

Abstract
Mitochondria are the essential site of aerobic energy production in eukaryotic cells. Reactive oxygen species (ROS) are an inevitable by-product of mitochondria metabolism and can cause mitochondrial DNA mutations and dysfunction. Mitochondrial damage can also be the consequence of disease processes. Therefore, maintaining a healthy population of mitochondria is essential to the well-being of cells. Autophagic delivery to lysosomes is the major degradative pathway in mitochondrial turnover, and we use the term mitophagy to refer to mitochondrial degradation by autophagy. Although long assumed to be a random process, increasing evidence indicates that mitophagy is a selective process. This review provides an overview of the process of mitophagy, the possible role of the mitochondrial permeability transitionin mitophagy and the importance of mitophagy in turnover of dysfunctional mitochondria.
Keywords: aging, apoptosis, autophagy, LC3, mitochondrial permeability transition, mtDNA, necrosis, photodamage, reactive oxygen species
Introduction
Mitochondria are the site of oxidative phosphorylation, which generates ATP coupled to electron transfer from respiratory substrates to oxygen by a series of oxidation-reduction reactions that pump protons across the mitochondrial inner membrane from the matrix space [1]. In mitochondria and submitochondrial particles as well as in intact cells, respiration produces reactive oxygen species (ROS) like H2O2 and superoxide anion (O2•-), especially if respiration is inhibited or otherwise disordered [2-5]. ROS derived from mitochondria can promote cytotoxicity and cell death [4;5].
As a major source of ROS production, mitochondria are especially prone to ROS damage. Such damage can induce the mitochondrial permeability transition (MPT) caused by opening of non-specific high conductance permeability transition (PT) pores in the mitochondrial inner membrane (see below). ATP depletion from uncoupling of oxidative phosphorylation then promotes necrotic cell death, whereas release of cytochrome c after mitochondrial swelling activates caspases and onset of apoptotic cell death [6;7].
ROS also attack nucleic acids and are thus genotoxic. A lack of histones in mitochondrial DNA (mtDNA) accounts, at least in part, for a 10- to 20-fold higher mutation rate of mtDNA compared to nuclear DNA [8]. Although oxidative stress and various disease processes cause mitochondrial damage and dysfunction, even normal mitochondria will accumulate enough oxidative “hits” over time to become damaged and possibly dangerous to the cell. Such defective mitochondria have the potential for futile ATP hydrolysis, accelerated production of ROS and release of proapoptotic proteins. Timely elimination of aged and dysfunctional mitochondria is essential to protect cells from the harm of disordered mitochondrial metabolism and release of proapoptotic proteins. The mechanism of mitochondrial turnover is predominantly autophagic sequestration and delivery to lysosomes for hydrolytic degradation, a process also called mitophagy. Here, we discuss recent evidence that suggests mitophagy is selective process that can specifically target dysfunctional mitochondria.
General Features of Autophagy
Autophagy is the process by which organelles and bits of cytoplasm are sequestered and subsequently delivered to lysosomes for hydrolytic digestion [9]. Autophagy is ongoing in nucleated cells and is typically activated by fasting and nutrient deprivation. In the liver particularly, glucagon promotes autophagy, whereas insulin negatively regulates it [10;11]. During fasting, autophagy is important for generating amino acids, fueling the tricarboxylic cycle, and maintaining ATP energy production. Autophagy also removes toxic protein aggregates and unneeded organelles. Both insufficient and excess autophagy seem capable of promoting cell injury [12]. Appropriate regulation of autophagy is thus essential for cellular well-being.
During autophagy, an isolation membrane forms a cup-shaped membranous structure called a phagophore or pre-autophagosome that eventually envelopes the autophagic target (Figure 1) [13-15]. The origin of isolation membranes is controversial. One proposed source is ribosome-free regions of the rough endoplasmic reticulum (ER), but others suggest that isolation membranes are novel structures devoid of Golgi and ER markers [14]. As isolation membranes envelop and seal around their targets, double– membrane vesicles called autophagosomes form. These autophagosomes then fuse with lysosomes to form autolysosomes, and their sequestered contents are degraded by lysosomal hydrolases and recycled.
Figure 1. Scheme of Mitophagy.
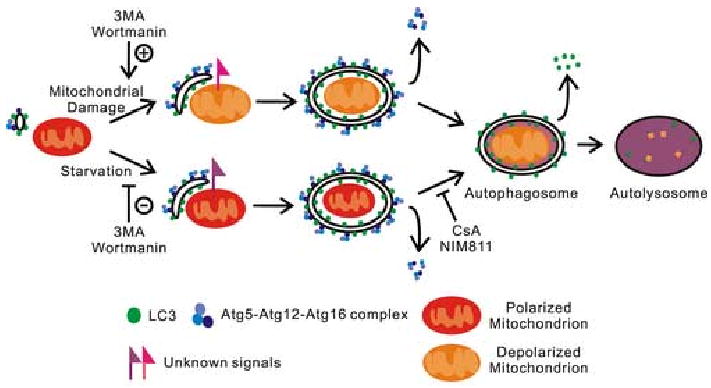
Atg12-Atg5-Atg16 and LC3 complexes localize to isolation membranes. In nutrient deprivation (starvation), isolation membranes target individual mitochondria by unknown signals in a process inhibited by the PI3K inhibitors, 3-methyladenine (3MA) and wortmannin. Isolation membranes completely envelop individual mitochondria to form double membrane vesicles (autophagosomes). After this sequestration, mitochondria depolarize in a CsA and NIM811 sensitive fashion, and Atg12-Atg5/Atg16 complexes are released from the autophagosomal surface. Autophagosomes then acidify and fuse with lysosomal vesicles to form autolysosomes. Lysosomal hydrolases digest the inner autophagosomal membrane and degrade LC3 trapped inside autophagosomes. Remaining LC3 on the surface of autophagosomes is released. After mitochondrial damage, mitochondria first depolarize and then are recognized and sequestered by isolation membranes recognizing unknown markers on the damaged mitochondria. 3MA and wortmannin do not inhibit this process but actually seem to augment it. In both pathways, sequestered mitochondria are completely digested and their molecular components recycled to the cytoplasm.
