

Abstract
Aim/hypothesis
We measured serum C-peptide in many individuals with chronic type 1 diabetes (T1D) and sought factors that might differentiate those with detectable C-peptide from those without it. Finding no differences, and in view of such subjects’ persistent anti-beta cell autoimmunity, we speculated that immunosuppression (to weaken the autoimmune attack) and euglycemia accompanying transplant-based treatments for type 1 diabetes (T1D) might promote recovery of T1D subject’s native pancreatic beta cell function.
Methods
We performed arginine stimulation tests in 3 islet transplant and 4 whole pancreas transplant recipients, and measured stimulated C-peptide in select venous sampling sites. We differentiated insulin secreted from the individual’s native pancreatic beta cells and from allografted beta cells based upon each sampling site’s C-peptide concentration and kinetics.
Results
Selective venous sampling demonstrated that despite long-standing T1D, all 7 beta cell allograft recipients displayed evidence that their native pancreas was a source for secreted C-peptide. Even so, if chronic immunosuppression coupled with near normal glycemia improved native pancreatic C-peptide production, the magnitude of the effect was quite small.
Conclusions/interpretation
While some native pancreatic beta cell function persists even years after disease onset in most with T1D, if prolonged euglycemia plus anti-rejection immunosuppressive therapy promotes improved the subjects’ native pancreatic insulin production, the effect is small. We may have underestimated pancreatic regenerative capacity by studying only a limited subject number, or by creating conditions (e.g. high circulating insulin concentrations, or immunosuppressive agents toxic to beta cells) that impair beta cell function.
Keywords: Type 1 diabetes, beta cell regeneration, islet transplantation, pancreas transplantation, C-peptide
INTRODUCTION
Many basic and clinical studies support the hypothesis that pancreatic beta cells either survive long term or that the pancreas may display an under-appreciated capacity for beta cell functional recovery in subjects with type 1 diabetes (T1D). For instance, several studies have reported that a significant subset of people with T1D continue to produce C-peptide for years after their diagnosis (1–7). Other observations supporting persistent beta-cell survival years after T1D onset include: (A) older autopsy studies reporting anti-insulin antibody staining pancreatic cells (8–12), (B) the high frequency (approaching 1%) of HLA-restricted anti-insulin specific T cells found in pancreas draining lymph nodes (i.e. the frequency of such anti-beta cell specific T cells is much lower at other sites (13)) which suggests a higher insulin antigen concentration at that pancreatic lymph node site (14), and (C) enduring anti-islet immune reactivity (anti-islet, anti-glutamic acid decarboxylase (GAD), and/or anti- insulinoma antigen 2 (IA2) antibodies) (15–17). The persistence of anti-beta cell specific auto-antibodies is notable since subjects with other autoimmune diseases (e.g. thyroid) typically lose their anti-tissue specific antibodies after the target tissue is removed (18). Collectively, these studies suggest that beta cells remain in the pancreas years after T1D onset despite continued evidence for an ongoing anti-beta cell immune response. Newer studies have expanded upon this earlier work reporting that a proportion of pancreatic beta cells undergo apoptosis, or co-stain for markers of cell division (Ki67), suggesting a regular slow rate of beta cell loss and replacement(19,20). Last, published and ongoing studies by several groups suggest that beta cell mass is regulated throughout life in humans, with diabetes perhaps representing an imbalance or failure of normal compensatory mechanisms (20–23).
Rodent studies that allow for more rigorous controls have shown that beta cell apoptosis and regeneration occur throughout life in mice and rats (24–28). Other rodent studies suggest that controlling the anti-islet immune response can restore euglycemia in rodents with autoimmune diabetes (29–32), and that beta cell proliferation is at least partially responsible for the effect (33).
Based upon these data, we asked whether ameliorating T1D-associated metabolic abnormalities and the anti-beta cell autoimmunity might promote a measurable improvement in native pancreatic beta cell function in subjects with the disease. Indeed, spontaneous recovery from apparent T1D has been suggested (34). Further, Japanese investigators reported biopsy data from a single pancreas allograft recipient that suggested transplant-associated immunosuppression and euglycemia may have promoted “beta-like” cells to recover within the subject’s native pancreas (35). In the present study, we therefore tested the hypothesis that chronic treatment of both autoimmunity and hyperglycemia might support the functional recovery of native pancreatic beta-like cells. We measured native pancreatic C-peptide production in humans who have undergone beta cell replacement therapy either in the form of islet or whole pancreas transplantation. In either instance, the patient’s native pancreas is left in place when the allogeneic beta cells are transplanted. We discriminated between native pancreatic- and transplant-associated C-peptide production using a novel “selective arginine stimulation” (SAS) technique that takes advantage of the physical distance between the islet or whole pancreas allograft and the recipient’s native pancreas.
METHODS
Subject Enrollment
All subjects interested in participating in the NIH clinical islet transplantation protocol underwent C-peptide testing performed either at the NIH Clinical Center Chemistry Laboratory or in other Clinical Laboratory Improvement Amendments (CLIA) approved referring hospitals or commercial laboratories. Subjects who had undergone islet transplantation at the NIH between December 2000 and June 2001 were recruited to undergo venous sampling. Details of the islet transplant protocol and description of outcomes have been reported previously (36).
