

Abstract
In most neurons postsynaptic [Ca2+]i changes result from synaptic activation opening voltage gated channels, ligand gated channels, or mobilizing Ca2+ release from intracellular stores. In addition to these changes that result directly from stimulation we found that in pyramidal cells there are spontaneous, rapid, Ca2+ release events, predominantly, but not exclusively localized at dendritic branch points. They are clearest on the main apical dendrite but also have been detected in the finer branches and in the soma. Typically they have a spatial extent at initiation of ~2 μm, a rise time of <15 ms, duration <100 ms, and amplitudes of 10–70% of that generated by a backpropagating action potential at the same location. These events are not caused by background electrical or synaptic activity. However, their rate can be increased by repetitive synaptic stimulation at moderate frequencies, mainly through metabotropic glutamate receptor mobilization of IP3. In addition, their frequency can be modulated by changes in membrane potential in the subthreshold range, predominantly by affecting Ca2+ entry through L-type channels. They resemble the elementary events (“sparks” and “puffs”) mediated by IP3 receptors and ryanodine receptors that have been described primarily in non-neuronal preparations. These spontaneous Ca2+ release events may be the fundamental units underlying some postsynaptic signaling cascades in mature neurons.
Introduction
At most synapses release of glutamate or other transmitters activates ionotropic receptors, which, either through direct opening of ligand gated channels or indirectly by subsequent opening of Ca2+ channels leads to Ca2+ entry through the plasma membrane. In addition, synaptically activated Ca2+ release from internal stores has been described in many neurons (Finch and Augustine, 1998; Takechi et al., 1998; Nakamura et al., 1999; Yeckel et al., 1999; Morikawa et al., 2000; Larkum et al., 2003; Stutzmann et al., 2003; Power and Sah, 2007). These increases follow the mobilization of IP3 by activation of Group I metabotropic receptors (mGluRs) and muscarinic receptors.
In several non-neuronal cell types [e.g., oocytes (Parker and Yao, 1996; Sun et al., 1998) and HeLa cells (Bootman et al., 1997)], in cultured PC12 cells and in some hippocampal cells in primary culture (Koizumi et al., 1999) randomly generated, small, localized, IP3-mediated Ca2+ release events (“puffs”) have been described. These puffs are usually generated experimentally by uncaging IP3 within the cell or by the application of a metabotropic agonist to the cell, although there are a few observations of spontaneous puffs (Parker and Yao, 1991). When these puffs occur close enough together in space and time they interact to generate large amplitude [Ca2+]i increases, often in the form of Ca2+ waves. The mechanisms underlying the generation of these puffs and waves in oocytes have been the subject of intense analysis (Dawson et al., 1999; Shuai et al., 2006).
A second kind of small Ca2+ release event, often called “sparks,” has been described in cardiac myocytes, some types of smooth muscle cells, and in dissociated dorsal root ganglion neurons (for review, see Cheng and Lederer, 2008). These events are thought to occur through Ca2+-induced Ca2+ release (CICR) by the activation of localized clusters of ryanodine receptors (RyRs). The frequency of these events is usually increased by membrane depolarization and by RyR agonists like caffeine, but do not generally lead to regenerative release.
Much slower spontaneous Ca2+ release events have been described in embryonic retinal ganglion cells in intact retina (Lohmann et al., 2002), hippocampal neurons in slice cultures (Lohmann et al., 2005; Lohmann and Bonhoeffer, 2008), and striatal neurons in acute slices (Osanai et al., 2006). Previously, spontaneous Ca2+ release events with the characteristics of puffs or sparks have not been observed in the dendrites of mature neurons.
Against this background we examined pyramidal cells in rat hippocampal slices with high speed imaging using high affinity Ca2+ indicators. We found that most CA1 pyramidal neurons express Ca2+ release events spontaneously at resting potential, primarily in the dendrites. They are highly localized, concentrated at branch points, much faster than events in embryonic or cultured cells, and are caused by release of Ca2+ from internal stores. They are insensitive to TTX or to blockade of spontaneous ionotropic synaptic transmission in the slice. Most interestingly, the frequency of these events can be modulated by membrane potential changes and by mGluR-mediated synaptic transmission. As with events in oocytes and HeLa cells, when the release events are induced by IP3 mobilizing mechanisms to occur at a high rate they coalesce to generate high amplitude Ca2+ waves (Nakamura et al., 1999). However, high rates of events induced by increasing [Ca2+]i or depolarization do not transition to Ca2+ waves.
Materials and Methods
Whole-cell recording.
Transverse hippocampal slices (300 μm thick) from 2- to 4-week-old Sprague Dawley rats were prepared as previously described (Nakamura et al., 1999). Animals were anesthetized with isoflurane and decapitated using procedures approved by the Institutional Animal Care and Use Committee of New York Medical College. Slices were cut in an ice-cold solution consisting of (in mm): 80 NaCl, 2.5 KCl, 0.29 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 75 sucrose, 10.1 glucose, 1.3 ascorbate, and 3 pyruvate. They were incubated for at least 1 h in solution consisting of (in mm): 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10.1 glucose, 1.3 ascorbate and 3 pyruvate, bubbled with a mixture of 95% O2–5% CO2, making the final pH 7.4. Normal ACSF (artificial CSF), composed of (in mm) 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 glucose, was used for recording.
Submerged slices were placed in a chamber mounted on a stage rigidly bolted to an air table and were viewed with a 60× water-immersion lens in an Olympus BX50WI microscope mounted on an X–Y translation stage. Somatic whole-cell recordings were made using patch pipettes pulled from 1.5 mm outer diameter thick-walled glass tubing (1511-M, Friedrich & Dimmock). Tight seals on CA1 pyramidal cell somata were made with the “blow and seal” technique using video-enhanced differential interference contrast optics to visualize the cells (Sakmann and Stuart, 1995). For most experiments the pipette solution contained (in mm): 130 potassium gluconate, 4 NaCl, 4 Mg-ATP, 0.3 Na-GTP, 7 dipotassium-phosphocreatine, and 10 Hepes, pH adjusted to 7.3 with KOH. Final osmolarity was 297 mOsm. This solution was supplemented with the high affinity Ca2+ indicator Oregon Green Bapta-1 (25–100 μm; Invitrogen). Synaptic stimulation was evoked with 200 μs pulses with glass electrodes placed on the slice ∼5–30 μm to the side of the main apical dendritic shaft and at varying distances from the soma. These electrodes were low resistance patch pipettes (<10 MΩ) filled with ACSF. Temperature in the chamber was maintained between 31 and 33°C. Caged IP3 (d-myo-inositol 1,4,5-trisphosphate, p4(5)-1-(2-nitrophenyl)ethyl ester) was purchased from EMD Biosciences. All other chemicals were obtained from Fisher Scientific or Sigma. Nifedipine and nimodipine were prepared as 10 mm stock solutions in ethanol and Bay K-8644 was prepared as a 10 mm stock solution in DMSO. These solutions were diluted to final values in ACSF at the time of the experiment.
Dynamic [Ca2+]i measurements.
For most experiments time-dependent [Ca2+]i measurements from different regions of the pyramidal neuron were made as previously described (Lasser-Ross et al., 1991; Nakamura et al., 2002). Briefly, a Photometrics Quantix cooled CCD camera, operated in the frame transfer mode, was mounted on the camera port of the microscope. Custom software (original version described by Lasser-Ross et al., 1991) controlled readout parameters and synchronization with electrical recordings. A second custom program was used to analyze and display the data. Pixels were binned in the camera to allow frame rates of 30–50 Hz. For most experiments we measured fluorescence changes of OGB-1 with single wavelength excitation (494 ± 10 nm) and emission at 536 ± 20 nm. For high speed experiments changes in fluorescence were acquired using a back-illuminated 80 × 80 pixel cooled CCD camera (NeuroCCD-SMQ; Red Shirt Imaging), controlled with Neuroplex software (Red Shirt Imaging). Images with this camera were acquired at 500 Hz. [Ca2+]i changes are expressed as ΔF/F where F is the fluorescence intensity when the cell is at rest and ΔF is the change in fluorescence during activity. Corrections were made for indicator bleaching during trials by subtracting a low-pass filtered signal measured under the same conditions when the cell was not stimulated. We did not correct for tissue autofluorescence. [Ca2+]i increases in different parts of the cell are displayed using either selected regions of interest (ROIs) or a pseudo “line scan” display (Nakamura et al., 2000).
Photolysis of caged IP3.
For some experiments caged IP3 (100 μm) was included in the patch pipette, and was allowed to diffuse throughout the cell after membrane rupture. Pulsed UV light centered at 365 nm from a light-emitting diode (UVILED, Rapp Optoelectronics) was focused through the objective via a 200 μm diameter quartz fiber optic light guide making a spot of ∼40 μm in diameter on the slice with the 60× objective lens. UV light intensity was regulated by changing the proportion of time the UV light was on when alternating the on-off cycle at 1000 Hz.