As first characterized in yeast, a machinery of genetically conserved autophagy-related proteins regulate and participate in autophagy [16]. These Atg proteins are grouped into different categories depending on their function, including 1) a pair of novel ubiquitin-like protein conjugating systems, the ATG12 and Atg8 systems, which produce vesicle extension and completion, 2) a Class III phosphatidyl choline-3-kinase (PI3K) complex which functions in vesicle nucleation, and 3) a serine-threonine kinase complex (Atg1, Atg13 and Atg17) involved in autophagic induction in yeast. The mammalian homologue of Atg 1 is recently identified as ULK1 (Unc-51-like kinase) [17].
During sequestration and formation of autophagosomes, an Atg12-Atg5 complex binds to Atg16, which translocates to the isolation membrane and functions as a linker involved in formation and elongation of the phagophore (Figure 1). An E1-like enzyme, Atg7, activates Atg12, which is transferred Atg10, an E2-like enzyme, and conjugated to Atg5 to form an autophagosomal precursor [16;18].
LC3 is a mammalian autophagosomal ortholog of yeast Atg8. In mammalian cells, newly synthesized ProLC3 is processed to its cytosolic form, LC3-I. Like Atg12, LC3-I is activated by Atg7, but is instead transferred to Atg3, a second E2-like enzyme, which cleaves 22 amino acids from the C-terminus to form LC3-II. Conjugation with a phospholipid (phosphatidyl ethanolamine in yeast) creates membrane-bound LC3-II [19;20]. LC3-II localizes selectively to forming and newly formed autophagosomes, making LC3-II a useful autophagosomal marker. Some LC3-II becomes entrapped on the inner surfaces of the double membrane autophagosomes. After fusion with lysosomes, this LC3-II is degraded. Surface LC3-II also disappears, most likely by breakdown of the phospholipid conjugate (Figure 1). Recently, a transgenic mouse strain was created that expresses a green fluorescent protein (GFP)-LC3-II fusion protein. In cells and tissues of this mouse, GFP fluorescence selectively identifies the membranes of forming and newly formed autophagosomes [21].
Molecular Control of Autophagy
Phosphoinositide 3-kinases (PI3Ks) phosphorylate phosphatidylinositol at position 3 of the inositol ring and play an important role in the regulation of autophagy [22]. PI3K inhibitors, such as 3-methyladenine, wortmannin, and LY294002, potently block autophagy. However, different classes of PI3K exert opposing effects on autophagy: Class III PI3K promotes sequestration of autophagic vacuoles, whereas class I PI3K inhibits autophagy. Class III PI3K/p150 associates with Beclin1, a mammalian homologue of Atg6 discovered by yeast two hybrid screening for its interaction with Bcl-2. Recruitment of PI3K-Beclin1 complexes together with Atg12-Atg5 is an initial step in autophagosome formation [23]. Mammalian target of rapamycin (mTor) is a kinase downstream of Class I PI3K whose activation suppresses autophagy. Rapamycin, which inhibits mTOR, induces autophagy apparently by activating protein phosphatase 2A (PP2A) [24]. PP2A also dephosphorylates proapoptotic BH3 only Bcl-2 family proteins, such as Bad and Bcl-2, that associate with mitochondria membranes [25] [26]. Thus, a functional relationship between autophagic proteins and mitochondrial proteins may exist.
Heterotrimeric guanine nucleotide-binding proteins are also involved in autophagy. Nonhydrolyzable GTP analogs, such as GTPγS, inhibit autophagy [27;28].
The Gα-interacting protein (GAIP) elicits autophagic sequestration by accelerating GTP hydrolysis and activating Gαi3. In addition, Rab24, Rab22, and Rab7, small GTP binding proteins that regulate vesicular transport, participate in processing of late autophagosomes [29;30]
Selective Autophagy
Whether autophagy is selective or non-selective has been controversial. Cytosolic enzymes with different half-lifes are sequestered at similar rates during autophagy, and autophagosomes often contain a variety of different cytoplasmic elements, including cytosolic proteins and organelles such as ER, peroxisomes and mitochondria [31;32]. Such findings led to the assumption that autophagy is a non-specific form of lysosomal degradation. However, more recent findings indicate that autophagy can be a selective process. The presence of peroxin 14 on peroxisomes is required for autophagic degradation of peroxisomes in yeast, a process now called pexophagy in recognition of its selectivity [33]. Some pathogens selectively regulate autophagy in mammalian cells for their survival. Shigella flexneri produces the protein, IscB, which inhibits the binding of bacterial VirG to Atg5, which would otherwise induce autophagy [34]. In this way, shigella escapes recognition for autophagic sequestration and elimination. In addition during the postnatal period, glycogen is selectively sequestered into autophagosomes to enhance glycolytic substrate generation after interruption of transplacental nutrition [35] [36]. Autophagosomes formed postnatally contain large amounts of glycogen and rarely contain mitochondria or other organelles [37].