Subjects with whole organ allografts were recruited from major medical institutions that perform pancreas transplantation. To be eligible, subjects had a clinical history consistent with T1D prior to their transplant, were 18 years of age or older, at least 4 years earlier had received a pancreas allograft with systemic (as opposed to portal) venous drainage, had no rejection episodes for at least one year prior to this study using a stable immunosuppressive regimen, and currently had insulin independent euglycemia. The allograft’s venous anastomosis to either the inferior vena cava or the iliac vein (rather than the superior mesenteric vein, which drains via the portal vein to the liver) was critical since it allowed cannulation from the femoral vein without transhepatic puncture, and it kept the pancreas allograft’s venous effluent clearly distinct from the native pancreas venous outflow. We limited our whole pancreas allograft recipient study to subjects who had their transplants for four years or more, reasoning that native pancreatic beta cell regeneration may be a slow process.
All protocols were approved by the NIDDK Institutional Review Board and all procedures were performed after obtaining written informed consent. This research was supported by the Intramural Research Program of the NIH, NIDDK and registered at www.clinicaltrials.gov (NCT00006505 and NCT00246844).
Subject Evaluation and Metabolic Testing
NIH islet transplant recipients (NCT00006505) received close clinical follow-up including frequent monitoring of immunosuppressant levels and every-three-month arginine stimulation tests prior to and through their enrollment in this venous sampling protocol (NCT00246844).
Whole pancreas recipients, who had received their allografts outside the NIH, were admitted to the NIH Clinical Center for a complete history, physical examination, basic laboratory evaluation, a standard 2-hour/75 gram oral glucose tolerance test, and standard peripheral arginine stimulation testing. Normal glucose tolerance was a prerequisite to proceed with the sampling study.
C-Peptide Assays and Routine Arginine Stimulation
At the NIH, C-peptide concentrations were determined using one of two competitive chemiluminescence assays.
When subjects were evaluated for transplant candidacy in 2000 and 2001, the Abbot Axsym assay was employed. Its lower limit of detection was 0.167 nmol/L. The normal range for C-peptide in healthy individuals was 0.30– 1.33 nmol/L, with an intra-assay precision of 3.4% at 1.45 nmol/L and an inter-assay precision of 7.7% and 8.3% at 0.373 nmol/L and 1.98 nmol/L respectively.
For the selective arginine stimulation test C-peptide measurements, a more sensitive assay was available (Roche Elecsys reagents). The lower limit of detection was 0.0033 nmol/L, the intra-assay precision was 1.3% and 2.6% at 0.105 and 0.0067 nmol/L respectively with an inter-assay precision of 3.1% and 2.4% at 0.104 nmol/L and 0.007 nmol/L respectively.
Routine arginine stimulation tests were performed after an overnight fast infusing 5 g of arginine hydrochloride 10% intravenously. Peripheral venous blood samples were drawn at time points: −0, 0, 2, 3, 4, 5, 7, and 10 minutes relative to the arginine injection (37).
Selective Arginine Stimulation (SAS)
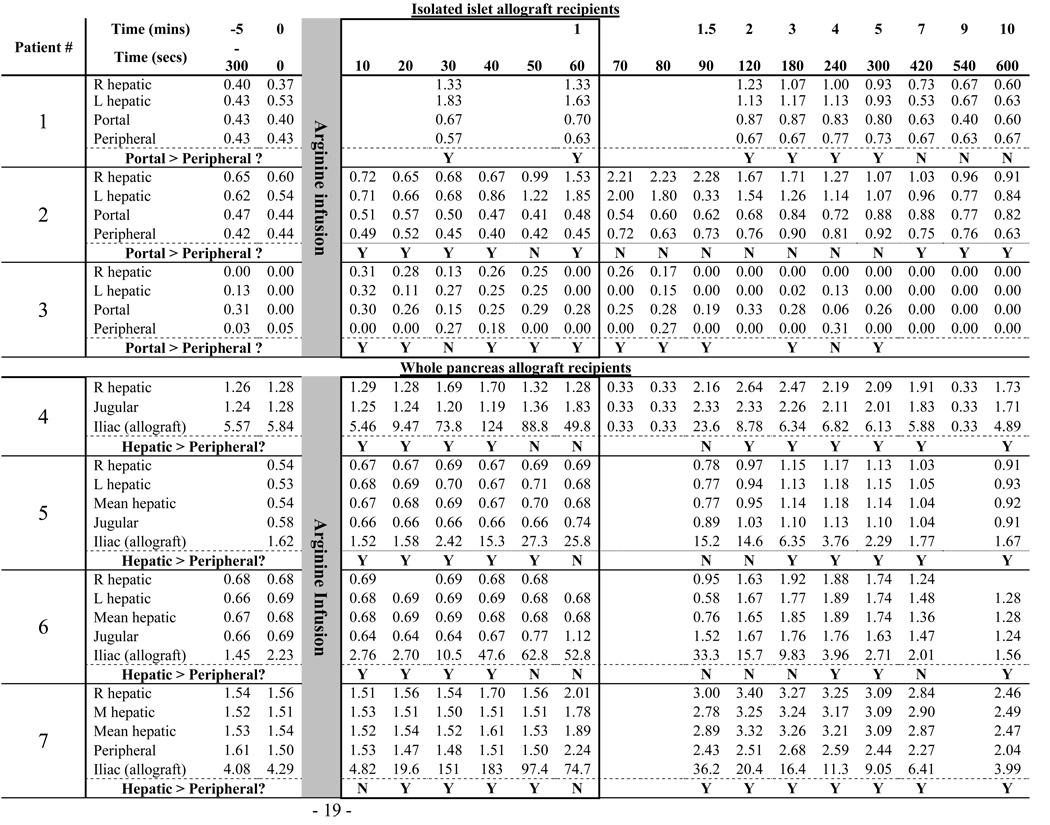
Unlike the standard peripheral arginine stimulation test that detects the individuals’ C-peptide response, the selective arginine stimulation (SAS) test discriminates between the C-peptide produced by the native pancreas as opposed to that produced by the transplanted allogeneic beta cells. The SAS technique is based on the facts that transplanted islets are placed in the recipient’s liver and whole pancreas allografts are placed in the pelvis; i.e. both are distant from the diabetic subject’s native pancreas which is not removed or altered during the transplant procedure. Thus, sampling blood immediately downstream from the different locations (liver, transplanted beta cells, and the native pancreas) after an arginine infusion identified the source of C-peptide secretion. (Figure 1). Assuming that arginine simultaneously reached the native pancreas and the allograft (islets or pancreas), we drew concurrent, sequential blood samples from the different venous sites (Figure 1A and B). For islet transplant recipients, the data interpretation was based on differences in C-peptide concentrations: any C-peptide measured in the portal vein (over the simultaneously sampled peripheral vein site concentration) represented native pancreatic C-peptide production; while any C-peptide in the hepatic vein (over the simultaneously sampled portal vein concentration) represented islet allograft production (Figure 1C). For whole pancreas transplant recipients, data interpretation was based on C-peptide kinetics such that, early after the arginine infusion, any C-peptide concentration changes appearing in hepatic vein samples relative to simultaneously sampled peripheral vein samples represented native pancreas hormone production. We reasoned that any C-peptide produced by the transplanted pancreas would first have to travel through the systemic circulation before it could be detected, after that circulation time, in the cannula positioned in the hepatic vein.
