

Abstract
 Ortho-TMS benzaldehyde, an effective bifunctional linchpin for Type II Anion Relay Chemistry (ARC), permits efficient multi-component union of a variety of nucleophiles and electrophiles, including the first example of a Pd-mediated ARC Type II process. To demonstrate the utility of the Type II ARC protocol, a “proof of concept” synthetic sequence was designed and implemented for construction of a focused library of “natural product-like” compounds.
Ortho-TMS benzaldehyde, an effective bifunctional linchpin for Type II Anion Relay Chemistry (ARC), permits efficient multi-component union of a variety of nucleophiles and electrophiles, including the first example of a Pd-mediated ARC Type II process. To demonstrate the utility of the Type II ARC protocol, a “proof of concept” synthetic sequence was designed and implemented for construction of a focused library of “natural product-like” compounds.
Keywords: Anion Relay Chemistry, Multi-component Coupling, Diversity-Oriented Synthesis, Dithiane Chemistry, Ortho-TMS Benzaldehyde
Nature, with beautiful elegance, constructs natural products often in iterative fashion with both superb efficiency and exquisite stereochemical control.[1] For more than 100 years chemists have atempted to mimic the elegance of Nature with laboratory syntheses. The shortcomings of many synthetic ventures however lie in the multi-step sequences, often leading to minimum structure augmentation, in conjunction with isolation and purification at each stage, leading to inefficient material advancement. Fortunately, multi-component reactions[2] hold considerable promise of alleviating this inefficiency by orchestrating the conversion of simple starting materials, in a single (one-pot) operation, to advanced intermediates of high structural complexity without the need for multiple isolations and purifications.[3] Towards this end, we recently turned to the development of multi-component reaction sequences exploiting Anion Relay Chemistry (ARC).[4]
At the highest level, the ARC tactic can be divided into two processes involving anion migration either through bonds or though space, the latter requiring the availability of a carrier to transport the negative charge. A simple example of “through-bond” anion relay is the well-known conjugate addition, through the π-system, with subsequent capture of the intermediate enolate. For “through-space” anion relay two possibilities exist involving σ-bonds (Type I and II), the difference residing in the resulting locus of the transposed anion.
Type I ARC is defined as a multi-component coupling protocol (Scheme 1), wherein a linchpin nucleophile reacts with an electrophile to generate an oxyanion that subsequently transfers (i.e., relays) the negative charge “back” to the initiating site, followed by reaction with a suitable electrophile. The Type II process, entails reaction of a nucleophile with a bifunctional electrophilic linchpin to generate an anionic species that relays the negative charge to a new (i.e., different) locus, which then reacts with a second electrophile. An important feature of the Type II ARC process is the potential for iteration with a sequential series of bifunctional linchpins, a process not disimilar to living polymerization. [5]
Scheme 1.

Type I and II Anion Relay Chemistry (ARC)
In 1997, based on the early work of Matsuda,[6] Tietze,[7] and Oshima,[8] we introduced an example of the Type I ARC process involving mono-silyl dithianes,[9] which through aegis of a solvent-controlled Brook rearrangement[10] led to the development of an effective three-component union protocol employing a wide variety of different second electrophiles. The utility of this process was subsequently demonstrated by completion of several natural product total syntheses, including (+)-spongistatins 1 and 2,[11] and more recently (-)-indolizidine 223AB and alkaloid (-)-205B, the latter two calling on an aziridine as the second electrophile.[12]
We extended the ARC concept, in 2006, to the Type II process, now employing bifunctional linchpins [cf. 1 and (-)-2], which in conjunction with solvent-controlled Brook rearrangements led to elaboration of a number of advanced intermediates utilized in the construction of a gorgonian sesquiterpene[4d] and the southern C(1)-C(25) fragment of the marine natural product spirastrellolide A (Figure 1).[4e]
Figure 1.