Increasing evidence indicates that autophagy of mitochondria also occurs selectively, and the term mitophagy has been suggested for this selective mitochondrial autophagy [38]. For example in yeast, an outer membrane protein, Uth1p, is required for efficient mitochondrial autophagy, but a corresponding mammalian protein is yet to be identified [39]. Our work, reviewed below, also strongly indicates that autophagy can show selectivity for mitochondria.
Characteristics and Possible Structure of Mitochondrial Permeability Transition Pores
Recent evidence suggests a possible involvement of the MPT in autophagy. In the MPT, opening of PT pores causes mitochondria to become permeable to all solutes up to a molecular mass of about 1500 Da, an event leading to mitochondrial depolarization and activation of the mitochondrial ATPase (ATP synthase operating in reverse) [40-43]. After the MPT, mitochondria undergo large amplitude swelling driven by colloid osmotic forces, which culminates in rupture of the outer membrane and release of proapoptotic mitochondrial intermembrane proteins into the cytosol, including cytochrome c, apoptosis inducing factor, Smac/Diablo and others. The immunosuppressant compound, cyclosporin A (CsA), and various of its analogs inhibit the MPT through interaction with cyclophilin D (CypD) [44;45].
In one model, PT pores are composed of the voltage dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocator (ANT) in the inner membrane and CypD in the matrix space (Figure 2A) [45-48]). Other proteins, such as creatine kinase (intermembrane space), hexokinase (outer membrane) and Bax (outer membrane), are also proposed to contribute to the composition of PT pores. However, the MPT still occurs in cells types like hepatocytes that lack creatine kinase and hexokinase and in ANT-deficient mitochondria isolated from conditional double ANT knockout mice [49]. Most recently, CypD knockout mice have been developed, and mitochondria from these mitochondria still display an MPT, but the MPT observed is insensitive to CsA and requires higher concentrations of calcium for induction [50].
Figure 2. Models of the Permeability Transition Pore.
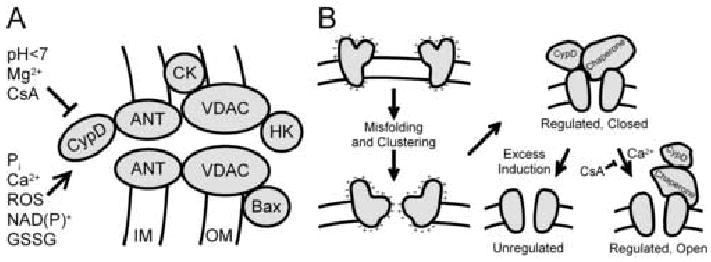
In one model (A), the PT pore is composed of ANT from the inner membrane (IM), CypD from the matrix, VDAC from the outer membrane (OM) and other proteins, including hexokinase (HK), creatine kinase (CK) and Bax, a proapoptotic Bcl2 family member. Ca2+, inorganic phosphate (Pi), ROS and oxidized pyridine nucleotides NAD(P)+ and glutathione (GSSG) promote PT pore opening, whereas CsA, Mg2+ and pH less than 7 inhibit opening. In an alternative model (B), PT pores form from misfolding and aggregation of damaged mitochondrial membrane proteins at hydrophilic surfaces facing the hydrophobic membrane bilayer. CypD and other chaperones bind to the nascent PT pores and block conductance of solutes through the aqueous channels formed by the protein clusters. High Ca2+ opens these regulated channels acting through CypD, an effect blocked by CsA. As misfolded protein clusters exceeds the number of chaperones available to regulate them, constitutively open unregulated channels form that are not inhibited by CsA.
An alternative model of the MPT has been proposed that accounts for these observations (Figure 2B) [51]. This model postulates that PT pores form as a consequence of misfolding of integral membrane proteins caused by ROS, reactive chemicals and other stresses. Becauses misfolding exposes hydrophilic surfaces to the hydrophobic membrane bilayer, the proteins aggregate at these hydrophilic surfaces to enclose channels that conduct all aqueous solutes smaller in size than the channel diameter. Since such permeabilizatoin would be catastrophic to mitochondrial function, chaperones have evolved, including cyclophilin D, that block conductance through these nascent channels. Other chaperones remain to be identified, although indirect evidence suggests that the small heat shock protein, Hsp25/27, and the Rieske iron sulfur protein may be involved [52;53]. When matrix calcium rises to high levels, PT pores open to induce the MPT, an effect mediated by CypD and blocked by CsA. When formation of nascent PT pores from misfolded protein aggregates exceeds the number of chaperones that can regulate and close these pores, an unregulated MPT occurs that is CsA-insensitive and calcium-independent. This change from a regulated to an unregulated PT pore occurs as the time and strength of MPT induction increases. On a molar basis, ANT is the most abundant inner membrane protein and thus is often a target of stresses causing protein misfolding. However, other proteins can also misfold, which explains MPT onset in ANT knockout mice. Since CypD participates in calcium sensing, the model also explains the greater requirement for calcium for the MPT in CypD deficient mitochondria. Lastly, the model explains why completely exogenous pore-forming peptides like mastoparan and alamethicin induce a CsA-sensitive and calcium-dependent MPT at low concentrations but CsA-insensitive and calcium-independent mitochondrial swelling at higher concentrations [51;54]. At low concentration, chaperones recognize the pore-forming peptides as misfolded protein aggregates and block their conductance, but as the chaperone supply becomes exhausted conductance can no longer be blocked, and mitochondrial swelling, depolarization and uncoupling ensue.