Results
Small amplitude, localized Ca2+ events in the dendrites
In previous experiments examining calcium signaling in pyramidal neurons investigators usually measured [Ca2+]i increases associated with particular electrical events like synaptic activation or action potentials. For these new experiments we first examined fluctuations in resting [Ca2+]i levels without stimulation in pyramidal neurons from the CA1 region in hippocampal slices from 2- to 4-week-old rats. In most cells (>300 examined) we observed sharp increases in Oregon-Green-BAPTA-1 (OGB-1) fluorescence at restricted places in the primary apical dendrites (Fig. 1A) and oblique dendrites and soma (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These were low frequency events, typically occurring at a rate of 1–3 events/s at any one location. Their amplitudes varied, but typical fluorescence changes (ΔF/F) were 5–35%. These were ∼10–70% of the amplitude of the fluorescence change from a single backpropagating action potential (bAP) recorded at the same locations in the same cell. Previous experiments (Helmchen et al., 1996) indicate that the amplitude of a bAP-evoked [Ca2+]i increase in the apical dendrites is ∼150 nm suggesting that typical event amplitudes were 20–100 nm. Because this kind of event previously was not observed in pyramidal neurons in slices we characterized them further.
Figure 1.
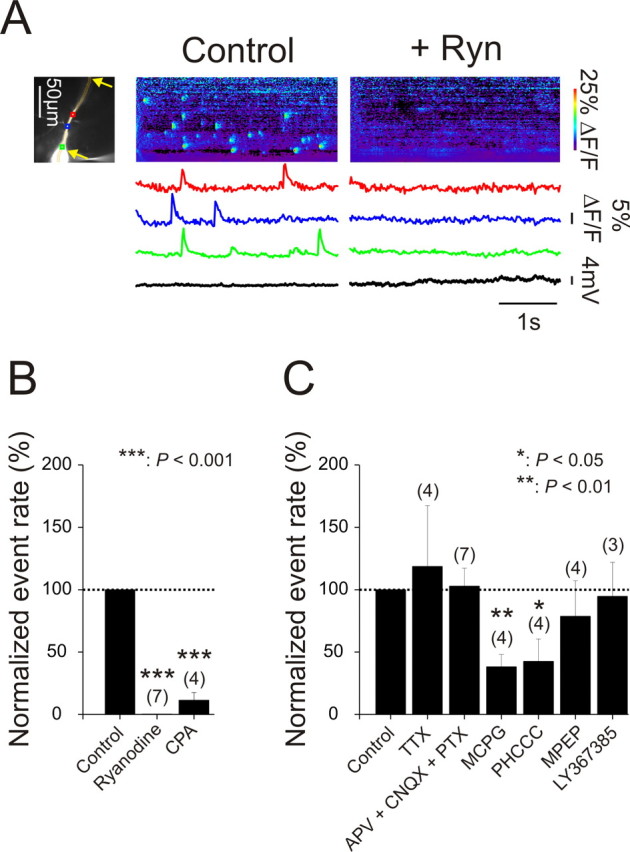
Spontaneous Ca2+ release events occur in localized regions of the dendrites of hippocampal pyramidal neurons. A, Pyramidal neuron filled with OGB-1. Three ROIs are marked. The string of wide pixels indicates the locations of the line scan images. Wide pixels were used to improve the S/N, and did not affect the locations or time courses of the Ca2+ signals compared with those recorded with narrower pixels. In normal ACSF (“Control”) spontaneous increases in [Ca2+]i were detected asynchronously at the three locations. The pseudocolor “line scan” image shows the increases at all locations along the dendrite. When 20 μm ryanodine was added to the ACSF (“+Ryn”), the spontaneous events were blocked. The voltage traces below show that there was no potential change at the times of the [Ca2+]i increases. Yellow arrows indicate limits of the dendritic region in this and other figures in which event rate was measured. B, Known blockers of Ca2+ release from internal stores strongly reduce the frequency of events. Several trials were recorded to determine the control frequency. Then the frequency was determined when ryanodine (20 μm) or CPA (20 μm) was added to the bath. C, Events do not result from background synaptic activity in the slice. Blockade of Na+ channels (by 1 μm TTX) or ionotropic synaptic transmission (by 100 μm APV, 10 μm CNQX, 100 μm picrotoxin) did not change event frequency. Some antagonists of mGluRs (MCPG, 1 mm; PHCCC, 100 μm) reduced event frequency but others (MPEP, 3 μm; LY367385, 100 μm) did not. No mGluR antagonist completely blocked events. Error bars in each column are SE and numbers of cells are shown in parentheses.
Events result from spontaneous Ca2+ release from internal stores
There was no corresponding change in membrane potential at the time of most of these elementary events. Therefore, it is likely they reflect Ca2+ release from internal stores. We tested this hypothesis by applying agents that are known to affect Ca2+ release. The frequency of spontaneous fluorescence changes were dramatically decreased by 20 μm ryanodine (0.2 ± 0.1% of control rate, N = 7 cells, p < 0.001, paired t test, Fig. 1A,B), and strongly reduced by 20 μm cyclopiazonic acid (CPA; 11.2 ± 6.2% of control rate, N = 4 cells, p < 0.001; paired t test, Fig. 1B). Ryanodine depletes Ca2+ stores by blocking the ryanodine receptor in the open state (Rousseau et al., 1987) and CPA blocks the ER Ca2+-ATPase (Seidler et al., 1989). The ryanodine experiment demonstrates that ryanodine receptors (RyRs) are active in these cells but does not prove that Ca2+ release is through these receptors since depleting the stores will prevent release by activation of IP3 receptors if both receptors are on the same compartment (Khodakhah and Armstrong, 1997). In an attempt to further characterize the pathway for release we included heparin (1–20 mg/ml) and ruthenium red (100 μm-1 mm), in the internal pipette solution in separate sets of experiments, or bath applied dantrolene (100 μm). Heparin is thought to block IP3 receptors (Ghosh et al., 1988; Kobayashi et al., 1988) and ruthenium red and dantrolene block RyRs in some preparations (Endo, 1977; Smith et al., 1985; Llano et al., 1994) although there is clear evidence that all these compounds are not specific (Ehrlich et al., 1994; Bengtson et al., 2004; MacMillan et al., 2005). Unfortunately, we could not get conclusive results in these experiments (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In earlier experiments (Nakamura et al., 1999, 2000) we found that heparin (1 mg/ml) blocked Ca2+ waves, whereas ruthenium red (100 μm) did not block Ca2+ waves, consistent with the interpretation of these waves as regenerative IP3-mediated events. But these small events may be less sensitive to these compounds or may involve a different mix of channels on the endoplasmic reticulum (ER).
Increasing intracellular IP3 enhances event generation
Evidence that these elementary events could result from activation of IP3 receptors came from experiments in which we directly evoked similar events by uncaging IP3 in the dendrites (Fig. 2A). After loading the cells with caged IP3 from the patch electrode on the soma we gave long flashes of UV light to a 40 μm diameter region of the dendrites. At moderate flash energies a large amplitude Ca2+ wave was evoked (Hong and Ross, 2007). However, when the flash energy was reduced the regenerative Ca2+ wave did not occur. Instead, smaller, graded fluorescence increases were detected with occasional sharp transients on top of these changes. At even lower energies only isolated events were detected but these occurred at a higher rate than observed in the control period before UV illumination, whereas the frequency of events in nearby unilluminated regions did not change (Fig. 2C). There were no changes in membrane potential corresponding to any of these responses. These isolated events were generated only over a narrow range of UV intensities. Similar results demonstrating events transitioning to large amplitude Ca2+ release waves following graded uncaging of IP3 previously were shown in Xenopus oocytes (Sun et al., 1998). Although these experiments show that release of IP3 alone can evoke these events they do not rule out a possible role for Ca2+ release through RyRs (see below). In oocytes it is clearer that IP3 receptors are the main Ca2+ release channels since functional RyRs are not found in those cells (Lechleiter and Clapham, 1992).
Figure 2.

Low level uncaging of IP3 or weak tetanic synaptic activity increase event rates but more intense stimulation evokes Ca2+ waves. A, [Ca2+]i changes evoked by uncaging IP3 in a localized dendritic region. Left panel shows an OGB-1 filled pyramidal neuron. The yellow circle indicates the approximate location of the uncaging flash. The red box indicates the ROI from where the fluorescence traces were taken. The wide pixels for the line scans in A and B were chosen to improve the S/N. The first panel (“High”) shows the response to a moderate intensity flash for 5 s, represented by the vertical band across the image and trace. The bright red zone at the beginning of the flash and the cutoff of the peak in the trace indicate a Ca2+ wave restricted approximately to the region of the UV flash. The true amplitude of the [Ca2+]i changes were much higher than the ranges shown for the image and trace because the Ca2+ indicator was close to saturation. Most of the artifact resulting from tissue autofluorescence caused by the UV flash was digitally subtracted. The second panel and trace (“Medium”) show the responses to a lower energy flash. There was a widespread low level increase in fluorescence punctuated by a few sharp transients. The third panel and trace (“Low”) show responses to an even lower energy flash. In this case the main response was an increase in elementary event frequency in the illuminated region. B, A similar set of responses to tetanic synaptic transmission at 20 Hz for 4 s at different stimulation currents (200 μs pulses). The position of the stimulating electrode is shown with the dotted arrow. C, Raster plots showing the time of events in the last panels of A and B. All events along the visible apical dendrite (Fig. 1A, see yellow arrows) are plotted. There was clearly an increase in event rate during the uncaging flash and during the tetanus. D, Histograms comparing event rates during unstimulated time periods and during stimulation. For both the uncaging and synaptic experiments the increase in event rate was statistically significant. Number of cells included in the samples is indicated above the bars.