Type II ARC Bifunctinal Linchpins Utilized in Natural Product Total Synthesis
The versatility of the Type II ARC tactic is limited however by the number of viable, readily available bifunctional linchpins that have been successfully implemented in the multi-component tactic. Convinced of the considerable synthetic utility of the ARC tactic, we initiated a program to design, synthesize and test the viability of several new bifunctional linchpins.[4] A central design element of any sucessful bifunctional linchpin comprises the appropriate placement of an electrophilic center relative to an Anion Stabilizing Group (ASG), such that intramolecular anion transfer is both feasible and effective (Scheme 1).
Building on our earlier successful incorporation of the dithiane,[9] nitrile[4d] and allyl[13] functionalities as ASGs in bifunctional linchpins, in conjunction with the recent work of Takeda,[13] Moser,[14] and Xian[15] we reasoned that ortho-TMS benzaldehyde (3) held considerable promise as an effective linchpin for the Type II ARC process. Specifically, the early work of Moser and co-workers[14] demonstrated that strong electron withdrawing groups, like a chromium tricarbonyl moiety,are required to facilitate the Brook rearrangement in aryl systems. The precedent of Takeda and co-workers[13] further suggested that additives such as copper (I) would facilitate the silyl-transfer process.
Accordingly, we explored a variety of reaction conditions, discovering that both low temperature and a less polar solvent than THF (-78 °C and Et2O) comprise a prerequisite for success (Table 1). We also observed that when either CuI or HMPA were independently added only minor amounts of the Brook-rearranged product (5) were isolated. However, when a mixture of HMPA and THF (1:1) containing 1.2 equivalents of CuI was added at -78 °C silyl migration was complete within 30 minutes (Entry 7). Presumably the additives in conjunction with HMPA promote silyl transfer by stabilizing the resultant aryl carbanion.
Table 1.
Additive Effect on the Aryl-Silyl Migration

| Entry | Solvent | Additive | Temperature/Time | Yield (4:5) |
|---|---|---|---|---|
| 1 | THF | - | -78°C/30min | 87%:0% |
| 2 | Et2O | - | -78°/30min | 88%:0% |
| 3 | Et2O | - | 0°/30min | 88%:0% |
| 4 | Et2O | CuI(1.2eq) | -78°/30min then RT/1h | 73%:<5% |
| 5 | Et2O | HMPA:THF = (1:1) | -78°/30min then RT/1h | 46%:24% |
| 6 | Et2O | CuI(0.6 eq), HMPA:Et2O = (1:1) | -78°/30min then RT/30 min | 0%:52% |
| 7 | Et2O | CuI(1.2 eq), HMPA:THF = (1:1) | -78°/30min then RT/30 min | 0%:74% |
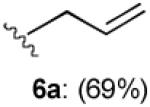
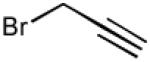
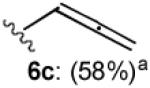
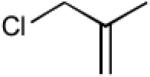
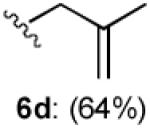
With optimized Brook rearrangement conditions in hand, we explored the versatility of linchpin 3 in the Type II ARC process with a variety of electrophiles. A series of carbon- and heteroatom-based electrophiles proved viable, generating multi-component products, 6a-g and 7 in modest to good yields (Table 2). Of particular significance, a multi-component palladium-mediated cross coupling reaction proved feasible employing n-BuLi, linchpin 3 and vinyl bromide with 3 mol% Pd(PPh3)4 to furnish 6g; the yield was 56% (Entry 8). To the best of our knowledge, this is the first report of a Pd-mediated Type II ARC process. Studies to extend this intriguing observation are ongoing in our Laboratory.
Table 2.