Mitophagy Induced by Nutrient Deprivation
A role of the MPT in mitophagy is implicated in cultured hepatocytes during nutrient deprivation. Autophagic stimulation of rat hepatocytes by serum deprivation and glucagon (a hormone released to the liver during fasting) increases the rate of spontaneous depolarization of mitochondria by 5-fold to about 1% of mitochondria per hour (Figure 3) [55]. These depolarized mitochondria move into acidic vacuoles, which also increase in number after nutrient deprivation. The acidic structures containing mitochondrial remnants are autophagosomes and autolysosomes, and serial imaging reveals an average mitochondrial digestion time of about 7 min after autophagic sequestration [56]. CsA, the MPT blocker, suppresses both mitochondrial depolarization during nutrient deprivation and the proliferation of autophagosomes and autolysosomes. Tacrolimus, an immunosuppressant that does not block the MPT, does not block autophagosomal proliferation, whereas NIM811, a CsA analog and MPT inhibitor that is not immunosuppressive, does block [55;56].
Figure 3. Mitochondrial Depolarization in Hepatocytes after Nutrient Deprivation.
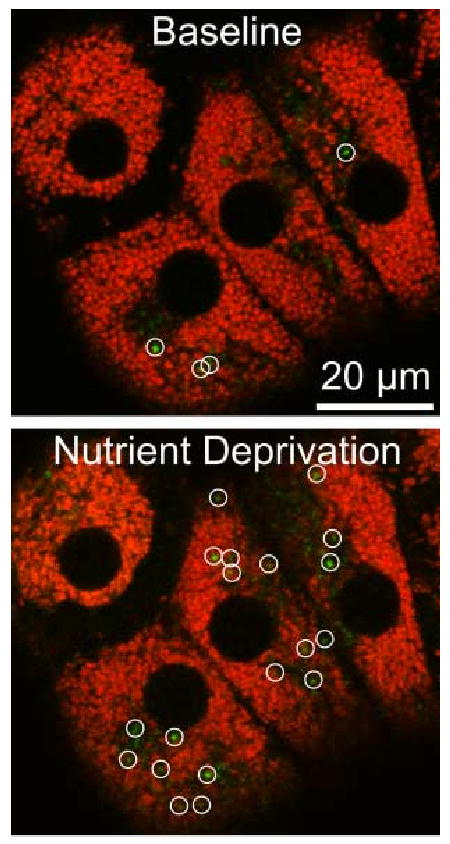
Cultured rat hepatocytes were loaded with MitoTracker Green and TMRM and imaged by confocal microscopy before (Baseline) and 60 min after (Nutrient Deprivation) changing from complete growth medium to a modified Krebs-Ringer buffer containing glucagon. Green-fluorescing structures (circles) in the overlay images are newly depolarized mitochondria.
Time-lapse confocal imaging of hepatocytes isolated from transgenic mice expressing GFP fused with LC3 [21] allows direct visualization of the progression of nutrient deprivation-induced mitophagy. In nutritionally replete culture medium, GFP-LC3 fluorescence is diffuse except for small (0.2-0.3 μm) dotted structures distributed randomly throughout the cytoplasm (Figure 4). After imposition of nutrient deprivation, such green dotted structures appear in close association with mitochondria and then form crescent structures surrounding mitochondria. These phagophores or pre-autophagosomes go on to sequester completely the individual mitochondria (Figure 4). Sequestration occurs in about 6 min from the first appearance of dots. In some instances, only a portion of an individual mitochondria becomes sequestered, which indicates that mitochondrial fission occurs coordinately with autophagosome formation. Partial mitochondrial sequestration can occur from both the ends and middle parts of mitochondria [57].
Figure 4. Mitophagy During Nutrient Deprivation in Hepatocytes from Transgenic GFP-LC3 Mice.