These events occur in most pyramidal neurons without stimulation. Spontaneous events could result from an intrinsic mechanism in the neurons or result from external activation in ambient conditions. To examine the second possibility we first applied blockers of ionotropic glutamate receptors (APV, 100 μm and CNQX, 10 μm) and GABAA receptors (picrotoxin, 100 μm). This mixture did not affect event frequency (control: 159 events over a total of 108 s in 7 cells; APV+CNQX+picrotoxin: 291 events during 192 s in the same 7 cells; percentage with blockers/control: 103 ± 15%, Fig. 1C). TTX (1 μm) also had no effect on event rate (percentage with TTX/control: 119 ± 49%, N = 4 cells, Fig. 1C) showing that background spiking in the slice did not directly or indirectly affect event generation.
In previous experiments (Nakamura et al., 1999, 2000; Larkum et al., 2003) we found that synaptically activated Ca2+ waves were mediated through activation of group I mGluRs (mGluR1 and mGluR5) since they were blocked by some inhibitors of these receptors. To test whether ambient glutamate in the slice was activating these receptors and generating localized events we applied several different blockers. MCPG (1 mm), a group I and II mGluR antagonist, and PHCCC (100 μm), a group I mGluR antagonist, decreased event frequency compared with control periods in the same cells (percentage with blockers/control: MCPG: 38 ± 10%, N = 4 cells; p < 0.01, PHCCC: 42 ± 18%, N = 4 cells, p < 0.05, paired t test, Fig. 1C). In contrast, neither MPEP (3 μm), an mGluR5 antagonist, nor LY367385 (100 μm), an mGluR1 antagonist, had a significant effect on event frequency (percentage with blockers/control: 79 ± 29% of control, N = 4 cells and 95 ± 27% of control, N = 3 cells, respectively, Fig. 1C).
The reasons for these different results are not clear. The concentration of ambient glutamate in slices is controversial. Recent experiments by Herman and Jahr (2007) argue that the ambient glutamate concentration is ∼25 nm, but other measurements (Sah et al., 1989; Meldrum, 2000; Bandrowski et al., 2003) suggest higher values. The Ki for activation of most mGluRs is in the micromolar range or higher (Conn and Pin, 1997). If this estimate is valid for mGluRs on pyramidal neurons then we would expect these neurons to be insensitive to ambient glutamate if the lower estimate for this concentration is correct, and, therefore, the antagonists should have no effect. One possible explanation for why some antagonists reduced event frequency is that the G-proteins coupled to mGluRs have a low level of tonic activity in the absence of glutamate and some antagonists, sometimes called “inverse agonists” (Ango et al., 2001; Milligan, 2003), on binding to the receptors, reduce this tonic activity level leading to a reduction in IP3 concentration and lowering of event frequency. The important point, however, is that no mGluR antagonist eliminated events. Therefore, at least some of the events must occur spontaneously in these neurons even without ambient glutamate in the slice.
Synaptic activation of mGluRs enhances event frequency
Although ambient glutamate activation of mGluRs is not necessary to generate spontaneous events it is possible that synaptically released glutamate could increase the event rate by activating these receptors. To test this idea we gave weak trains of stimulating pulses at 20 Hz through a patch electrode containing ACSF placed in the stratum radiatum near the apical dendrite. The bath solution contained 100 μm APV, 10 μm CNQX, and 100 μm picrotoxin to block ionotropic glutamate and GABAA receptors. At moderate stimulation intensities, below the threshold for spike generation in normal ACSF (tested before applying the ionotropic blockers), the tetanus evoked a large amplitude Ca2+ wave in a restricted region of the dendrites (Fig. 2B). When the stimulation intensity was lowered we recorded a slow, nonregenerative fluorescence increase with occasional sharp transients. At even lower stimulation intensities we mostly detected event-like transients, which occurred at a higher rate than during periods without stimulation (Fig. 2D). None of these stimuli led to membrane depolarization because of the presence of ionotropic receptor blockers in the bath, suggesting that voltage dependent event activation was not involved. This pattern of response closely resembles the pattern observed when the UV flash intensity was lowered in cells filled with caged IP3 (Fig. 2A). This correlation strongly suggests that low intensity synaptic stimulation mobilizes a minimal concentration of IP3 that releases Ca2+ from disconnected event initiation sites. As with the experiments with caged IP3, we found that there was only a narrow range of stimulation intensities that increased event frequency before reaching threshold for wave generation.
Modulation of event frequency by membrane potential changes and Ca2+ entry through L-type channels
Although the generation of these events by uncaging of IP3 suggests they have properties resembling puffs, other properties more closely resemble those found in sparks, which are generated by RyR-mediated Ca2+ release (Cheng and Lederer, 2008). In particular, we found that event frequency was modulated by changes in membrane potential. Figure 3A shows a cell that had spontaneous events at rest (−63 mV) at four obvious locations. Depolarizing the soma to −55 mV (just subthreshold for spiking) with current injection increased the event rate; hyperpolarizing to −70 mV blocked most of the events; returning to −63 mV restored the basal event rate. Summarizing results from many cells (see legend for details) shows that depolarizing ∼6 mV from rest increased event frequency to 250 ± 78% of control rate (N = 7 cells, p < 0.01) and hyperpolarizing by 15 mV lowered the frequency to 12.7 ± 3.3% of control rate (N = 18 cells, p < 0.05, Fig. 3C). Transiently depolarizing the cell to subthreshold potential (Fig. 3B) immediately enhanced event frequency. This figure also makes it clear that the increase in [Ca2+]i from entry through voltage gated Ca2+ channels and the enhancement of event frequency are separate processes. The increase in [Ca2+]i from entry through Ca2+ channels occurs everywhere and synchronously, whereas events occur stochastically and at restricted locations. Together these experiments show that event frequency is rapidly and sensitively modulated by changes in potential around resting level.
Figure 3.
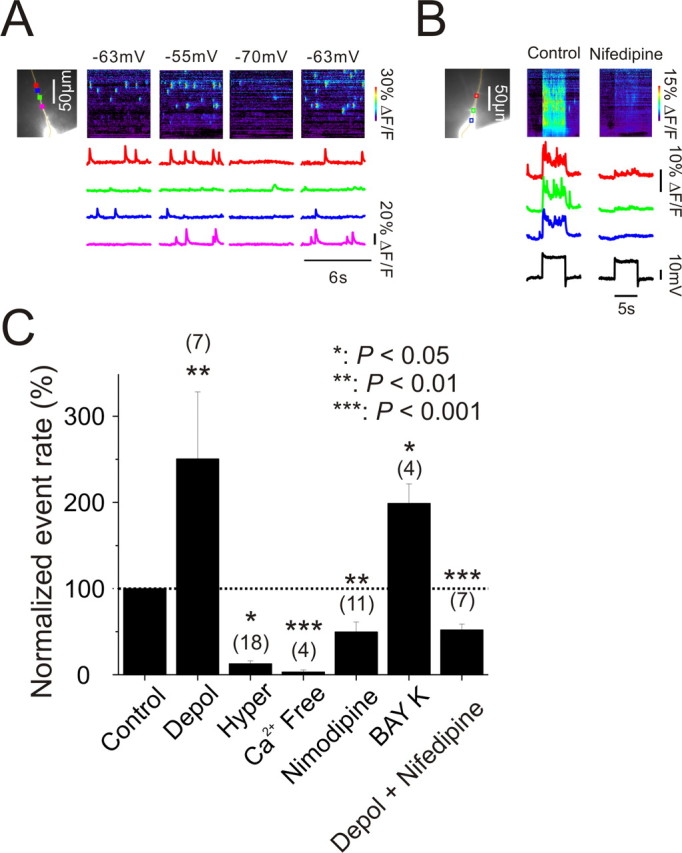
Changes in membrane potential rapidly modulate elementary event frequency by regulating [Ca2+]i by changing Ca2+ entry through L-type Ca2+ channels. A, Representative recordings showing that event rates increased when the cell was depolarized from −63 mV to −55 mV and reduced when the cell was hyperpolarized to −70 mV. The return to −63 mV restored the basal frequency. B, Transient depolarization rapidly enhanced event rate and increased [Ca2+]i at all locations. Both these effects were strongly reduced by 20 μm nifedipine. C, Average response at three potentials [Control: −60 mV ± 0.8 mV, N = 10 cells; depolarized (Depol): −54.6 ± 1.3 mV, N = 7 cells; hyperpolarized (Hyper): −75.1 ± 2.0 mV, N = 18 cells]. Depolarization increased event rate by 150%; hyperpolarization and Ca2+ free (0 mm CaCl2 and 4 mm MgCl2) almost completely eliminated the events; addition of 10 μm nimodipine reduced event rate by 50% and addition of 10 μm Bay K-8644 doubled the event rate. In the presence of nifedipine there was no effect of depolarization on the event rate.
One possible explanation of these experiments is that the event rate was affected by changes in resting [Ca2+]i levels caused by changes in tonic voltage gated Ca2+ entry. Indeed, as previously reported by Magee et al. (1996), membrane hyperpolarization lowered resting [Ca2+]i in addition to reducing event rate (data not shown). We also found that replacing normal ACSF with a solution containing 0 mm CaCl2 and 4 mm MgCl2 strongly reduced the event rate (3.2 ± 2.3% of control rate, N = 4 cells, p < 0.001, paired t test, Fig. 3C). Although we did not measure the change in [Ca2+]i directly, other groups have reported that a similar change to 0 mm CaCl2 lowers resting [Ca2+]i by ∼25% (Wanaverbecq et al., 2003).