Figure 1.

Location of venous cannulas used for SAS and interpretation with transplanted beta cells represented by dots. In the whole pancreas transplant subjects, cannulas were placed in the hepatic, iliac, and jugular veins. Arginine stimulates both pancreata simultaneously. (A) Any C-peptide produced by the native pancreas is detected in the hepatic veins (yellow stars) before the C-peptide from the transplanted organ re-circulates (which will be detected by the jugular cannula). (B) In the case of islet transplant subjects, C-peptide found in the portal vein (green star) can be compared to the peripheral vein. Any early increase in the portal vein can be attributed to native pancreatic C-peptide production. (C) A mathematical representation to interpret the various sampling sites ([CP] = C-Peptide concentration).
All subjects were admitted to the NIH clinical center for SAS. After an overnight fast with intravenous fluids, subjects were taken to the interventional radiology suite and given conscious sedation with fentanyl and midazolam. Ultrasound and fluoroscopic guidance was employed to place catheters with multiple side-holes deep in the right and left hepatic veins (when possible) and the left jugular vein. The hepatic veins were percutaneously accessed from the right and left jugular veins; the catheter’s deep placement prevented any contamination from the inferior vena cava where C-peptide from the pancreas allograft would appear.
Gaining access to the veins just downstream from the allograft differed for whole pancreas and isolated islet allograft recipients. For isolated islet transplant recipients, a transhepatic catheter was placed through the liver parenchyma into the portal vein. For pancreas transplant subjects, a catheter was placed via the ipsilateral femoral vein into the allograft’s venous anastomosis.
After all cannula placements were confirmed, and with the patient remaining within the interventional radiology suite, 5g of arginine 10% was injected over 30 seconds through a peripheral vein, and blood (approximately 2 milliliters/sample) was drawn from each site simultaneously up to every ten seconds (see Table 2 for specifics relating to each subject). The catheters were not flushed between samples, and dead-space for each cannula was similar (less than 1.0 ml). After all samples were collected, cannulae were removed and hemostasis was achieved by applying local pressure. Serum was separated from the samples after centrifugation at 2000 rpm for 20 minutes at 4°C, and C-peptide concentrations were determined by the NIH Clinical Center laboratory.
 |
Data Analysis
For subjects presenting for possible enrollment in the islet transplant protocol, we evaluated various clinical variables for an association with measurable C-peptide concentrations greater than 0.167 nmol/L by using either unpaired Student’s t or Fisher’s exact tests at a significance level of p < 0.05. Bonferroni correction was applied for multiple comparisons. We determined the source of any measured C-peptide as described above (Figure 1C). For the beta cell allograft recipients, we reasoned that if the native pancreas made no C-peptide, then the C-peptide concentration in blood sampled downstream of that pancreas should be the same as that in blood sampled simultaneously from a central or peripheral venous site. In such a scenario, then C-peptide concentrations downstream from the native pancreas should be greater than simultaneously sampled peripheral vein concentrations only 50% of the time (i.e. pure chance). We applied standard binomial probability testing (akin to the probability test applied to coin tosses) to assess the probability of C-peptide concentrations downstream of the native pancreas being greater than simultaneously sampled peripheral vein concentrations.
RESULTS
C-Peptide in a general population with T1D
C-peptide values were assessed in 141 potential subjects for the islet transplant protocol. 104 measurements were performed by the NIH Clinical Center and 37 by outside centers. Consistent with previous reports, 38 of the 141 patients (27%) had detectable C-peptide concentrations ≥ 0.167 nmol/L. Of the 103 patients with C-peptide concentration ≤ 0.167 nmol/L, 51 underwent standard arginine stimulation testing. An additional 16 patients were found to have C-peptide concentrations of ≥ 0.167 nmol/L. Thus, the incidence of a positive arginine test result in patients without measurable basal levels was 16/51 (31.4%), consistent with other reports showing C-peptide responses following a glucagon-challenge in 23% of 105 patients with long-standing T1D (17). All together, at least 54 patients of the 141 individuals we evaluated (38.3%) had either basal or arginine stimulated C-peptide levels of at least 0.167 nmol/L.