Three-Component Linchpin Coupling of Ortho-TMS Benzaldehyde with a Series of Electrophiles
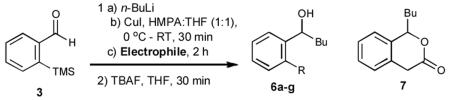
| Entry | Electrophile | R |
|---|---|---|
| 1 |
|
|
| 2 |
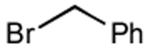
|
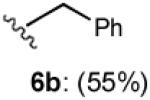
|
| 3 |
|
|
| 4 |
|
|
| 5 | PhS-SPh |
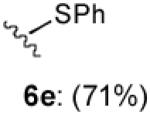
|
| 6 | Bu3SnCl |
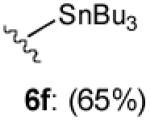
|
| 7 |
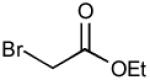
|
7: (50%)c |
| 8 |

|
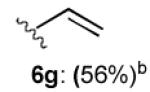
|
a) propargyl bromide, 12 h
b) vinyl bromide, 3 mol % Pd(PPh3)4, THF
2% HCI in EtOH was used to deprotect the TMS group, 1 h
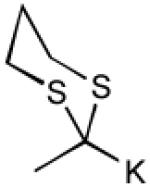
To interrogate further the versatility of linchpin 3, a series of nucleophiles were employed (Table 3). Addition of the lithium anions derived from methyl, allyl, vinyl, and phenyl halides[16] as well as 2-methyl-1,3-dithiane[17] furnished 8a-e with good efficiency (ca > 60%) after Brook rearrangement and subsequent allylation. Turning next to explore the Type II ARC process in an iterative venue, we applied the optimized Type II conditions to a four-component union sequence employing linchpins (-)-2 and 3 (Scheme 2). Pleasingly, a 63% yield of the coupled product (9) was obtained albeit, not surprising, with poor diastereoselectivity (1.25:1) at the carbanol site. Removal of the TMS-group with methanolic potassium carbonate[18] followed by chromatographic seperation of the resulting diastereomers furnished alcohols (-)-10a and (+)-10b with an overall yield of 59% from 2-methyl-1,3-dithiane. Single crystal X-ray analysis defined the relative and absolute stereogenicity of (-)-10a, the latter via the anomalous dispersion technique. With both diastereomers of 10 secured, we advanced each isomer towards a “proof of concept” sequence for construction of “natural product-like” libraries.
Table 3.
Three-Component Linchpin Coupling of Ortho-TMS

| Entry | Nucleophile | Product |
|---|---|---|
| 1 | MeLi |
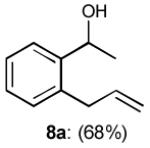
|
| 2 |

|
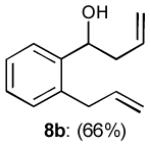
|
| 3 |

|
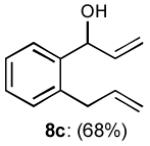
|
| 4 | PhLi |
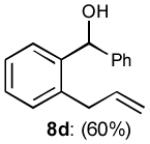
|
| 5 |
|
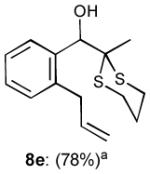
|
a) t-BuLi, KOt-Bu, -78 °C, 30 min,
Scheme 2.

Optimized Four-Component Linchpin Union Implementing Ortho-TMS Benzaldehyde
Union of both alcohols (10a,b) with 4-pentenoic acid, followed by ring-closing metathesis (RCM), implementing the recent conditions of Grubbs and co-workers,[19] provided macrolides (-)-12a and (+)-12b in high yield (Scheme 3). It should be noted that a wide variety of commerically available acids or alkyl halides hold the promise of a diverse series of macrolides and cyclic ethers, respectively. Removal of both dithiane moieties employing the Corey-Erickson protocol (NCS and AgNO3 in MeCN:H2O)[20] led to diketones (-)-13a and (+)-13b in 65 and 67% yields, which in turn were subjected to acid-mediated removal of the silyl groups[14] to provide the epimeric “natural product-like” targets. From the perspective of diversity-oriented synthesis, macrolactones (-)-14a and (+)-14b were constructed employing linchpins (-)-2 and 3 in the Type II ARC process in 13% and 12% overall yield, respectively. Currently, we are reducing this “proof of concept” sequence to practice by exploiting a variety of nucleophiles, electrophiles, and acids with (-)-2 and 3 to create an array of new “natural product-like” chemical entities (i.e., five points of diversity) for the NIH Roadmap Program.[21]
Scheme 3.