These results suggest that Ca2+ entry through voltage gated Ca2+ channels might be the mechanism for event frequency modulation by changes in membrane potential. Magee et al. (1996) found that L-type Ca2+ channels were the main source of tonic Ca2+ entry in pyramidal neurons. Following their lead we applied nimodipine (10 μm), an L-type Ca2+ channel blocker, to the slice and found that this compound strongly reduced the resting event rate by ∼50% (50 ± 12% of control rate, N = 11 cells, p < 0.01, paired t test, Fig. 3C). Furthermore, addition of Bay K-8644 (10 μm), an L-type Ca2+ channel agonist, strongly enhanced event frequency to 199 ± 23% of control rate (N = 4 cells, p < 0.05, paired t test, Fig. 3C). These Bay K-8644-evoked events were completely blocked by ryanodine (N = 2 cells; data not shown) showing that they also resulted from Ca2+ release from stores. We also tested mibefradil (10 μm), a partial R/T channel blocker, and found it had no effect on event frequency. Together these results suggest that tonic voltage gated Ca2+ entry through L-type channels is an important regulator of event frequency.
To assess the effect of dihydropyridines on the change in event frequency caused by changes in membrane potential we measured the relative event rate at depolarized potentials in normal ACSF and with 20 μm nifedipine (a different L-type Ca2+ channel blocker) added (Fig. 3C). Since nifedipine reduced this rate by ∼50% (52 ± 7% of control rate, N = 7 cells, p < 0.001, paired t test, Fig. 3C) this implies that nifedipine almost completely blocked the depolarization induced increase in event frequency.
In the experiments in which we enhanced event frequency by membrane depolarization or by the addition of Bay K-8644 it appeared that the enhancement occurred both by increasing the rate at locations in which events were previously detected and by inducing them at locations in which they were not detected previously. We cannot be certain of this conclusion since we did not obtain control records of sufficiently long duration to be sure that events never occurred at some locations. However, this observation does suggest that there is a spectrum of sensitivities to [Ca2+]i in the regulation of event frequency.
Although these properties are consistent with an interpretation of these events as sparks we note that IP3 receptors are activated by both Ca2+ and IP3 (Iino, 1990; Bezprozvanny et al., 1991), and IP3 receptor channel open probabilities are very sensitive to changes in [Ca2+]i near resting levels (for review, see Foskett et al., 2007). Therefore, these experiments do not clearly categorize these events.
Caffeine increases elementary event frequency
Another test for spark-like properties is to see whether the events can by enhanced by direct RyR agonists like caffeine (McPherson et al., 1991). When we bath applied caffeine (5 mm) we found that the event rate was strongly enhanced after 2 min and declined to a low level after 5 min (Fig. 4). During parallel experiments with no caffeine (ACSF) the event rate declined over a similar time interval, but there was no enhancement at 2 min. We are not certain why there was a decline during the ACSF control, but a likely possibility is that we were damaging the cells with the long exposure times of this experiment. Therefore, some of the decline in the response after caffeine application may also be the result of cell damage and not store depletion. Nevertheless, the strong enhancement at 2 min is consistent with caffeine acting on RyRs to release Ca2+ from the ER in discrete packets.
Figure 4.
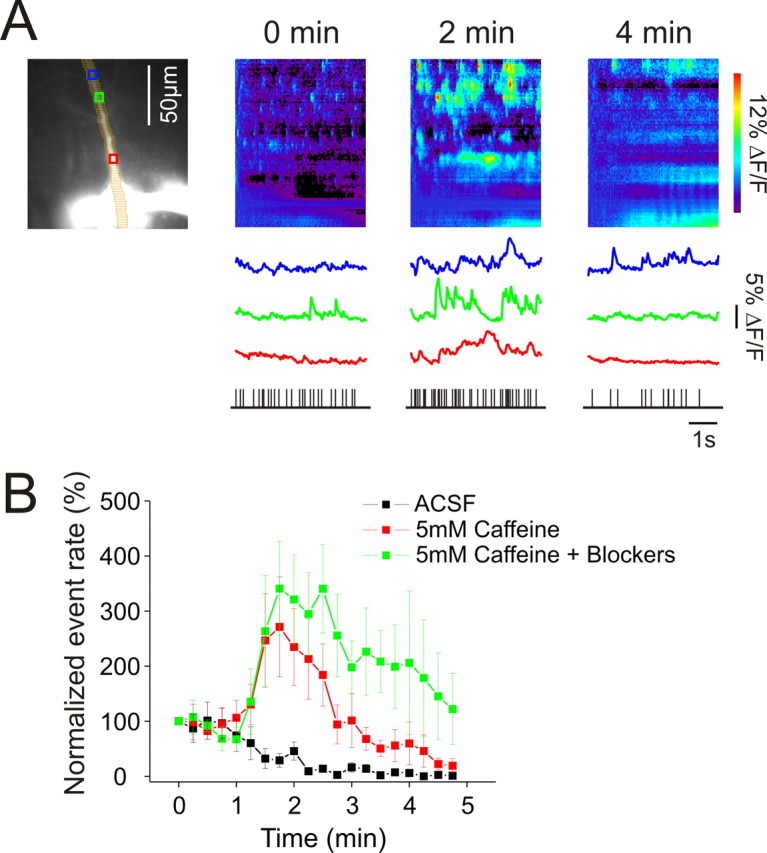
Caffeine transiently increases puff rate in dendrites. A, Left panel, Pyramidal neuron filled with 50 μm OGB-1 with pixels and ROIs marked. Starting at 0 min 5 mm caffeine began to enter the recording chamber and remained in the chamber for the course of the experiment. Fluorescence recordings of 6 s duration were taken every 15 s. Selected recordings at 0, 2, and 4 min are shown. The bottom raster plots show the times of events in the dendritic region during these recordings. B, Summary of experiments. Data from five cells were averaged for each condition. In the experiments with blockers (100 μm APV, 10 μm CNQX, 1 μm TTX, and 3 μm MPEP), these compounds were added to the bath several minutes before caffeine was added. For the caffeine experiments (with or without blockers) each cell was from a separate slice. The blockers did not prevent the increase in event rate suggesting that caffeine acted directly on the pyramidal neuron.
One weakness of this experiment is that caffeine is known to act on RyRs in presynaptic terminals leading to tonic transmitter release (Lelli et al., 2003). In our slice experiments this released glutamate could act on postsynaptic receptors, mobilizing Ca2+ release. To control for this indirect action of caffeine we repeated the experiment in slices that were preincubated with TTX (1 μm), APV (100 μm), CNQX (10 μm), and MPEP (3 μm) to block electrical activity in the slice and postsynaptic activation of ionotropic receptors and mGluR5s. As previously shown in Figure 1 these inhibitors had no effect on the spontaneous event rate. In these slices caffeine had exactly the same effect as in slices without the blockers (Fig. 4B), suggesting that caffeine was enhancing Ca2+ release events by directly acting on RyRs in the recorded pyramidal neurons.
Events are localized events that occur repeatedly at the same location, often at branch points
Because events seem to occur stochastically it is possible that their locations could also be random, unrelated to any specific molecular or morphological organization in the cell. To examine this issue more precisely we marked the center of many release events in parts of the dendrites that were in focus. We positioned small boxes (ROI, 2 μm × 2 μm) over these locations and determined the spatial parameters of these events. The first observation was that events occurred repeatedly at the same location but generally did not occur at nearby sites, even those only 4 μm away (Fig. 5A, typical of 5 analyzed cells). The second observation was that events occurred predominantly, but not exclusively at branch points (Fig. 5B) as previously observed in cultured PC12 cells (Koizumi et al., 1999). During 54 s of recordings from 6 cells 18 events occurred at branch points (within 2 μm) and 9 at locations that did not appear to be branch points. The true distribution is actually more biased toward branch points than these numbers suggest since branch points only represent a small part of the main dendrite. It is possible that the frequency at branch points may have been even higher since we could not be sure we detected all branches even though we changed focus above and below the main dendrite to look for these processes. These features suggest that there is a morphological feature, e.g., a cluster of IP3 receptors (Parker et al., 1996) or RyRs, concentrated at branch points, through which Ca2+ is preferentially released.
Figure 5.