We analyzed multiple clinical variables to find potential patient characteristics associated with detectable C-peptide (Table 1). Only disease duration achieved apparent statistical significance. Those with detectable C-peptide had a disease duration of 19.2 ± 11.8 years versus 26.2 ± 13.1 years for those without detectable C-peptide, (p = 0.004). However, after Bonferroni correction for the fifteen variables studied, the p-value of disease duration was not less than the corrected α value of 0.003 and can therefore not be considered statistically significant.
Table 1.
Clinical variables that were tested in subjects with C-peptide production on random serum samples. Data are presented as means (standard deviation).
| Positive C-Peptide |
Negative C-Peptide |
||
|---|---|---|---|
| Number of Subjects | 38 | 103 | |
| Gender (Female) | 24 | 59 | p=0.57 |
| Body Mass Index | 25.0 (3.78) | 24.5 (3.35) | p=0.41 |
| Age of diabetes onset | 16.7 (10.6) | 16.7 (9.90) | p=0.98 |
| Diabetes duration in years | 19.2 (11.8) | 26.2 (13.1) | p=0.0045 |
| Hemoglobin A1c | 8.19% (2.27) | 7.95 (1.42) | p=0.45 |
| Positive anti-GAD antibody | 61.9% | 63.9% | p=1.00 |
| LDL Cholesterol | 111 (31.6) | 112 (28.3) | p=0.89 |
| HDL Cholesterol | 66.0 (21.1) | 63.5 (17.0) | p=0.51 |
| Triglycerides | 93.4 (56.8) | 81.5 (51.9) | p=0.29 |
| Current insulin pump use | 39.5% | 37.9% | p=0.85 |
| DKA Episodes (lifetime) | 3.59 (6.79) | 3.72 (11.4) | p=0.95 |
| Blood glucose for symptoms of hypoglycemia | 54.7 (17.4) | 51.9 (15.9) | p=0.41 |
| Nephropathy | 50.0% | 40.2% | p=0.34 |
| Neuropathy | 42.1% | 35.3% | p=0.56 |
| Retinopathy | 57.9% | 57.8% | p=1.00 |
We next sought to assess whether any of the measured circulating C-peptide was secreted from the patient’s pancreas since non-pancreatic insulin synthesis has been reported (38–40)
SAS in islet allograft recipients
Three subjects with successful islet transplantation clinical outcomes (i.e. insulin independent euglycemia for at least one year following the procedures) agreed to undergo selective arginine stimulation. Of note, none of the three had measurable peripheral vein C-peptide concentrations (basal or stimulated) of at least 0.167 nmol/L prior to receiving their isolated islet allograft. Patients #1 and 2 were both insulin independent at the time of the SAS procedure. Both were more than 2 years after their last islet allograft infusion and as per protocol, were maintained on their immunosuppressive medications (tacrolimus and sirolimus). Patient 3 was studied several months after discontinuing all immunosuppressive agents due to immunosuppressive agent related complications (41), and at the time of the SAS procedure had returned to her pre-transplant injected insulin requirements.
C-peptide concentrations measured in the portal vein of the first insulin independent subject were consistently higher than those simultaneously sampled from the subject’s peripheral vein site during the first 5 minutes after the arginine injection (Table 2, and Figure 2A). The second insulin independent islet recipient (Subject #2) showed no obvious difference (Table 2 and Figure 2B) in the C-peptide concentrations measured in the portal and peripheral vein samples. Even so, we noted that 5 of the 6 C-peptide concentrations measured in the subject’s portal vein were slightly higher than simultaneously measured peripheral vein concentrations. The probability of that happening by chance, using binomial probabilities, is p= 0.094 suggesting some potential native pancreatic C-peptide production. The third islet transplant recipient, studied 11 months after discontinuing all immunosuppression (Subject #3)(41), also displayed a consistent “step-up” in portal vein compared to simultaneously sampled peripheral vein C-peptide concentrations, though the C-peptide concentrations were very low (Table 2). Again, while the magnitude of the step-up in subject #3 was extremely small, it was persistent in 10 of 12 samples taken from 10 – 300 seconds post-arginine injection (binomial probabilities p=0.016). We reasoned from these data that at least 2 and perhaps all 3 islet allograft recipients displayed evidence of native pancreatic C-peptide production. We therefore sought to study successful pancreas allograft recipients to ask if longer-term (at least 4 years) immunosuppression and euglycemia might promote clinically significant native pancreatic insulin production.
Figure 2.

Stimulated C-peptide responses in two insulin indepent recipients of isolated islet allografts. (A) Subject #1 displayed a significant “step-up” in portal vein C-peptide concentrations (the area shaded gray represents the excess portal vein C-peptide concentration relative to simultaneously sampled peripheral vein samples), (B) while subject #2 had nearly identical C-peptide concentrations at all time points.
SAS in whole pancreas allograft recipients
All four whole pancreas allograft recipients had also received a concurrent kidney allograft due to T1D-associated kidney failure (Table 3). All were diagnosed with T1D during childhood, and were continuously treated with insulin from the time of their diagnosis until their pancreas transplant. Unfortunately, their C-peptide secretion had not been tested prior to the simultaneous pancreas-kidney transplantation. Post transplant, all subjects had insulin-independent euglycemia, as documented by normal HgA1c values. Immunosuppression to prevent allograft rejection consisted of chronic calcineurin inhibitor use (tacrolimus or cyclosporine) in conjunction with either mycophenolate mofetil or low dose glucocorticoid.