“Proof of Concept” Library of “Natural Product-Like” Compounds Utilizing Type II ARC
In summary, we have developed and implemented ortho-TMS benzaldehyde (3) as an effective bifunctional linchpin to expand the synthetic versatility of the Type II ARC tactic. In addition, we uncovered a first of kind Pd-mediated cross coupling process applicable to the ARC tactic. Finally, the synthesis of (-)-14a and (+)-14b comprises a “proof of concept” sequence to “natural product-like” compounds.
Experimental Section
To a precooled (-78 °C) solution of 2-methyl-1,3-dithiane (1.24 g, 9.2 mmol, 1.2 equiv.) in THF (15 mL) was added KO-t-Bu (1.0 M in THF, 9.7 mL, 9.7 mmol, 1.26 equiv.) and t-BuLi (1.7 M in pentane, 5.7 mL, 9.7 mmol, 1.26 equiv.). The resulting solution was stirred at -78 °C for 30 min, and a solution of epoxide (-)-2 (2.24 g, 7.7 mmol, 1.0 equiv.) in THF (15 mL) was added. The mixture was stirred for 20 min at -78 °C and then a solution of aldehyde 3 (1.64 g, 9.2 mmol, 1.2 equiv.) in THF (15 mL) was added via cannula. After stirring for 30 min at -78 °C, the resulting solution was transferred via cannula to a mixture of CuI (142.6 mg, 0.75 mmol, 1.2 equiv.) and HMPA/THF (10 mL/10 mL) at 0 °C and then warmed to ambient temperature and stirred for 30 min. Allyl bromide (2.0 mL, 23.1 mmol, 3.0 equiv.) was next added at ambient temperature and after 1 h, the reaction was quenched with saturated aqueous NH4Cl solution (100 mL). The resulting mixture was then extracted with Et2O (100 mL × 3) and the organic layers were combined, washed with brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo. Flash chromatography on silica gel (Et2O/hexane; 1/50), provided a 1.25:1 diastereomeric mixture of 9 (3.13 g, 4.87 mmol, 63%) as pale yellow oil. Rf 0.8 (hexane/Et2O = 10/1).
At ambient temperature, a methanolic (40 mL) solution of 9 (1.94 g, 3.02 mmol, 1.0 equiv.) was added K2CO3 (4.17, 30.2 mmol, 10.0 equiv.) and stirred overnight. The reaction was diluted with H2O (100 mL) and extracted with Et2O (50 mL × 3). The organic layers were combined, washed with brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. Flash chromatography on silica gel, (Et2O/hexane; 1/10) provided two separable diastereomers (-)-10a as solid and (+)-10b as pale yellow oil (1.62 g, 2.84 mmol, 94%). Rf 0.2 (hexane/Et2O = 10/1).