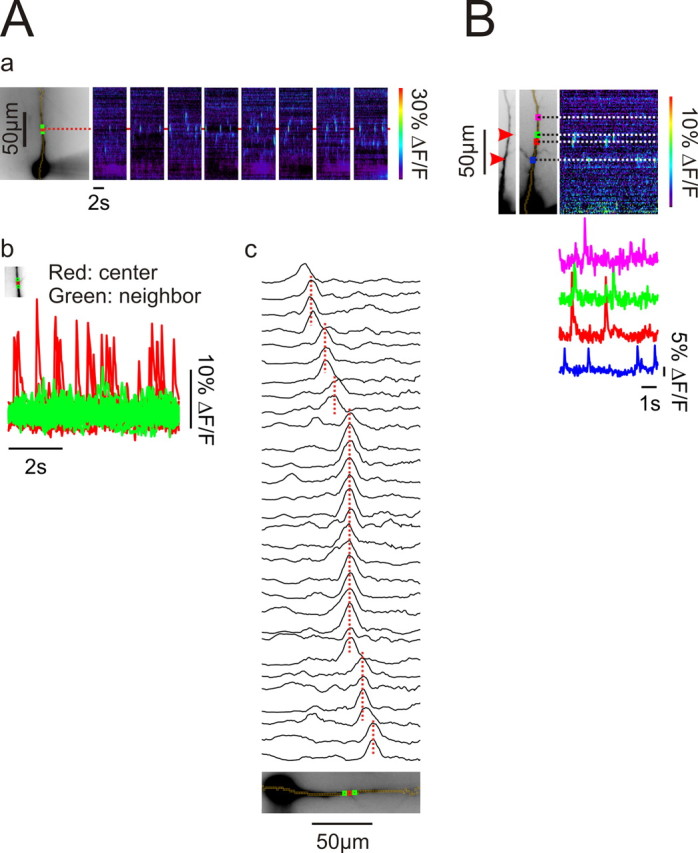
Events occur repeatedly at the same locations, primarily branch points in the dendrites. Aa, Spontaneous events detected over a series of 6 s intervals. The center of one event is marked with a red ROI and two neighboring ROIs are indicated in green. Ab, The red and green traces show the superimposed fluorescence changes at these locations over 8 intervals. The largest signals were always detected from the red ROI. In this cell the red ROI was at a branch point. Ac, Fluorescence intensity line scans along the pixel sequence at the times of the peak response for individual events. The scans have been organized to show events with the same peak location next to each other. There are many events, especially the ones highlighted in red in (Ab), that always occur at the same place. Scans were low-pass filtered in the spatial dimension. Typical result for five analyzed cells. B, The locations of the centers of several events are shown in relationship to the branching pattern of the apical dendrite. In this cell two events were clearly at branch points (red arrows). Two other events were not obviously at branch points but slight darkenings at those locations might indicate some specialization.
Spontaneous Ca2+ events are very rapid, localized events of characteristic amplitude that primarily spread by diffusion
Examination of typical events (Fig. 1) suggests that events spread over a length of 5–10 μm along the dendrite. This length could reflect the size of an IP3 receptor or RyR cluster releasing Ca2+ simultaneously along this length, or it could be a short Ca2+ wave initiated at a single location, or it could reflect Ca2+ released from a short length that then diffused further. These possibilities are not exclusive, e.g., the Ca2+ released at the edges of a short wave might diffuse further along the dendrite.
To examine these possibilities we recorded the fluorescence changes from a group of spontaneous events with a high speed camera operating at 500 Hz and compared these fluorescence changes to those caused by a single bAP. We did not analyze events evoked by depolarization, caffeine, or caged IP3. It is possible that those events have different characteristics although superficially they appear similar. The spontaneous signals were recorded at the same locations as those from bAPs, often on the same sweep. Therefore, indicator concentration and pixel location were the same for both kinds of events. Figure 6A shows typical OGB-1 fluorescence changes from a single bAP and several events at a location near a branch point. Both signals had fast rise times and fast decay times. Interestingly, the event transients seemed to recover faster than the bAP generated transients. Analysis of many events showed that the rise time (10–90%) of the fluorescence signal from events was slightly longer than from a bAP (event: 10.9 ± 1.8 ms, N = 28 events, 4 cells; bAP: 6.1 ± 1.9 ms, N = 40 events, 10 cells; p < 0.001, t test). These values were insensitive to indicator concentration in the range of 25–100 μm. The spike signal rise time was consistent with the value of 4.7 ± 0.3 ms recently measured with high speed 2-photon scanning microscopy (Cornelisse et al., 2007). The average event rise time of 10.9 ms is as fast as has been measured in oocytes or any other cell type (Demuro and Parker, 2008). This high speed may reflect the properties of these events in pyramidal neurons or it may result from the fact that the thin dendrite was entirely in focus permitting a clear measurement at the site of origin; events in most other cell types are more 3-dimensional.
Figure 6.
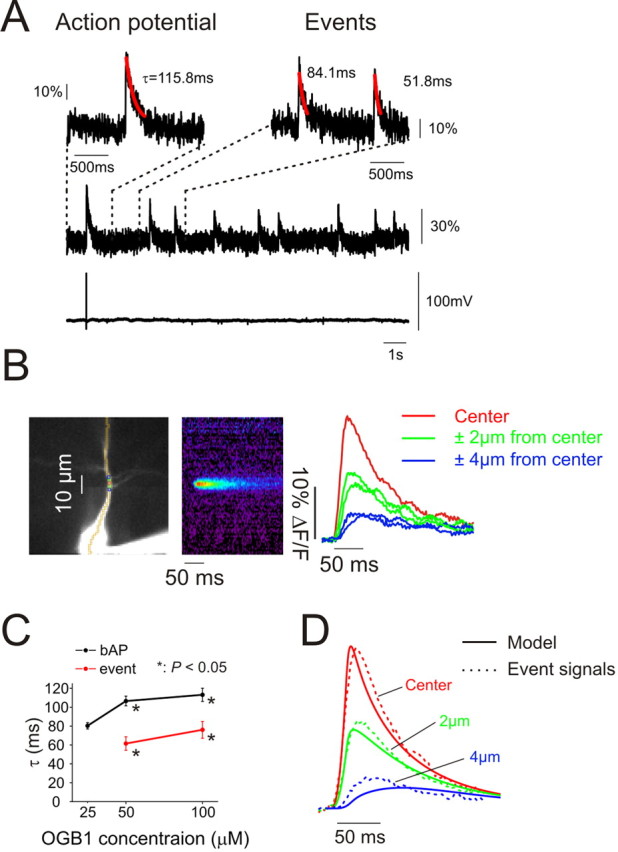
Events are very rapid Ca2+ transients whose recovery time is accelerated by diffusion along the dendrite. A, High speed (500 Hz) measurements of Ca2+ transients from a bAP and a series on spontaneous events measured at the location of the events on the dendrite. The electrical recording below shows the single bAP with no change at the time of the events. The traces above show regions of the signal on an expanded time scale with best fit exponentials to the recovery time course. B, Averaged records of event signals from nearby locations. The data were recorded at 500 Hz and spontaneous event signals (N = 11 events) were aligned at the time of the start of the rising phase of the fluorescence transient. The resulting traces were filtered with a 3-point interpolation algorithm. The pseudocolor image shows the “line scan” of the averaged signal. The traces show the signal at the center and neighboring (±2 μm, ±4 μm) locations. C, Time constants of the recovery phase of the two signals measured with different concentrations of OGB-1 in the pipette. The rates were slightly faster at lower concentrations but the difference between event signals and bAP traces remained (*p < 0.05). D, Traces from equivalent locations from a computational model (Fig. 7) that assumed that Ca2+ was released from a short segment in the dendrites and removed by a pump and by diffusion. There is a good correspondence between the measured and modeled signals.
The event decay time was much faster than decay time of the bAP-evoked fluorescence transient (Fig. 6A,C) (event: 61.5 ± 7.2 ms, N = 12 events, 4 cells; bAP: 106.6 ± 5.1 ms, N = 12 events, 4 cells; p < 0.001, t test). These measurements were made with 50 μm OGB-1 in the pipette. The difference in decay times also was observed using 100 μm OGB-1 (event: 76 ± 8.9 ms, N = 18 events, 6 cells; bAP: 113.1 ± 7 ms, N = 10 events, 4 cells; p < 0.01, t test). We tried to measure these times using 25 μm OGB-1, where we expected the least buffering (Helmchen et al., 1996) but the signal-to-noise ratio (S/N) for the events was poor; only recovery times from bAPs were measured at this indicator concentration.
To examine why event signals decay faster than bAP signals we averaged the signals from groups of events from the same location to determine their space-time structure. The sharp edge of the rising signal at the center of the event provided a good reference point for aligning individual signals. We reasoned that if we chose the wrong initiation time or if the location of the event moved slightly it would blur the spatial and temporal properties of the signal; the true signal could only be sharper. Figure 6B shows one example of a signal generated by averaging 11 events. At the center the rise time (10–90%) was 10 ms. Two micrometers away on either side the signal rose more slowly. The peak was more rounded and the amplitude smaller than at the center. Four micrometers away the signal was still smaller and slower. Similar observations were made on three other groups of events in other cells with an average rise time at the center of 12 ± 2.1 ms. Since these values are close to the mean for signals without averaging the blurring effect was small.