Table 3.
| Subject | Gender | Allograft Type |
Age | Age of Diabetes Onset (years) |
Time since Transplant (years) |
Insulin Independence1 |
BMI (kg/m2) |
Arginine Stimulation Index2 |
HLA | GAD65 Antibody |
Immunosuppression3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | Islet | 42 | 30 | 2.42 | Yes | 20.1 | 1.80 | A01,02 B07,13 Cw6,7 DR04,07 DQ02,08 |
Positive | Sirolimus 3 mg QD Tacrolimus 1.5 mg daily |
| 2 | F | Islet | 52 | 13 | 2.17 | Yes | 23.7 | 1.37 | A02,68 B08,40 Cw 03,07 DR 03,13 DQ 02,06 |
Positive | Sirolimus 4 mg QD Tacrolimus 3.5 mg daily |
| 3 | F | Islet | 57 | 7 | 2.75 | No | 20.9 | N/A | A02,68 B08,60 Cw 03,07 DR13,17 DQ 06,02 |
Positive | None |
| 4 | M | Pancreas | 51 | 1.5 | 4.8 | Yes | 32.8 | 1.52 | A01,02 B08,15 Cw03,07 DR 03,04 DQ 02,03 |
Negative | Tacrolimus 1mg BID MMF 500mg BID Prednisone 5mg QD |
| 5 | F | Pancreas | 44 | 13 | 8 | Yes | 22.9 | 2.25 | A24,26 B27,55 Cw01,03 DR01,04 DQ03,05 |
Positive | Tacrolimus 3mg BID |
| 6 | F | Pancreas | 50 | 15 | 10 | Yes | 28.3 | 3.14 | A02,11 B1402,15 Cw03,08 DR01,04 DQ03,05 |
Negative | Cyclosporine 125mg BID MMF 750mg BID |
| 7 | F | Pancreas | 58 | 15 | 10 | Yes | 28.4 | 1.88 | A24,32 B35,39 Cw02,07 DR04,07 DQ02,03 |
Negative | Cyclosporine 125mg BID MMF 500mg BID Prednisone 5mg QD |
Demographics of islet and pancreas transplant recipients.
Insulin independence was at the time of sampling.
Arginine Stimulation Index was defined as the ratio of the peak C-peptide over the average baseline (−10 and 0 min).
QD = once daily, BID = twice daily, MMF = Mycophenolate Mofetil.
Each pancreas allograft recipient displayed normal glucose tolerance (data not shown) and robust arginine stimulation indices consistent with the strong allograft C-peptide signal during SAS (Figure 3). As shown in Table 2 and Figure 3, the allograft venous samples (measured in the iliac vein) showed rapid C-peptide secretion within 20–30 seconds after initiating the arginine infusion with peak concentrations (ranging from 27.3 to 183 nmol/L) reached within 50 seconds. Peripheral vein C-peptide concentrations (measured in the jugular vein) increased approximately 60 – 90 seconds after arginine infusion, and the concentrations were much lower (owing to dilution in the peripheral circulation). Therefore, we interpreted any C-peptide measured in the hepatic veins (that drain the native pancreas) within the first 60 seconds after arginine infusion to reflect hormone produced in the native pancreas (see Figure 1).
Figure 3.

C-peptide results from all sampling sites in pancreas allograft subjects showing that the vast majority of circulating C-peptide originated from the allograft
Because of a small left liver lobe with no easily accessible left hepatic vein (a normal anatomical variation), for subject #4 (Table 2, and Figure 3A and Figure 4A) we were able to measure only the right hepatic vein C-peptide concentrations. Allograft-produced C-peptide became detectable in the jugular vein 60 seconds after the arginine infusion, then in the hepatic vein 90 seconds after the arginine infusion. However, within 30 seconds following the arginine injection, a transient C-peptide increase was measured in the hepatic vein. We interpret this result as representing native pancreatic C-peptide production (i.e. 1.27 nmol/L in the jugular vein compared with 1.70 nmol/L in the hepatic vein blood).
Figure 4.

C-peptide concentration from the hepatic and peripheral sampling sites (excluding the iliac vein results shown in Figure 3 and which show C-peptide eminating from the pancreas allograft) in pancreas allograft subjects early after the arginine infusion. Each subject displays minimally elevated level C-peptide concentrations when hepatic veins samples are compared with concurrent peripheral vein blood samples.
For subjects #5, #6, and #7 (Table 2 and respectively Figure 3B–D and Figure 4B–D), cannulae were successfully placed in both right and left hepatic veins, and mean hepatic C-peptide levels were consistently elevated over peripheral levels, but only marginally so (< 7% increase in all patients between times 10 – 50 seconds). While the absolute hepatic vein C-peptide concentrations were only slightly higher than the simultaneously sampled central vein concentrations, 14 of 18 relevant time points sampled within the first minute following the arginine infusion showed higher C-peptide concentrations in the hepatic vein samples (binomial probabilities p= 0.01). Even so, the difference between the two areas under the curve (for the first 60 seconds following the arginine infusion) was only 1.05, 1.35, and 2.28 nmol·sec/ml, respectively for the 3 subjects. Of note, subject #5 rejected her pancreas allograft 3 months after sampling and returned to complete insulin dependence. Taking all 4 pancreas allograft recipient data into account, hepatic vein C-peptide concentrations were greater than simultaneously sampled peripheral vein concentrations in 17 of the 24 comparisons within the first 60 seconds following arginine infusion (see Table 2: binomial probability p = 0.021). Taking all 7 beta cell allograft recipient data into account (i.e. combining data from the isolated islet and whole pancreas recipients), C-peptide concentrations measured downstream of the native pancreas were greater than simultaneously sampled peripheral vein concentrations in 29 of the 38 comparisons within the first 60 seconds following arginine infusion (see Table 2: binomial probability p = 0.00059).