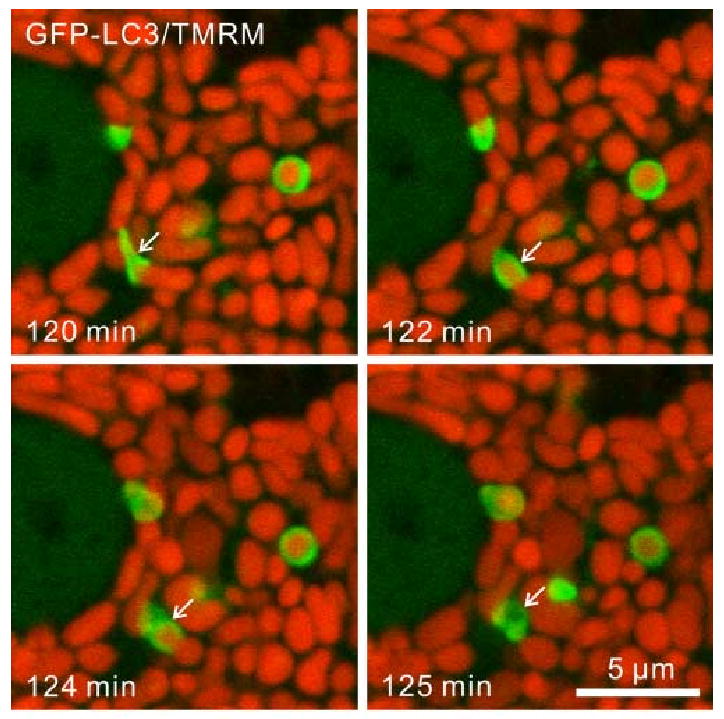
Hepatocytes were loaded with TMRM to monitor mitochondrial membrane potential and subjected to nutrient deprivation in modified Krebs-Ringer buffer containing glucagon. In the time-lapse confocal images, note green-fluorescing GFP-LC3 enveloping and then sequestering a red-fluorescing mitochondrion (arrows). After sequestration, the mitochondrion loses its red fluorescence, indicating depolarization.
After ring closure, mitochondria depolarize in about 10 min, as revealed by the release of the potential-indicating fluorophore, tetramethylrhodamine methylester (TMRM) (Figure 4). As mitochondria inside autophagosomes depolarize, acidification occurs as revealed by uptake of LysoTracker Red (LTR). After vesicle acidification, GFP-LC3 fluorescence is released or degraded. Typically, fusion with lysosomal precursors occurs at about this time, leading to formation of autolysosomes. Preliminary data indicates that CsA does not block sequestration and formation of GFP-LC3-labeled autophagosomes during nutrient deprivation-induced mitophagy. Rather, CsA may block mitochondrial depolarization and acidification occurring after sequestration. Taken together, these results show that LC3-containing membranes sequester polarized mitochondria during nutrient deprivation-induced mitophagy. Mitochondrial depolarization apparently linked to the MPT then follows completion of autophagosome formation. With acidification and fusion with lysosomal precursors the autophagosomes become autolysosomes [55-57].
Mitophagy after Photodamage
Autophagic processes have long been proposed to remove damaged and dysfunctional mitochondria. Direct experimental confirmation of a role of autophagy in removing damaged mitochondria comes in experiments in which selected mitochondria inside living hepatocytes are subjected to laser-induced photodamage [58]. When portions of GFP-LC3-expressing hepatocyes cells containing 5 to 10 mitochondria are exposed to a pulse of 488-nm light from an argon laser, mitochondrial depolarization occurs in a light dose-dependent fashion, as documented by release of TMRM (Figure 5). At lower doses of light, mitochondria depolarize transiently but subsequently recover TMRM fluorescence within a few minutes. After a greater exposure, mitochondrial depolarization becomes irreversible. After irreversible but not reversible photodamage, green GFP-LC3 fluorescence begins to envelop and completely encircle depolarized mitochondria after about 30 min (Figure 5). This photodamage-induced mitophagy is not dependent on TMRM as a photosensitizer, since photodamage also induces mitophagy in the absence of TMRM. After formation of these autophagosomes, or more specifically mitophagosomes, acidification occurs, as shown by uptake of LTR.
Figure 5. Photodamage-induced Mitophagy.
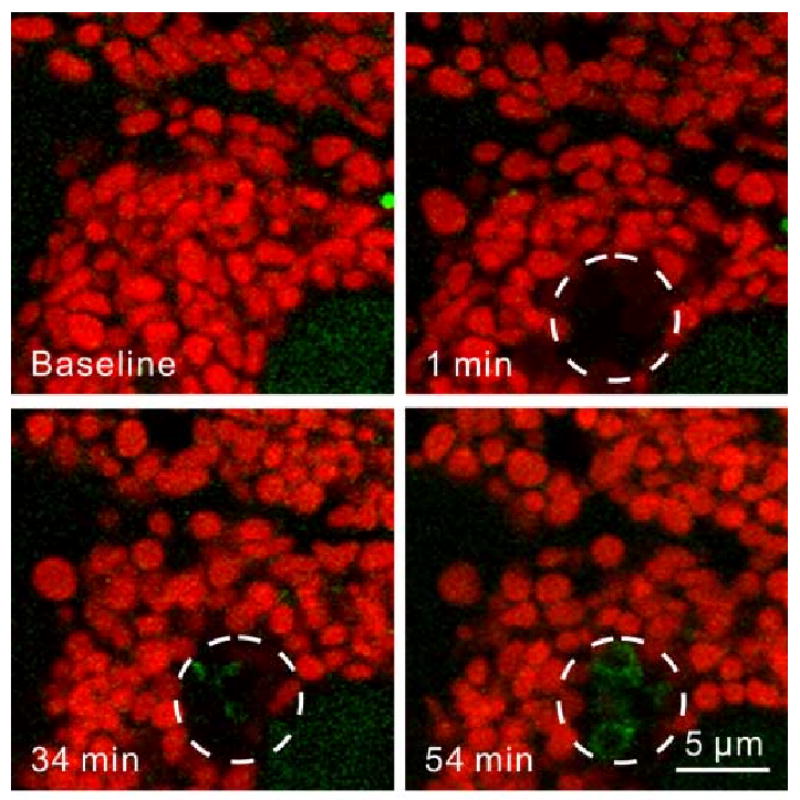
GFP-LC3 transgenic hepatocytes were loaded with TMRM (Baseline), and 5 to 10 mitochondria were exposed to a pulse of 488-nm laser light at full power (circles). Mitochondria lost red TMRM within a minute, indicating mitochondrial depolarization. After 30 min, green GFP-LC3 fluorescence localized to the region where mitochondria were damaged. After 55 min, the photodamaged mitochondria were sequestered into GFP-LC3-labeled autophagosomes, which indicated that the damaged mitochondria were selectively targeted for mitophagy.