These waveforms appeared to have the characteristics of diffusion away from a central source rather than active propagation in which wave shape is relatively invariant (Nakamura et al., 2002). To test this hypothesis we constructed a computer model, described in more detail in the legend of Figure 7, of event Ca2+ dynamics in the dendrites using the NEURON simulation environment (Hines, 1998) and compared this model to the averaged data from the four sets of events. We modeled Ca2+ release into a ∼2 μm long dendritic segment with no radial structure. Diffusion in the radial direction should be close to equilibrium in <10 ms and therefore was ignored in this model. Ca2+ was removed by a pump and was allowed to diffuse axially along the dendrite. The pump rate constant was determined by fitting the removal of Ca2+ after a bAP (Fig. 7A). Since this transient is uniform along the dendrite there is no axial diffusion after Ca2+ entry. Once the pump rate was set we determined the duration of the Ca2+ release pulse (∼20 ms) by fitting the rise time of the event Ca2+ signal (Fig. 7B). The rise time was not sensitive to other parameters. Therefore, there were only two remaining free parameters in the model for event Ca2+ dynamics, the diffusion constant for Ca2+ in cytoplasm and the spatial extent of the IP3 receptor or RyR cluster. The diffusion constant was previously estimated to be D = 0.014 μm2/ms (Kushmerick and Podolsky, 1969) but is possibly greater than this value in the dendrite because of buffered diffusion by the indicator (Shuai et al., 2006). We tested models varying D in the range of 0.01–0.06 μm2/ms while varying the size of the receptor cluster in small amounts ∼2 μm in length (Fig. 7C,D). Figure 6D compares the modeled and recorded transients at the center of the event and at two close locations when D = 0.03 μm2/ms. The best fit was achieved when the event size was slightly larger than 2 μm in length. This is probably an overestimate since light scattering in the slice, the size of the pixels, and the averaging process all increase the apparent size of the event. The close correspondence of the model to the data show that the faster recovery time for events compared with bAP transients primarily results from Ca2+ diffusing along the dendrite. It also suggests that these events result from localized and fast Ca2+ release from a cluster of receptors, similar to the mechanisms suggested to occur in oocytes (Sun et al., 1998) and cardiac myocytes (Cheng and Lederer, 2008). We did not need to include any specialized removal mechanism like a local pump into the ER to account for the event time course.
Figure 7.
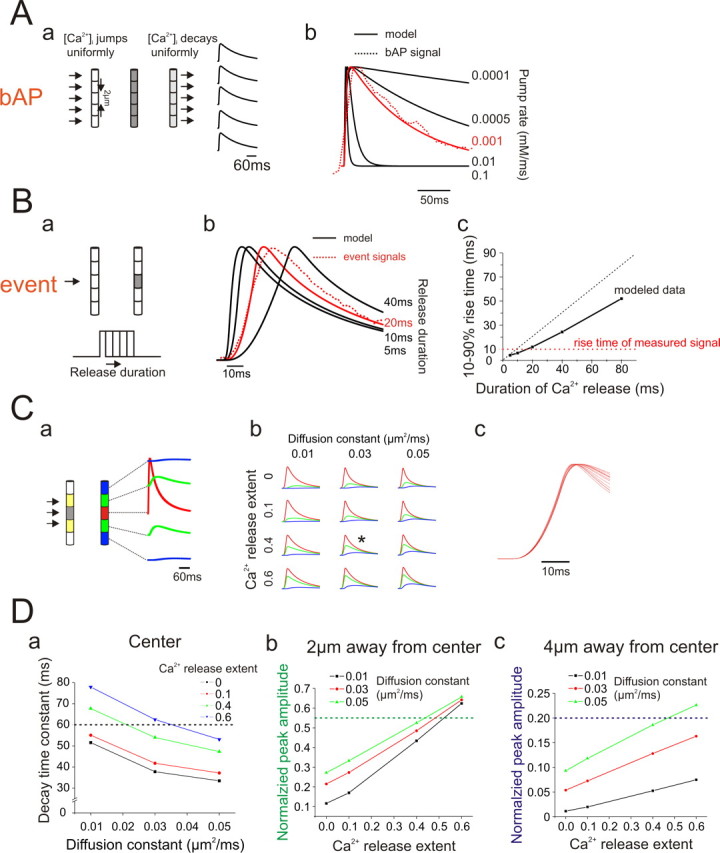
Computational model of Ca2+ release and removal for a single event using the NEURON simulation environment. A, Model of Ca2+ entry and removal after the generation of a single bAP. Aa, Outline of the model. Ca2+ entry was assumed to be uniform along a length of the apical dendrite and was removed by a pump that was uniformly distributed along the dendrite. Ca2+ entry was driven by a single bAP activated by Na+ and K+ channels in the dendrites using standard Hodgkin–Huxley kinetics built into NEURON. Ca2+ entry was through a mixture to L-type and T-type Ca2+ channels distributed uniformly along the dendrite. The details of this part of the model are not critical. Ab, The pump rate was assumed to be proportional to the instantaneous difference between the dynamic [Ca2+]i level and the resting [Ca2+]i level. A pump rate of 0.001 mm/ms provided a close fit to the bAP transient recorded at 500 Hz and was used for the rest of the simulation. B, Fitting the rising phase of an event. Ba, An event was simulated as Ca2+ entry through a small length of the plasma membrane. The model had no radial structure so computationally this was equivalent to Ca2+ release from stores. The initial spatial extent of release was assumed to be 2 μm. Ca2+ was assumed to be removed only by the pump and by diffusion. The initial diffusion constant was set to D = 0.03 μm2/ms. Bb, Using these parameters the time course of [Ca2+]i at the center of the event was simulated for Ca2+ release of varying durations. Constant release for 20 ms provided a close fit to the recorded rising phase. Varying the diffusion constant or the spatial extent of release had no effect of this parameter (see Cc). Bc, Predicted 10–90% event rise times for entry of varying durations. Note that the rise time is always significantly less than the duration of Ca2+ release (dotted black line shows equality). This difference is caused by the accelerating removal of Ca2+ by the pump and diffusion during the rising phase of the event. C, Fitting the time course and amplitude of the event at different locations. Ca, To fit the measured transients we varied the diffusion constant (D) and the spatial extent of release. Release was assumed to extend over a 6 μm length, but the magnitude in the two outer 2 μm segments (yellow) was assumed to be less than in the center (gray segment). The ratio of this magnitude to the magnitude at the center (“Ca2+ release extent”) was a free parameter. The model simulated the time course at these five locations. Cb, Examples of event simulations varying D and Ca2+ release extent. Changing these parameters affects the relative amplitudes at different locations. The variations in the removal time constant are not immediately obvious in this figure. The simulation with the asterisk (*) had the best fit to the data and is shown in Figure 6D. Cc, Overlay of the rising phase of the central time course of the nine simulations shown in Cb demonstrating that varying these parameters had no effect on the rising phase of the event. D, Details of model parameter selection. Da, The time constant (τ) of the recovery phase in the center of the event was determined in event models in which the diffusion constant and Ca2+ release extent were varied. The target value of 60 ms from the high speed measurements is shown as a dotted line. Very localized release (Ca2+ release extent of 0.0 or 0.1) was ruled out. Db, The normalized amplitude of the transient 2 μm from the center also was modeled as a function of these two parameters. Again, low values of Ca2+ release extent were eliminated since they could not generate values close to the measurement ratio of 0.55. Dc, Similarly, low values of Ca2+ release extent could not generate normalized peak amplitudes close to the target value of 0.2 at locations 4 μm away from the center of the event. From these models it appears that Ca2+ release extents of 0.4–0.6 and diffusion constants of 0.03–0.05 μm2/ms will fit the data. Traces using 0.4 and 0.03 μm2/ms are overlaid on top of the measured traces in Figure 6D. The match appears to be very good.
If the elementary events often occur repetitively at the same location suggesting a structural basis for their generation, then we might expect that event amplitudes would be constant. To test this assumption we measured the peak amplitude of the same series of events recorded with the 500 Hz camera. The high sampling rate ensured that we did not miss the peaks of these events. Supplemental Figure 3, available at www.jneurosci.org as supplemental material, shows the amplitude histogram of four of these event series. Because they were measured in different cells we normalized the amplitudes to the signals from the bAPs recorded at the same locations in these neurons. The histograms show that for each event the amplitudes were clustered around a modal value; the distributions do not appear random. However the values are not so tightly clustered that a strong conclusion about the mechanism underlying event generation can be inferred. There is some uncertainty about the true peak amplitude because events are localized events. As with [Ca2+]i changes in spines after synaptic activation and changes under the membrane after the opening of voltage gated channels, we expect steep local gradients around the channels releasing Ca2+ from the ER. We estimated the peak fluorescence change by calculating the average value within a 2 μm × 2 μm pixel. Most likely the values near the release channels are significantly higher than this average.
Discussion
Events are spontaneous
The core observation of this study is that fast, localized, Ca2+ release events normally occur spontaneously in the dendrites of pyramidal neurons. Spontaneous release events have not previously been reported in these cells, possibly because the imaging systems were not tuned to detect rapid, localized, stochastic events. This study focused on events in 2- to 4-week-old pyramidal neurons from the CA1 region of the rat hippocampus. But, in trial experiments we detected similar events from pyramidal neurons in the hippocampal CA3 region, in the somatosensory neocortex and in CA1 pyramidal neurons from 6-week-old animals (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Therefore, we expect that these kinds of events will be found in neurons from other regions of the CNS. Events of similar characteristics have been observed in many cell types, like oocytes (Sun et al., 1998), HeLa cells (Bootman et al., 1997), cultured differentiated PC12 cells, cultured pyramidal neurons (Koizumi et al., 1999), and recently in the somata of dissociated DRG cells (Ouyang et al., 2005). This is the first description of such events in adult dendrites. Lohmann et al. (2005) described local Ca2+ release events in the dendrites of hippocampal neurons in embryonic slice cultures. These events appear to regulate filopodia outgrowth and may play a role in synaptogenesis. However, they differed in several significant ways from the events observed in our experiments. First, they lasted much longer (typically 4.1 s compared with 0.07 s) and were primarily located at sites of synaptic contact rather than branch points. Second, they were primarily synaptically activated. In contrast, neither TTX nor any blocker of ionotropic receptors had an effect on event frequency in our experiments.