DISCUSSION
Diabetes therapy would be revolutionized by any intervention that promotes growth of functional beta cells in the pancreas of a patient with T1D. We thus sought to test: 1) whether insulin secretion persists in a convincing number of persons with long standing T1D and if so 2) whether modulating the autoimmunity and controlling the metabolic milieu might support beta cell functional recovery. We found that 38 of the 141 (27%) subjects with long-term T1D had random C-peptide concentrations of at least 0.167 nmol/L, and that another third (16 of 51) with random circulating C peptide concentrations less than 0.167 nmol/L could still produce C-peptide of at least 0.167 nmol/L following arginine stimulation. Overall, these data suggest that pancreatic insulin function persists in over a third of individuals with long-standing T1D, but is minimal and often detectable only after appropriate stimulation and with sensitive C-peptide assays.
The observed endogenous insulin secretion in patients with long-standing T1D is consistent with previous reports (2–7), and inspired us to look for native pancreas beta cell regeneration in islet transplant recipients. Finding evidence for some native pancreas C-peptide production following several months of euglycemia and immunosuppression in at least 2 and possibly all 3 islet allograft recipients prompted our evaluation of subjects with longer term beta cell replacement and immunosuppression. Using the SAS method, which discriminates between native and allograft C-peptide production, patients with whole pancreas allografts offered a unique opportunity to test the possibility. We hypothesized that long-term treatment for autoimmunity and beta cell failure would provide the best environment for native islet recovery. However, our results do not support a robust recovery of beta cell function in these individuals. In total, while all seven beta cell allograft recipients displayed slightly higher C-peptide concentrations in the native pancreatic venous effluent compared to simultaneously sampled peripheral vein blood, the concentration gradient was very small, we cannot state definitively that the gradient improved relative to what may have been measured pre-transplant, and we saw no clear dose-response relationship. That is, despite more than 8 years of immunosuppression and metabolic control enjoyed by whole pancreas allograft recipients 5, 6, and 7, the C-peptide produced by their native pancreas was mimimal.
While these data are provocative, we recognize several limitations. For instance, our estimates regarding the proportion of individuals with long-standing T1D with continued C-peptide production (even though the subjects met established clinical T1D diagnostic criteria) may have been biased by the subjects’ referral for research protocol participation (e.g. such patients may treat their diabetes differently). Further, our attempt to correlate clinical characteristics with detectable C-peptide may not have encompassed relevant variables during screening (e.g. HLA type), or might require a larger patient population because of a weak association. Also, several studies have reported that cells other than the pancreas can produce insulin under certain circumstances, so the C-peptide may not be a product of residual beta cells (38–40), though data presented herein suggests that at least some of that C-peptide is produced by the pancreas.
Additional caveats with regard to our allograft studies include the fact that we studied a limited subject number. Therefore, while we can conclude that native pancreas does not commonly “heal” to produce very much C-peptide following a successful beta cell allograft (and associated chronic immunosuppression and controlled metabolic parameters), occasional recovery cannot be ruled out. It is also possible that patients with more advanced diabetes complications (each of the subjects we studied had long-standing T1D prior to their transplant) might have a reduced beta cell regenerative capacity. Furthermore, we looked for native pancreatic beta cell function by using arginine to stimulate insulin and C-peptide release. While arginine stimulation is known to produce a strong and rapid secretory response from normal native and allograft pancreata (42), “regenerated” or immature native pancreatic beta cells may not respond to such a stimulus (43). In addition, careful follow-up of our islet allograft recipients has documented that exogenous insulin can diminish in vivo islet secretory responses to arginine (unpublished observation). Thus, the supra-physiological circulating insulin concentrations typically found in pancreas allograft recipients, and in particular those with systemically drained pancreas allografts, may have suppressed the native pancreatic beta cell regenerative response. Another consideration is that mild hyperglycemia may be an important stimulant for beta cell regeneration or recovery. Animal studies suggest that glucose sensing through glucokinase and insulin receptor substrate – 2 may be important for compensatory beta cell hyperplasia (33,44). It is possible that good, but imperfect, glucose control can stimulate beta cell growth and account for the persistent C-peptide levels found in patients with long standing T1D. Finally, current immunosuppressive regimens (relying upon calcineurin inhibitors, anti-metabolites, and glucocorticoids) may have directly prevented native pancreatic beta cell regeneration. Of the four pancreas transplant subjects, the one with the strongest response (subject #4) also had the shortest exposure to calcineurin inhibitor (4.5 years). Animal data support this observation. For instance, recent rodent studies suggest that calcineurin/nuclear factor of activated T cells signal blockade prevents beta cell growth and function (45). Also, since the effectiveness of these immunosuppressive agents for autoimmunity is poorly understood, chronic insulitis may still be inhibiting beta cell regeneration (46,47). If so, then therapies directed towards beta cell regeneration, beta cell replacement, or beta cell repopulation will require new and less toxic immunosuppressive regimens (48).
Our present study suggests that vigorous beta cell regeneration does not occur in subjects with long-term T1D after islet or pancreas transplantation. While beta cell regeneration can not be completely ruled out, these data suggest that the process is not robust given allograft-mediated restoration of glucose metabolism and currently available immunosuppression designed to abrogate both the auto-and allo-immune processes. The C-peptide assays performed by most clinical departments employ kits that detect C-peptide levels at or near the normal range, i.e. lower limits of detection of 0.167 nmol/l while the normal range for C peptide in healthy individuals is 0.30–1.33 nmol/L. Using these standard assays, very low basal C peptide levels are often not detected and therefore any pancreatic islet regeneration restoring C peptide production to low but perhaps still significant levels will not be observed. Future studies aimed at restoring pancreas function should always use the more sensitive C-peptide assays. Therapies using other methods of immunosuppression (e.g. calcineurin inhibitor and steroid sparing), and agents to stimulate beta cell replication may yet still prove successful.