(-)-10a: [α]20D -0.90 (c 1.0 CDCl3); Rf 0.2 (hexane/diethyl ether = 10/1). IR (film) 3434 (m), 2927 (s), 2855 (s), 1471 (m), 1254 (s), 1090 (s), 1028 (s), 775 (s) cm-1; 1H NMR (500 MHz, CDCl3) δ 7.78-7.76 (m, 1H), 7.25-7.22 (m, 2H), 7.17-7.16 (m, 1H), 6.04-5.96 (m, 1H), 5.29 (s, 1H), 5.09 (dd, J = 10.0 and 1.5 Hz, 1H), 5.03 (dd, J = 17.0 and 2.0 Hz, 1H), 4.59-4.55 (m, 1H), 3.81 (dd, J = 16.0 and 6.5 Hz, 1H), 3.63 (br, 1H), 3.55 (dd, J = 16.0 and 6.0 Hz, 1H), 3.12-3.06 (m, 1H), 3.03-2.94 (m, 2H), 2.85-2.62 (m, 6H), 2.44 (dd, J = 15.5 and 6.5 Hz, 1H), 2.05-1.81 (m, 6H), 1.73 (s, 3H), 0.83 (s, 9H), 0.12 (s, 3H), -0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 138.9, 137.5, 137.0, 129.6, 129.5, 128.0, 125.6, 115.9, 73.3, 68.4, 58.3, 50.7, 48.5, 46.3, 37.9, 29.5, 27.6, 27.1, 26.7, 26.2, 26.1, 25.2, 24.0, 18.1, -3.35, -3.4; high resolution mass spectrum (ES+) m/z 593.2061 [(M+Na)+; calcd for C28H46O2S4SiNa: 593.2048]. (+)-10b: [α]20D +18.6 (c 0.5 CDCl3); IR (film) 3440 (m), 2952 (s), 2856 (s), 1468 (m), 1254 (s), 1092 (s), 1026 (s), 775 (s) cm-1; 1H NMR (500 MHz, CDCl3) δ 7.75-7.74 (m, 1H), 7.26-7.20 (m, 2H), 7.18-7.17 (m, 1H), 6.04-5.96 (m, 1H), 5.26 (s, 1H), 5.09 (dd, J = 10.5 and 1.5 Hz, 1H), 5.04 (dd, J = 17.0 and 1.5 Hz, 1H), 4.60-4.56 (m, 1H), 3.92 (d, J = 3.5 Hz, 1H), 3.79 (dd, J = 16.0 and 6.5 Hz, 1H), 3.58 (dd, J = 16.0 and 6.5 Hz, 1H), 3.06-3.01 (m, 1H), 2.97-2.92 (m, 1H), 2.91-2.67 (m, 7H), 2.44 (dd, J = 15.5 and 5.0 Hz, 1H), 2.32 (dd, J = 15.5 and 6.0 Hz, 1H), 1.96 (dd, J = 8.0 and 3.5 Hz, 1H), 1.94-1.80 (m, 4H), 1.72 (s, 3H), 0.91 (s, 9H), 0.22 (s, 3H), 0.20 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 138.6, 137.5, 137.1, 129.6, 129.3, 127.8, 125.4, 116.0, 73.0, 68.9, 58.3, 49.9, 48.0, 45.0, 37.7, 29.3, 27.6, 27.0, 26.6, 26.2, 26.1, 25.1, 24.2, 18.1, -3.4; high resolution mass spectrum (ES+) m/z 593.2049 [(M+Na)+; calcd for C28H46O2S4SiNa: 593.2048].
Acknowledgments
This work was funded by grants from the National Institute of Health (GM-029028 and GM-081253) and a fellowship from the University of Pennsylvania (WMW).