Elementary events primarily occur at branch points in the dendrites
We found that events occur repetitively at the same locations in the dendrites and that these locations were frequently (but not exclusively) at branch points suggesting that event sites were related to the locations of particular molecules or structures. A similar preference for branch points was observed in cultured PC12 cells (Koizumi et al., 1999). In oocytes puff sites were suggested to be the sites of IP3 receptor clusters (Parker et al., 1996). Related to this proposal Hertle and Yeckel (2007) found concentrations of type I IP3 receptors at branch points in CA1 pyramidal neurons but they also found these receptors at many other locations in the cells. Some earlier experiments suggested that localized Ca2+ release could occur in spines in response to synaptic stimulation (Emptage et al., 1999). These results are controversial (Kovalchuk et al., 2000; Sabatini et al., 2002). In preliminary experiments we detected spontaneous Ca2+ release events on oblique dendrites where spine density is highest (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, our current experiments did not have the spatial resolution to determine whether these events occur at the sites of synaptic activation.
Previously, Nakamura et al. (2002) found that synaptically activated Ca2+ waves in pyramidal neurons preferentially initiated at branch points. In oocytes Ca2+ waves grow from the synergistic interaction of puffs (Sun et al., 1998). If a similar organization exists in pyramidal cell dendrites then a distribution that places most elementary events at branch points probably explains the initiation of Ca2+ waves at the same locations.
It is not clear whether the location of events at branch points has a specific physiological consequence. Koizumi et al. (1999) suggested that conductances activated by event associated [Ca2+]i increases at this location might provide an explanation for branch point failure of action potential propagation. We have seen no evidence of branch point failure that could not also be explained by an impedance mismatch at that location.
Event frequency can be modulated
The second important conclusion is that event frequency can be modulated by changes in membrane potential and by mGluR-mediated synaptic activation. Modulation by changes in membrane potential was particularly dramatic. Hyperpolarization by <10 mV shut down event activity and depolarization by similar amounts (and still subthreshold for spike generation) enhanced event frequency by >150%. The modulation occurred quickly. The frequency change certainly occurred within 200 ms of the step change in potential and may have been quicker except that events normally do not occur at a high enough frequency to enable precise measurements of this time. The modulation appears to result from changes in resting [Ca2+]i caused by turning on and off voltage gated Ca2+ channels, in particular L-type channels that are known to be open at rest in these cells (Magee et al., 1996). Both L-type channel antagonists and the L-type channel agonist Bay K-8644 had strong effects on event frequency. Membrane potential changes and L-channel-mediated Ca2+ entry have a similar effect on Ca2+ sparks in cardiac myocytes (Cheng and Lederer, 2008). Interestingly, membrane hyperpolarization has the opposite effect on puff frequency in oocytes (Yao and Parker, 1994), primarily because in these cells most Ca2+ entry is through channels open at negative potentials.
Since event frequency can be modulated rapidly by small changes in membrane potential we would expect that several normally occurring physiological events could change event frequency. For example, the sustained depolarization after trains of EPSPs or during cortical “up states” (Sanchez-Vives and McCormick, 2000) could increase event rate. Similarly, trains of bAPs, by providing a sustained increased [Ca2+]i level in the dendrites (Helmchen et al., 1996) could enhance event frequency. In contrast, the slow GABAB-mediated IPSP or the spike-evoked slow AHP, both of which are prominent in pyramidal neurons, might reduce the event rate. In addition to modulating the frequency of events in the dendrites these potential changes should change the rates in the soma where, in preliminary experiments (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), we have detected spontaneous Ca2+ transients. In contrast, events generated by synaptic mobilization of IP3 would not be expected to occur in the soma since there are few glutamatergic synaptic contacts on the cell body (Watanabe et al., 2006).
Low intensity tetanic synaptic stimulation increased event frequency in a manner very similar to the increase in event frequency observed after uncaging of IP3. In both cases an increase in event frequency was observed in only a narrow range of intensities; above that level a graded, continuous increase in [Ca2+]i was observed before leading to a fully regenerative Ca2+ wave at higher intensities. The continuous increase may reflect many events overlapping (Sun et al., 1998) or may reflect Ca2+ coming out of IP3 receptors that are not organized into clusters. More precise experiments will be needed to make this discrimination.
Although raising [Ca2+]i and raising IP3 concentration both increased event frequency there was an important difference between these two modulatory effects. Even when high event frequencies were generated with depolarization or in the presence of Bay K-8644, Ca2+ waves were not generated. In contrast it was hard to avoid Ca2+ wave generation when IP3 concentration was raised. One possible explanation is that wave generation requires a higher concentration of IP3 than is normally found in these cells at rest while resting [Ca2+]i is high enough to be an effective cofactor when IP3 concentration is elevated. Another possibility is that events can be generated by two different mechanisms, one mediated by IP3 receptors and the second mediated through the activation of RyRs. These events, like “sparks” in cardiac myocytes (Cheng and Lederer, 2008), can be directly activated by increases in [Ca2+]i without a requirement for IP3 as a cofactor. The sensitivity of event rates to caffeine is consistent with this possibility. There are some clear examples of cells that seem to express both puffs and sparks (Gordienko and Bolton, 2002).
Potential physiological consequences of event generation
In many cells in which similar elementary events have been observed it has been hard to establish a specific consequence of their occurrence. In most cases they are seen as the building blocks for Ca2+ waves or regenerative CICR. The most common suggested roles for Ca2+ waves are as local developmental signals (Lohmann and Bonhoeffer, 2008) or as activators of gene expression (Li et al., 1998; Hardingham et al., 2001). Recently, Ca2+ waves have been implicated in a form of long term potentiation (Fernández de Sevilla et al., 2008). It is possible that Ca2+ events could contribute to each of these mechanisms by themselves without becoming regenerative. Presumably, these contributions would be more subtle, resulting in smaller changes and with more spatially restricted expression. Events could also modulate membrane potential by activating Ca2+ sensitive conductances. In other neurons spontaneous miniature outward currents (SMOCs), attributed to opening of BK or SK type channels by Ca2+ released from internal stores, have been detected (Merriam et al., 1999; Mitra and Slaughter, 2002). To our knowledge, this kind of event has not been observed in CA1 pyramidal neurons, possibly because K+ channels in the somatic region are weakly activated by Ca2+ (Gu et al., 2008). On a more macroscopic scale there is clear pharmacological evidence that Ca2+ release from RyR sensitive stores can contribute to the activation of the K+ conductance underlying the slow AHP in CA3 pyramidal neurons (Tanabe et al., 1998) and vagal neurons (Sah and McLachlan, 1991), and contributes to endocannabinoid-mediated depolarization suppression of inhibition (DSI) (Isokawa and Alger, 2006). RyR activation also modulates IP3-mediated Ca2+ release in mouse cortical pyramidal neurons (Stutzmann et al., 2006).
An interesting characteristic of these elementary events is that they are stochastic like the opening of membrane ion channels. They occur spontaneously but their rates can be modulated. The location and time of an individual event is not closely connected to a specific generating event. This suggests that their significance lies in their population behavior, either as a group generating a Ca2+ wave or possibly over time through the accumulated action of individual [Ca2+]i increases.
Footnotes
This work was supported in part by National Institutes of Health Grant NS016295. We thank Nechama Lasser-Ross for computer programming and Rishikesh Narayanan for comments on this manuscript.
