. doi: 10.1002/ejic.200700178. [DOI] [Google Scholar]
- 31.G. Tircsó, E. Tircsóné Benyó, A. D. Sherry, unpublished results 2007.
- 32.Zhang SR, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Acc Chem Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- 33.Zhang SR, Wu KC, Sherry AD. J Am Chem Soc. 2002;124:4226–4227. doi: 10.1021/ja017133k. [DOI] [PubMed] [Google Scholar]
- 34.Lincoln SF, Merbach AE. Advances in Inorganic Chemistry. Academic Press; New York: 1995. pp. 1–87. [Google Scholar]
- 35.Kumar K, Tweedle MF. Inorg Chem. 1993;32:4193–4199. [Google Scholar]
- 36.Sherry AD, Ren J, Huskens J, Brucher E, Toth E, Geraldes CFCG, Castro MMCA, Cacheris WP. Inorg Chem. 1996;35:4604–4612. [Google Scholar]
- 37.Liler M. J Chem Soc Perkin Trans 2. 1971:334–338. [Google Scholar]
- 38.Pulukkody KP, Norman TJ, Parker D, Royle L, Broan CJ. J Chem Soc Perkin Trans 2. 1993:605–620. [Google Scholar]
- 39.Burai L, Kiraly R, Lazar I, Brucher E. Eur J Inorg Chem. 2001:813–820. [Google Scholar]
- 40.Irving HM, Miles MG, Pettit LD. Analytica Chim Acta. 1967;38:475–488. [Google Scholar]
- 41.Zekany L, Nagypal I. In: Computational Methods for the Determination of Formation Constants. Leggett DJ, editor. Plenum Press; New York: 1985. p. 291. [Google Scholar]
- 42.Schwarzenbach G, Flaschka H. Complexometric Titrations. 2. English, Barnes and Noble; New York: 1969. p. 490. [Google Scholar]
- 43.http://www.micromath.com/products.php?p=scientist.