Acknowledgements
The authors would like to thank Jasline Jesson for her help in the pancreas transplant sampling protocol. We also would like to thank Patricia Swanson and Terri Wakefield for their help in protocol preparation and management, and Janet Lee, Frank Leopardi, and Pamela Brooks for their assistance in obtaining and processing of samples. This research was supported by the Intramural Research Program of the NIDDK/NIH and the Clinical Center/NIH
Abbreviations
- SAS
selective arginine stimulation
Reference List
- 1.The Diabetes Control and Complications Trial Research Group. Effect of intensive therapy on residual beta-cell function in patients with type 1 diabetes in the diabetes control and complications trial. A randomized, controlled trial. Ann Intern Med. 1998;128(7):517–523. doi: 10.7326/0003-4819-128-7-199804010-00001. [see comments] [DOI] [PubMed] [Google Scholar]
- 2.Madsbad S. Factors of importance for residual beta-cell function in type I diabetes mellitus. A review. Acta Med Scand Suppl. 1983;671:61–67. doi: 10.1111/j.0954-6820.1983.tb08549.x. [DOI] [PubMed] [Google Scholar]
- 3.Madsbad S, Faber OK, Binder C, et al. Prevalence of residual beta-cell function in insulin-dependent diabetics in relation to age at onset and duration of diabetes. Diabetes. 1978;27 Suppl 1:262–264. doi: 10.2337/diab.27.1.s262. [DOI] [PubMed] [Google Scholar]
- 4.Eff C, Faber O, Deckert T. Persistent insulin secretion, assessed by plasma C-peptide estimation in long-term juvenile diabetics with a low insulin requirement. Diabetologia. 1978;15(3):169–172. doi: 10.1007/BF00421234. [DOI] [PubMed] [Google Scholar]
- 5.Scholin A, Bjorklund L, Borg H, et al. Islet antibodies and remaining beta-cell function 8 years after diagnosis of diabetes in young adults: a prospective follow-up of the nationwide Diabetes Incidence Study in Sweden. J Intern Med. 2004;255(3):384–391. doi: 10.1046/j.1365-2796.2003.01273.x. [DOI] [PubMed] [Google Scholar]
- 6.Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26(3):832–836. doi: 10.2337/diacare.26.3.832. [DOI] [PubMed] [Google Scholar]
- 7.Madsbad S. Prevalence of residual B cell function and its metabolic consequences in Type 1 (insulin-dependent) diabetes. Diabetologia. 1983;24(3):141–147. doi: 10.1007/BF00250151. [DOI] [PubMed] [Google Scholar]
- 8.Gepts W, De Mey J. Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes. 1978;27 Suppl 1:251–261. doi: 10.2337/diab.27.1.s251. [DOI] [PubMed] [Google Scholar]
- 9.Stefan Y, Orci L, Malaisse-Lagae F, et al. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes. 1982;31(8 Pt 1):694–700. doi: 10.2337/diab.31.8.694. [DOI] [PubMed] [Google Scholar]
- 10.Lohr M, Kloppel G. Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia. 1987;30(10):757–762. doi: 10.1007/BF00275740. [DOI] [PubMed] [Google Scholar]
- 11.Foulis AK, Liddle CN, Farquharson MA, et al. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29(5):267–274. doi: 10.1007/BF00452061. [DOI] [PubMed] [Google Scholar]
- 12.Gepts W. Islet morphology in type I diabetes. Behring Inst Mitt. 1984;75:39–41. [PubMed] [Google Scholar]
- 13.Pinkse GG, Tysma OH, Bergen CA, et al. Autoreactive CD8 T cells associated with beta cell destruction in type 1 diabetes. Proc Natl Acad Sci U S A. 2005;102(51):18425–18430. doi: 10.1073/pnas.0508621102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kent SC, Chen Y, Bregoli L, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435(7039):224–228. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 15.Yokota I, Matsuda J, Naito E, et al. Comparison of GAD and ICA512/IA-2 antibodies at and after the onset of IDDM. Diabetes Care. 1998;21(1):49–52. doi: 10.2337/diacare.21.1.49. [DOI] [PubMed] [Google Scholar]
- 16.Simone E, Eisenbarth GS. Chronic autoimmunity of type I diabetes. Horm Metab Res. 1996;28(7):332–336. doi: 10.1055/s-2007-979808. [DOI] [PubMed] [Google Scholar]
- 17.Jaeger C, Allendorfer J, Hatziagelaki E, et al. Persistent GAD 65 antibodies in longstanding IDDM are not associated with residual beta-cell function, neuropathy or HLA-DR status. Horm Metab Res. 1997;29(10):510–515. doi: 10.1055/s-2007-979091. [DOI] [PubMed] [Google Scholar]
- 18.Chiovato L, Latrofa F, Braverman LE, et al. Disappearance of humoral thyroid autoimmunity after complete removal of thyroid antigens. Ann Intern Med. 2003;139(5 Pt 1):346–351. doi: 10.7326/0003-4819-139-5_part_1-200309020-00010. [DOI] [PubMed] [Google Scholar]
- 19.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008 doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meier JJ, Bhushan A, Butler AE, et al. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48(11):2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 21.Butler AE, Janson J, Bonner-Weir S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 22.Meier JJ, Lin JC, Butler AE, et al. Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia. 2006;49(8):1838–1844. doi: 10.1007/s00125-006-0308-2. [DOI] [PubMed] [Google Scholar]