Unlike autophagy induced by nutrient deprivation, photodamage-induced mitophagy is not blocked by PI3K inhibition with 3-methyladenine (10 mM) or wortmannin (100 nM) [58]. Rather, PI3K inhibition appears to augment GFP-LC3 association with photodamaged mitochondria. These findings suggests that light activates mitophagy downstream of PI3K signaling. The results again suggest an involvement of the MPT in autophagy, since previous work shows that photodynamic damage to mitocondria is likely mediated by MPT onset [59;60]. However, photostimulation of mitophagy appears to bypass the classical upstream PI3K signaling pathways involved in nutrient deprivation-induced autophagy (Figure 1).
Mitophagy and Cell Death
Controversy exists as to whether autophagy promotes or prevents cell death [12;61;62]. If autophagy removes damaged mitochondria that would otherwise activate caspases and apoptosis, then autophagy should be protective. In agreement, disruption of autophagic processing and/or lysosomal function promotes caspase-dependent cell death [62;62;63]. However, excessive and dysregulated autophagy may promote cell death, since enzymes leaking from lysosomes/autolysosomes, such as cathepsins and other hydrolases, can initiate mitochondrial permeabilization, caspase activation and apoptosis, and in certain instances deletion of autophagy genes decreases apoptosis [63]. Indeed, autophagy is often a prominent feature of programmed cell death to the extent that autophagic cell death has been proposed as a distinct mode of cell death [62;64].
The MPT provides a common pathway leading to mitophagy, apoptosis and necrosis (Figure 6). With low intensity stresses, limited MPT onset may only increase mitophagy to rid cells of damaged mitochondria as a repair mechanism. With increasing stress, mitophagy may no longer contain proapoptotic factors being released from mitochondria undergoing the MPT, in which case apoptosis begins to occur. Additionally, an overburdened autophagic apparatus may release lysosomal enzymes and possibly other factors to promote cell death signaling. Lastly when extreme stress causes MPT onset in virtually all cellular mitochondria, ATP levels plummet. Because of bioenergetic failure, neither autophagy nor apoptosis can progress, and only necrotic cell death ensues. The progression from mitophagic repair to apoptosis and then to necrosis after increasing MPT onset has been termed necrapoptosis (Figure 6). Interventions that modulate the extent of MPT onset after stresses therefore affect the relative amount of autophagy, apoptosis and necrosis that follows [6;65].
Figure 6. Progression of Mitophagy, Apoptosis and Necrosis.
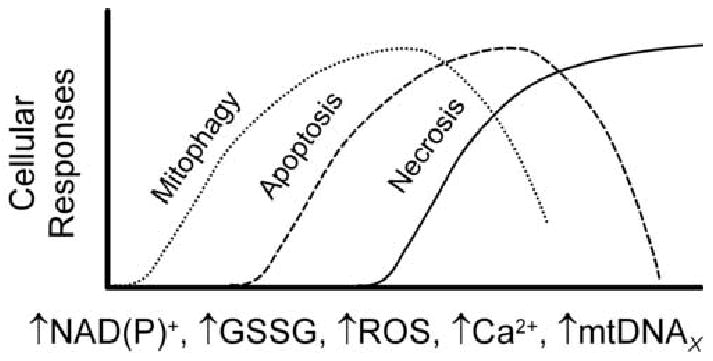
Inducers of the MPT include NAD(P)+, GSSG, ROS, Ca2+, and mutations of mtDNA (mtDNAX). As the MPT involves an increasing proportion of mitochondria, cellular responses progress from mitophagy, a repair mechanism, to apoptosis driven by release of cytochrome c and other proapoptotic factors from mitochondria, and finally to necrosis due to ATP depletion. As ATP becomes depleted, mitophagy and apoptosis become inhibited.
Mitophagy in Aging
Aging seems to affect mitochondria particularly. Because of mitochondrial ROS generation, protein damage occurs in mitochondria, and mutations of mtDNA accumulate. mtDNA is more susceptible to oxidative damage than nuclear DNA since histones are not present in mitochondria to protect mtDNA and because DNA repair mechanisms in mitochondria are less robust than in the nucleus [8;66]. Moreover, virtually all mtDNA is transcriptionally active compared to 2 or 3% of nuclear DNA, which also makes mtDNA relatively more vulnerable to damage. mtDNA mutations lead to synthesis of abnormal mitochondrial proteins or block synthesis altogether, which further exacerbates mitochondrial dysfunction. Thus in postmitotic cells of aged organisms, morphological abnormalities of mitochondria are often observed, including swelling, loss of cristae, and destruction of inner membranes [67-69]. Moreover, ATP production and respiration in mitochondria from aged animals are lower than in mitochondria from young animals.