References
- Ango F, Prézeau L, Muller T, Tu JC, Xiao B, Worley PF, Pin JP, Bockaert J, Fagni L. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature. 2001;411:962–965. doi: 10.1038/35082096. [DOI] [PubMed] [Google Scholar]
- Bandrowski AE, Huguenard JR, Prince DA. Baseline glutamate levels affect group I and II mGluRs in layer V pyramidal neurons of rat sensorimotor cortex. J Neurophysiol. 2003;89:1308–1316. doi: 10.1152/jn.00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson CP, Tozzi A, Bernardi G, Mercuri NB. Transient receptor potential-like channels mediate metabotropic glutamate receptor EPSCs in rat dopamine neurones. J Physiol. 2004;555:323–330. doi: 10.1113/jphysiol.2003.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bootman M, Niggli E, Berridge M, Lipp P. Imaging the hierarchical Ca2+ signalling system in HeLa cells. J Physiol. 1997;499:307–314. doi: 10.1113/jphysiol.1997.sp021928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium Sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Cornelisse LN, van Elburg RA, Meredith RM, Yuste R, Mansvelder HD. High speed two-photon imaging of calcium dynamics in dendritic spines: consequences for spine calcium kinetics and buffer capacity. PLoS ONE. 2007;2:e1073. doi: 10.1371/journal.pone.0001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson SP, Keizer J, Pearson JE. Fire-diffuse-fire model of dynamics of intracellular calcium waves. Proc Natl Acad Sci U S A. 1999;96:6060–6063. doi: 10.1073/pnas.96.11.6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I. Multi-dimensional resolution of elementary Ca2+ signals by simultaneous multi-focal imaging. Cell Calcium. 2008;43:367–374. doi: 10.1016/j.ceca.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Endo M. Calcium release from the sarcoplasmic reticulum. Physiol Rev. 1977;57:71–108. doi: 10.1152/physrev.1977.57.1.71. [DOI] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Núñez A, Borde M, Malinow R, Buño W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. J Neurosci. 2008;28:1469–1478. doi: 10.1523/JNEUROSCI.2723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J Biol Chem. 1988;263:11075–11079. [PubMed] [Google Scholar]
- Gordienko DV, Bolton TB. Crosstalk between ryanodine receptors and IP3 receptors as a factor shaping spontaneous Ca2+-release events in rabbit portal vein myocytes. J Physiol. 2002;542:743–762. doi: 10.1113/jphysiol.2001.015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Hu H, Vervaeke K, Storm JF. SK (KCa2) Channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J Neurophysiol. 2008;100:2589–2604. doi: 10.1152/jn.90433.2008. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines ML. The neurosimulator NEURON. In: Koch C, Segev I, editors. Methods in neuronal modeling. Cambridge, MA: MIT; 1998. pp. 129–136. [Google Scholar]
- Hong M, Ross WN. Priming of intracellular calcium stores in rat CA1 pyramidal neurons. J Physiol. 2007;584:75–87. doi: 10.1113/jphysiol.2007.137661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J Gen Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M, Alger BE. Ryanodine receptor regulates endogenous cannabinoid mobilization in the hippocampus. J Neurophysiol. 2006;95:3001–3011. doi: 10.1152/jn.00975.2005. [DOI] [PubMed] [Google Scholar]
- Khodakhah K, Armstrong CM. Inositol trisphosphate and ryanodine receptors share a common functional Ca2+ pool in cerebellar Purkinje neurons. Biophys J. 1997;73:3349–3357. doi: 10.1016/S0006-3495(97)78359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Somlyo AV, Somlyo AP. Heparin inhibits the inositol 1,4,5-trisphosphate-dependent, but not the independent, calcium release induced by guanine nucleotide in vascular smooth muscle. Biochem Biophys Res Commun. 1988;153:625–631. doi: 10.1016/s0006-291x(88)81141-0. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Bootman MD, Bobanovic LK, Schell MJ, Berridge MJ, Lipp P. Characterization of elementary Ca2+ release signals in NGF-differentiated PC12 cells and hippocampal neurons. Neuron. 1999;22:125–137. doi: 10.1016/s0896-6273(00)80684-4. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick MJ, Podolsky RJ. Ionic mobility in muscle cells. Science. 1969;166:1297–1298. doi: 10.1126/science.166.3910.1297. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Watanabe S, Nakamura T, Lasser-Ross N, Ross WN. Synaptically activated Ca2+ waves in layer 2/3 and layer 5 rat neocortical pyramidal neurons. J Physiol. 2003;549:471–488. doi: 10.1113/jphysiol.2002.037614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser-Ross N, Miyakawa H, Lev-Ram V, Young SR, Ross WN. High time resolution fluorescence imaging with a CCD camera. J Neurosci Methods. 1991;36:253–261. doi: 10.1016/0165-0270(91)90051-z. [DOI] [PubMed] [Google Scholar]
- Lechleiter JD, Clapham DE. Molecular mechanisms of intracellular calcium excitability in X. laevis oocytes. Cell. 1992;69:283–294. doi: 10.1016/0092-8674(92)90409-6. [DOI] [PubMed] [Google Scholar]
- Lelli A, Perin P, Martini M, Ciubotaru CD, Prigioni I, Valli P, Rossi ML, Mammano F. Presynaptic calcium stores modulate afferent release in vestibular hair cells. J Neurosci. 2003;23:6894–6903. doi: 10.1523/JNEUROSCI.23-17-06894.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Llopis J, Whitney M, Zlokarnik G, Tsien RY. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 1998;392:936–941. doi: 10.1038/31965. [DOI] [PubMed] [Google Scholar]
- Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Bonhoeffer T. A role for local calcium signaling in rapid synaptic partner selection by dendritic filopodia. Neuron. 2008;59:253–260. doi: 10.1016/j.neuron.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Myhr KL, Wong RO. Transmitter-evoked local calcium release stabilizes developing dendrites. Nature. 2002;418:177–181. doi: 10.1038/nature00850. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Finski A, Bonhoeffer T. Local calcium transients regulate the spontaneous motility of dendritic filopodia. Nat Neurosci. 2005;8:305–312. doi: 10.1038/nn1406. [DOI] [PubMed] [Google Scholar]
- MacMillan D, Chalmers S, Muir TC, McCarron JG. IP3-mediated Ca2+ increases do not involve the ryanodine receptor, but ryanodine receptor antagonists reduce IP3-mediated Ca2+ increases in guinea-pig colonic smooth muscle cells. J Physiol. 2005;569:533–544. doi: 10.1113/jphysiol.2005.096529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Avery RB, Christie BR, Johnston D. Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:3460–3470. doi: 10.1152/jn.1996.76.5.3460. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Kim YK, Valdivia H, Knudson CM, Takekura H, Franzini-Armstrong C, Coronado R, Campbell KP. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron. 1991;7:17–25. doi: 10.1016/0896-6273(91)90070-g. [DOI] [PubMed] [Google Scholar]
- Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- Merriam LA, Scornik FS, Parsons RL. Ca2+-induced Ca2+ release activates spontaneous miniature outward currents (SMOCs) in parasympathetic cardiac neurons. J Neurophysiol. 1999;82:540–550. doi: 10.1152/jn.1999.82.2.540. [DOI] [PubMed] [Google Scholar]
- Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol. 2003;64:1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- Mitra P, Slaughter MM. Mechanism of generation of spontaneous miniature outward currents (SMOCs) in retinal amacrine cells. J Gen Physiol. 2002;119:355–372. doi: 10.1085/jgp.20028478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci. 2000;20:RC103. doi: 10.1523/JNEUROSCI.20-20-j0003.2000. (1-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Lasser-Ross N, Nakamura K, Ross WN. Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol. 2002;543:465–480. doi: 10.1113/jphysiol.2002.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osanai M, Yamada N, Yagi T. Long-lasting spontaneous calcium transients in the striatal cells. Neurosci Lett. 2006;402:81–85. doi: 10.1016/j.neulet.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Ouyang K, Zheng H, Qin X, Zhang C, Yang D, Wang X, Wu C, Zhou Z, Cheng H. Ca2+ sparks and secretion in dorsal root ganglion neurons. Proc Natl Acad Sci U S A. 2005;102:12259–12264. doi: 10.1073/pnas.0408494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker I, Yao Y. Regenerative release of calcium from functionally discrete subcellular stores by inositol trisphosphate. Proc Biol Sci. 1991;246:269–274. doi: 10.1098/rspb.1991.0154. [DOI] [PubMed] [Google Scholar]
- Parker I, Yao Y. Ca2+ transients associated with openings of inositol trisphosphate-gated channels in Xenopus oocytes. J Physiol. 1996;15:663–668. doi: 10.1113/jphysiol.1996.sp021247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker I, Choi J, Yao Y. Elementary events of InsP3-induced Ca2+ liberation in Xenopus oocytes: hot spots, puffs and blips. Cell Calcium. 1996;20:105–121. doi: 10.1016/s0143-4160(96)90100-1. [DOI] [PubMed] [Google Scholar]
- Power JM, Sah P. Distribution of IP3-mediated calcium responses and their role in nuclear signalling in rat basolateral amygdala neurons. J Physiol. 2007;580:835–857. doi: 10.1113/jphysiol.2006.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau E, Smith JS, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. Am J Physiol. 1987;253:C364–C368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Ca2+-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca2+-activated Ca2+ release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-z. [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science. 1989;246:815–818. doi: 10.1126/science.2573153. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Stuart G. Patch-pipette recordings from the soma, dendrites, and axon of neurons in brain slices. In: Sakmann B, Neher E, editors. Single channel recording. Ed 2. New York: Plenum; 1995. pp. 199–211. [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Shuai J, Rose HJ, Parker I. The number and spatial distribution of IP3 receptors underlying calcium puffs in Xenopus oocytes. Biophys J. 2006;91:4033–4044. doi: 10.1529/biophysj.106.088880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Coronado R, Meissner G. Sarcoplasmic reticulum contains adenine nucleotide-activated calcium channels. Nature. 1985;316:446–449. doi: 10.1038/316446a0. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, LaFerla FM, Parker I. Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci. 2003;23:758–765. doi: 10.1523/JNEUROSCI.23-03-00758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XP, Callamaras N, Marchant JS, Parker I. A continuum of InsP3-mediated elementary Ca2+ signalling events in Xenopus oocytes. J Physiol. 1998;509:67–80. doi: 10.1111/j.1469-7793.1998.067bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Gähwiler BH, Gerber U. L-Type Ca2+ channels mediate the slow Ca2+-dependent afterhyperpolarization current in rat CA3 pyramidal cells in vitro. J Neurophysiol. 1998;80:2268–2273. doi: 10.1152/jn.1998.80.5.2268. [DOI] [PubMed] [Google Scholar]
- Wanaverbecq N, Marsh SJ, Al-Qatari M, Brown DA. The plasma membrane calcium-ATPase as a major mechanism for intracellular calcium regulation in neurones from the rat superior cervical ganglion. J Physiol. 2003;550:83–101. doi: 10.1113/jphysiol.2002.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hong M, Lasser-Ross N, Ross WN. Modulation of calcium wave propagation in the dendrites and to the soma of rat hippocampal pyramidal neurons. J Physiol. 2006;575:455–468. doi: 10.1113/jphysiol.2006.114231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Parker I. Ca2+ influx modulation of temporal and spatial patterns of inositol trisphosphate-mediated Ca2+ liberation in Xenopus oocytes. J Physiol. 1994;476:17–28. [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]