References
- [1].a) Cane DE, Walsh CT, Khosla C. Science. 1998;282:63. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]; b) Walsh CT. ChemBiochem. 2002;3:125. doi: 10.1002/1439-7633(20020301)3:2/3<124::AID-CBIC124>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- [2].Dömling A, Ugi I. Angew. Chem. Intl. Ed. 2000;39:3168. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- [3].a) Bertz SH. J. Am. Chem. Soc. 1981;103:3599. [Google Scholar]; b) Bertz SH, Sommer TJ. Chem. Comm. 1997:2409. [Google Scholar]; c) Wender PA, Croatt MP, Witulski B. Tetrahedron. 2006;62:7505. and references cited therein. [Google Scholar]
- [4].a) Smith AB, III, Duffey MO. Synlett. 2004:1363. [Google Scholar]; b) Smith AB, III, Xian M. J. Am. Chem. Soc. 2006;128:66. doi: 10.1021/ja057059w. [DOI] [PubMed] [Google Scholar]; c) Smith AB, III, Xian M, Kim W-S, Kim D-S. J. Am. Chem. Soc. 2006;128:12368. doi: 10.1021/ja065033e. [DOI] [PubMed] [Google Scholar]; d) Smith AB, III, Kim D-S, Xian M. Org. Lett. 2007;9:3307. doi: 10.1021/ol071281j. [DOI] [PubMed] [Google Scholar]; e) Smith AB, III, Kim D-S, Xian M. Org. Lett. 2007;9:3311. doi: 10.1021/ol071282b. [DOI] [PubMed] [Google Scholar]
- [5].Hirao A, Nakahama S. Acta Polymerica. 1998;48:208. [Google Scholar]
- [6].Matsuda I, Murata S, Ishii Y. J. Chem. Soc. Perk. Trans. 1. 1979;1:26. [Google Scholar]
- [7].Tietze LF, Geissler H, Gewart JA, Jakobi U. Synlett. 1994:511. [Google Scholar]
- [8].Shinokubo H, Miura K, Oshima K, Utimoto K. Tetrahedron. 1996;52:503. [Google Scholar]
- [9].Smith AB, III, Boldi AM. J. Am. Chem. Soc. 1997;119:6925. [Google Scholar]
- [10].a) Brook AG. J. Am. Chem. Soc. 1958;80:1886. [Google Scholar]; b) Moser WH. Tetrahedron. 2001;57:2065. [Google Scholar]
- [11].a) Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew. Chem., Int. Ed. 2001;40:191. [PubMed] [Google Scholar]; b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew. Chem., Int. Ed. 2001;40:196. [PubMed] [Google Scholar]
- [12].a) Smith AB, III, Kim D-S. Org. Lett. 2004;6:1493. doi: 10.1021/ol049601b. [DOI] [PubMed] [Google Scholar]; b) Smith AB, III, Kim D-S. Org. Lett. 2005;7:3247. doi: 10.1021/ol0510264. [DOI] [PubMed] [Google Scholar]; c) Smith AB, III, Kim D-S. J. Org. Chem. 2006;71:2547. doi: 10.1021/jo052314g. [DOI] [PubMed] [Google Scholar]
- [13].Taguchi H, Takami K, Tsubouchi A, Takeda T. Tetrahedron Lett. 2004;45:429. [Google Scholar]
- [14].a) Moser WH, Endsley KE, Colyer JT. Org. Lett. 2000;2:717. doi: 10.1021/ol0056022. [DOI] [PubMed] [Google Scholar]; b) Moser WH, Zhang J, Lecher CS, Frazier TL, Pink M. Org. Lett. 2002;4:1981. doi: 10.1021/ol025729m. [DOI] [PubMed] [Google Scholar]
- [15].Devarie-Baez NO, Shuhler BJ, Wang H, Xian M. Org. Lett. 2007;9:4655. doi: 10.1021/ol702149c. [DOI] [PubMed] [Google Scholar]
- [16].Dunne KS, Lee SE, Gouverneur V. J. Organometallic. Chem. 2006;691:5246. [Google Scholar]
- [17].Schlosser M, Strunk S. Tetrahderon Lett. 1984;25:741. [Google Scholar]
- [18].Hurst DT, McInnes AG. Can. J. Chem. 1965;43:2004. [Google Scholar]
- [19].Grubbs RH. Tetrahedron. 2004;60:7117. [Google Scholar]; also see:Blackwell HE, O'Leary DJ, Chatterjee AK, Washenfelder RA, Bussmann DA, Grubbs RH. J. Am. Chem. Soc. 2000;21:442..
- [20].Corey EJ, Erickson BW. J. Org. Chem. 1971;36:3553. [Google Scholar]
- [21].Huryn DM, Cosford NDP. Ann. Rep. in Med. Chem. 2007;42:401. doi: 10.1016/S0065-7743(07)42026-7. also see http://nihroadmap.nih.gov/ [DOI] [PMC free article] [PubMed]