Mitochondria of non-proliferating tissues like heart, brain, liver, and kidney constantly turnover with a half-life of 10 to 25 days. Mitochondrial biogenesis occurs by fission of pre-existing mitochondria analogously to bacterial division, but loss of mitochondria during turnover is primarily due to mitophagy [70;71]. Such mitophagy may important to the elimination of dysfunctional mitochondria and mutated mtDNA. Certain mtDNA mutations may decrease recognition signals for mitophagy and therefore accumulate with age [38;72]. For example, a mtDNA mutation causing respiratory defects leading to decreased ROS generation might make a mitochondrion less likely to be targeted for mitophagy, which would promote retention and amplification of the mutation.
Many studies show that mtDNA mutations accumulate with age at an accelerating rate, a phenomenon that may represent an age-related diminishment of autophagic activity [73]. Indeed, formation and processing of autophagosomes diminish with aging [69;74]. Another link between mitophagy and aging is Uth1p. Deletion of Uth1p leads to a selective defect in mitophagy and decreased longevity in yeast during nutrient deprivation [39;75-77]. Uth1p also confers resistance to superoxide- and H2O2-induced injuries [39]. Caloric restriction is well known to increase longevity in rodents and other animals [73;78]. Such caloric restriction is an inducer of autophagy, and increased longevity might thus be due, at least in part, to enhanced removal of oxidatively damaged mitochondria and their mutated mtDNA. These hypotheses and speculations relating mitophagy and aging need further investigation.
Conclusion
In normal physiology, cells utilize autophagy to get rid themselves of damaged, dysfunctional and superfluous cytoplasmic components to maintain cellular homeostasis and adjust to changing physiological demands. In this respect, mitochondrial degradation by autophagy (mitophagy) may play an essential role in maintaining mitochondrial functional and genetic integrity. However, there is a need for a better understanding of the regulatory pathways that control mitophagy and the specific signals and markers that target individual mitochondria for autophagic degradation. Such information will likely lead to new insights into the aging phenomena and the pathogenesis of different diseases.
Footnotes
This work was supported, in part, by Grants 2-R01 DK37034, 1 P01 DK59340 and C06 RR015455 from the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 2.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 3.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the eletron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–411. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 4.Gores GJ, Flarsheim CE, Dawson TL, Nieminen AL, Herman B, Lemasters JJ. Swelling, reductive stress, and cell death during chemical hypoxia in hepatocytes. Am J Physiol. 1989;257:C347–C354. doi: 10.1152/ajpcell.1989.257.2.C347. [DOI] [PubMed] [Google Scholar]
- 5.Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:C961–C967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- 6.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 7.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 8.Yakes FM, Van HB. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 10.Arstila AU, Trump BF. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am J Pathol. 1968;53:687–733. [PMC free article] [PubMed] [Google Scholar]
- 11.Schworer CM, Mortimore GE. Glucagon-induced autophagy and proteolysis in rat liver: mediation by selective deprivation of intracellular amino acids. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:3169–3173. doi: 10.1073/pnas.76.7.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seglen PO, Berg TO, Blankson H, Fengsrud M, Holen I, Stromhaug PE. Structural aspects of autophagy. Adv Exp Med Biol. 1996;389:103–111. doi: 10.1007/978-1-4613-0335-0_12. 103-11. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reggiori F. 1. Membrane origin for autophagy. Curr Top Dev Biol. 2006;74:1–30. doi: 10.1016/S0070-2153(06)74001-7. 1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–236. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 18.Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of ATG5*ATG16, a complex essential for autophagy. J Biol Chem. 2006 doi: 10.1074/jbc.M609876200. [DOI] [PubMed] [Google Scholar]
- 19.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med. 2006;27:411–425. doi: 10.1016/j.mam.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3- kinase--Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J. 2003;376:577–586. doi: 10.1042/BJ20030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 25.Simizu S, Tamura Y, Osada H. Dephosphorylation of Bcl-2 by protein phosphatase 2A results in apoptosis resistance. Cancer Sci. 2004;95:266–270. doi: 10.1111/j.1349-7006.2004.tb02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van HC, Goris J. Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta. 2003;1640:97–104. doi: 10.1016/s0167-4889(03)00029-6. [DOI] [PubMed] [Google Scholar]
- 27.Kadowaki M, Venerando R, Miotto G, Mortimore GE. Mechanism of autophagy in permeabilized hepatocytes: evidence for regulation by GTP binding proteins. Adv Exp Med Biol. 1996;389:113–119. 113-9. [PubMed] [Google Scholar]
- 28.Kadowaki M, Venerando R, Miotto G, Mortimore GE. Mechanism of autophagy in permeabilized hepatocytes: evidence for regulation by GTP binding proteins. Adv Exp Med Biol. 1996;389:113–119. [PubMed] [Google Scholar]
- 29.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 30.Petiot A, Pattingre S, Arico S, Meley D, Codogno P. Diversity of signaling controls of macroautophagy in mammalian cells. Cell Struct Funct. 2002;27:431–441. doi: 10.1247/csf.27.431. [DOI] [PubMed] [Google Scholar]