- 23.Yoon KH, Ko SH, Cho JH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88(5):2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 24.Teta M, Rankin MM, Long SY, et al. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12(5):817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 25.Kushner JA. Beta-cell growth: an unusual paradigm of organogenesis that is cyclin D2/Cdk4 dependent. Cell Cycle. 2006;5(3):234–237. doi: 10.4161/cc.5.3.2399. [DOI] [PubMed] [Google Scholar]
- 26.Teta M, Long SY, Wartschow LM, et al. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54(9):2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 27.Bonner-Weir S. beta-cell turnover: its assessment and implications. Diabetes. 2001;50(Suppl 1):S20–S24. doi: 10.2337/diabetes.50.2007.s20. [DOI] [PubMed] [Google Scholar]
- 28.Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol. 2000;24(3):297–302. doi: 10.1677/jme.0.0240297. [DOI] [PubMed] [Google Scholar]
- 29.Sherry NA, Kushner JA, Glandt M, et al. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55(12):3238–3245. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- 30.Zorina TD, Subbotin VM, Bertera S, et al. Recovery of the endogenous beta cell function in the NOD model of autoimmune diabetes. Stem Cells. 2003;21(4):377–388. doi: 10.1634/stemcells.21-4-377. [DOI] [PubMed] [Google Scholar]
- 31.von Herrath MG, Wolfe T, Mohrle U, et al. Protection from type 1 diabetes in the face of high levels of activated autoaggressive lymphocytes in a viral transgenic mouse model crossed to the SV129 strain. Diabetes. 2001;50(12):2700–2708. doi: 10.2337/diabetes.50.12.2700. [DOI] [PubMed] [Google Scholar]
- 32.Ryu S, Kodama S, Ryu K, et al. Reversal of established autoimmune diabetes by restoration of endogenous beta cell function. J Clin Invest. 2001;108(1):63–72. doi: 10.1172/JCI12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pechhold K, Koczwara K, Zhu X, et al. Blood glucose levels regulate pancreatic beta-cell proliferation during experimentally-induced and spontaneous autoimmune diabetes in mice. PLoS ONE. doi: 10.1371/journal.pone.0004827. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karges B, Durinovic-Bello I, Heinze E, et al. Complete long-term recovery of beta-cell function in autoimmune type 1 diabetes after insulin treatment. Diabetes Care. 2004;27(5):1207–1208. doi: 10.2337/diacare.27.5.1207. [DOI] [PubMed] [Google Scholar]
- 35.Kuroda A, Yamasaki Y, Imagawa A. Beta-cell regeneration in a patient with type 1 diabetes mellitus who was receiving immunosuppressive therapy. Ann Intern Med. 2003;139(10):W81. doi: 10.7326/0003-4819-139-10-200311180-00030-w1. [DOI] [PubMed] [Google Scholar]
- 36.Hirshberg B, Rother KI, Digon BJ, et al. Solitary islet transplantation for type 1 diabetes mellitus using steroid sparing immunosuppression: The NIH experience. Diabetes Care. 2003;26:3288–3295. doi: 10.2337/diacare.26.12.3288. [DOI] [PubMed] [Google Scholar]
- 37.Teuscher AU, Kendall DM, Smets YF, et al. Successful islet autotransplantation in humans: functional insulin secretory reserve as an estimate of surviving islet cell mass. Diabetes. 1998;47(3):324–330. doi: 10.2337/diabetes.47.3.324. [DOI] [PubMed] [Google Scholar]
- 38.Pugliese A, Zeller M, Fernandez A, Jr., et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet. 1997;15(3):293–297. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- 39.Narendran P, Neale AM, Lee BH, et al. Proinsulin is encoded by an RNA splice variant in human blood myeloid cells. Proc Natl Acad Sci U S A. 2006;103(44):16430–16435. doi: 10.1073/pnas.0607380103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kojima H, Fujimiya M, Matsumura K, et al. Extrapancreatic insulin-producing cells in multiple organs in diabetes. Proc Natl Acad Sci U S A. 2004;101(8):2458–2463. doi: 10.1073/pnas.0308690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Digon BJ, III, Rother KI, Hirshberg B, Harlan DM. Sirolimus-induced interstitial pneumonitis in an islet transplant recipient. Diabetes Care. 2003;26(11):3191. doi: 10.2337/diacare.26.11.3191. [DOI] [PubMed] [Google Scholar]
- 42.Ganda OP, Srikanta S, Brink SJ, et al. Differential sensitivity to beta-cell secretagogues in "early," type I diabetes mellitus. Diabetes. 1984;33(6):516–521. doi: 10.2337/diab.33.6.516. [DOI] [PubMed] [Google Scholar]
- 43.D'Amour KA, Bang AG, Eliazer S, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24(11):1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 44.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117(1):246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443(7109):345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 46.Meier JJ, Ritzel RA, Maedler K, et al. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia. 2006;49(1):83–89. doi: 10.1007/s00125-005-0069-3. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C, Todorov I, Lin CL, et al. Elimination of insulitis and augmentation of islet beta cell regeneration via induction of chimerism in overtly diabetic NOD mice. Proc Natl Acad Sci U S A. 2007;104(7):2337–2342. doi: 10.1073/pnas.0611101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu EH, Siegel RM, Harlan DM, O'Shea JJ. T cell-directed therapies: lessons learned and future prospects. Nat Immunol. 2007;8(1):25–30. doi: 10.1038/ni1429. [DOI] [PubMed] [Google Scholar]