- 31.Arstila A, Shelburne JD, Trump BF. Studies on cellular autophagocytosis. A histochemical study on sequential alterations of mitochondria in the glucagon-induced autophagic vacuoles of rat liver. Lab Invest. 1972;27:317–323. [PubMed] [Google Scholar]
- 32.Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol. 1990;111:941–953. doi: 10.1083/jcb.111.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellu AR, Komori M, van dK I, Kiel JA, Veenhuis M. Peroxisome biogenesis and selective degradation converge at Pex14p. J Biol Chem. 2001;276:44570–44574. doi: 10.1074/jbc.M107599200. [DOI] [PubMed] [Google Scholar]
- 34.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 35.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 36.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 37.Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy in glucose homeostasis. Pathol Res Pract. 2006;202:631–638. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 39.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 40.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- 41.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 42.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 43.Forte M, Bernardi P. Genetic dissection of the permeability transition pore. J Bioenerg Biomembr. 2005;37:121–128. doi: 10.1007/s10863-005-6565-9. [DOI] [PubMed] [Google Scholar]
- 44.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 45.Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large- amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 47.Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- 48.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 49.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J BiolChem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 51.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 52.He L, Lemasters JJ. Heat shock suppresses the permeability transition in rat liver mitochondria. J Biol Chem. 2003;278:16755–16760. doi: 10.1074/jbc.M300153200. [DOI] [PubMed] [Google Scholar]
- 53.He L, Lemasters JJ. Dephosphorylation of the Rieske iron-sulfur protein after induction of the mitochondrial permeability transition. Biochem Biophys Res Commun. 2005;334:829–837. doi: 10.1016/j.bbrc.2005.06.170. [DOI] [PubMed] [Google Scholar]
- 54.Pfeiffer DR, Gudz TI, Novgorodov SA, Erdahl WL. The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J Biol Chem. 1995;270:4923–4932. doi: 10.1074/jbc.270.9.4923. [DOI] [PubMed] [Google Scholar]
- 55.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim I, Rodriguez-Enriquez S, Mizushima N, Ohsumi Y, Lemasters JJ. Autophagic degradation of mitochondria in GFP-LC3 transgenic mouse hepatocytes after nutrient deprivation. Hepatology. 2004;40(Suppl 1):291A. Abstract. [Google Scholar]
- 58.Kim I, Mizushima N, Lemasters JJ. Selective removal of damaged mitochondria by autophagy (mitophagy) Hepatology. 2006;44(Suppl 1):241A. Abstract. [Google Scholar]
- 59.Lam M, Oleinick NL, Nieminen AL. Photodynamic therapy-induced apoptosis in epidermoid carcinoma cells. Reactive oxygen species and mitochondrial inner membrane permeabilization. J Biol Chem. 2001;276:47379–47386. doi: 10.1074/jbc.M107678200. [DOI] [PubMed] [Google Scholar]
- 60.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 61.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 62.Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- 63.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12 2:1528–1534. doi: 10.1038/sj.cdd.4401777. 1528-34. [DOI] [PubMed] [Google Scholar]
- 65.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 66.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 67.Ermini M. Ageing changes in mammalian skeletal muscle: biochemical studies. Gerontology. 1976;22:301–316. doi: 10.1159/000212145. [DOI] [PubMed] [Google Scholar]
- 68.Beregi E, Regius O, Huttl T, Gobl Z. Age-related changes in the skeletal muscle cells. Z Gerontol. 1988;21:83–86. [PubMed] [Google Scholar]
- 69.Terman A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology. 1995;41 2:319–326. doi: 10.1159/000213753. 319-26. [DOI] [PubMed] [Google Scholar]
- 70.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- 71.Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. 289-333. [DOI] [PubMed] [Google Scholar]
- 72.de Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. BioEssays. 1997;19:161–166. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 73.Bergamini E. Autophagy: a cell repair mechanism that retards ageing and age- associated diseases and can be intensified pharmacologically. Mol Aspects Med. 2006;27:403–410. doi: 10.1016/j.mam.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 74.Terman A. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology. 1995;41 2:319–326. doi: 10.1159/000213753. [DOI] [PubMed] [Google Scholar]
- 75.Camougrand N, Kissova I, Velours G, Manon S. Uth1p: a yeast mitochondrial protein at the crossroads of stress, degradation and cell death. FEMS Yeast Res. 2004;5:133–140. doi: 10.1016/j.femsyr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 76.Kennedy BK, Guarente L. Genetic analysis of aging in Saccharomyces cerevisiae. Trends Genet. 1996;12:355–359. [PubMed] [Google Scholar]
- 77.Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–496. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- 78.Bergamini E, Cavallini G, Donati A, Gori Z. The anti-ageing effects of caloric restriction may involve stimulation of macroautophagy and lysosomal degradation, and can be intensified pharmacologically. Biomed Pharmacother. 2003;57:203–208. doi: 10.1016/s0753-3322(03)00048-9. [DOI] [PubMed] [Google Scholar]