

Abstract
This study was initiated to find small molecule ligands that would induce a functional response when docked with neurotrophin Trk receptors. “Minimalist” mimics of β-turns were designed for this purpose. These mimics are: (i) rigid, yet easily folded into turn-like conformations, and (ii) readily accessible from amino acids bearing most of the natural side chains. Gram quantities of sixteen of these turn mimics were prepared, then assembled into 152 fluorescein-labeled bivalent peptidomimetics via a solution-phase combinatorial method. Fluorescence-based screening of these molecules using cells transfected with the Trk receptors identified 10 potential ligands of TrkC, the receptor for neurotrophin-3 (NT-3). Analogs of these bivalent peptidomimetics with biotin replacing the fluorescein label were then prepared and tested to confirm that binding was not due to the fluorescein. Several assays were conducted to find the mode of action of these biotinylated compounds. Thus, direct binding, survival and neuritogenic, and biochemical signal transduction assays showed 8 of the original 10 hits were agonistic ligands binding to the ectodomain of TrkC. Remarkably, some peptidomimetics afford discrete signals leading to either cell survival or neuritogenic differentiation. The significance of this work is three fold. First, we succeeded in finding small, selective, proteolytically stable ligands for the TrkC receptor; there are very few of these in the literature. Second, we show that it is possible to activate distinct and biased signaling pathways with ligands binding at the ectodomain of wild type receptors. Third, the discovery that some peptidomimetics initiate different modes of cell signaling increases their potential as pharmacological probes and therapeutic leads.
The neurotrophins are dimeric protein growth factors that help regulate the peripheral and central nervous systems and other tissues that express the Trk and p75 neurotrophin receptors. (1,2) Trk receptors are relatively selective for, and bind with high affinity (∼Kd 10-11 M) to the neurotrophin. Nerve growth factor (NGF) docks with TrkA, Brain Derived Neurotrophic Factor (BDNF) interacts with TrkB(3), and Neurotrophin-3 (NT-3) interacts preferentially with TrkC but can also bind TrkA and TrkB (Figure 1a). Ligand binding induces phosphorylation (pTyr) of Trk receptors and associated signaling partners and activation of the neurotrophic biological signals: cell growth, cell survival under stress (trophic activity), and/or cellular differentiation (neuritogenic or neurogenic activity).(4-6)
Figure 1.


a Generic-turn structure; b lead compound D3 mimics one β-turn that interacts with TrkA in a functional way; c the design of our second generation, triazole-based, monovalent β-turn mimics; d some salient features of the triazole monovalent mimics featured in this paper.
There is also a second receptor for the neurotrophins, p75, a member of the tumor necrosis factor (TNF) receptor superfamily. All the neurotrophins also bind the p75 receptor, but with lesser affinities (10-9 to 10-11 M) than for the Trk receptors. (7-9) The p75 receptor can transduce signals which are either pro-survival, pro-apoptotic, or pro-differentiation, (10,11) and it regulates Trk function and ligand docking preference.(12,13) The biology of p75 is complex and the cellular responses to ligands that bind both p75 and the Trk receptors are difficult to predict. Consequently, peptidomimetic ligands that target Trk receptors and exclude p75 are highly desirable.
Hypothesizing that β-turn structures (Figure 1b) on the periphery of the neurotrophin dimer are important as the ligand hot-spots for binding to Trk receptors, we(14-17) and others(18-21) prepared and tested disulfide linked cyclic peptides with sequences corresponding to the neurotrophin turn regions. These adopt β-turn conformations similar to regions of the proteins from which they were designed,(22,23) bind selectively to the appropriate Trk receptors, and function as agonists or antagonists.
Our subsequent efforts to identify small molecules that mimic or disrupt the interaction between the neurotrophins and their receptors(24) led to a set of “first generation” β-turn mimics like D3 (Figure 1b).(25-28) Compound D3 showed interesting characteristics as a partial agonist acting at the TrkA receptor.(16,28) Further, the compound induced significant improvement of memory/learning in cognitively impaired aged rats when administered in vivo.(29) Monovalent compounds such as this only mimic one hot-spot, but a method was developed to assemble these into bivalent mimics of two proximal turn regions.(30,31) These bivalent mimics bind the Trk receptors more strongly than the corresponding monovalent ones, and elicit functional cellular responses associated with activation of Trk receptors by neurotrophins.
Meanwhile, other researchers in the field have reported a few small molecule Trk(32-37) and p75(38-41) ligands. In a few cases, these compounds were found by screening natural products or compound libraries. In others, compounds with neurotrophin-like activities have been identified but it is not clear that they are acting via binding to the Trk receptors or by some other mechanism.(42-45)
The next phase of our work was to design even less peptidic turn mimics A (Figure 1c) that could be prepared in solution, in gram quantities, and assembled into bivalent molecules in solution (Figure 1d).(46) One potentially limiting feature of this design is that the R1 side-chain is derived from alkynes rather than amino acids, and it can be inconvenient to obtain alkynes where R1 corresponds to an amino acid side-chain. Further, even though some of these mimics bound to cells transfected with the Trk receptors (determined via FACS assays) the compounds tested so far do not have significant cellular activities.
The focus of this paper is the new β-turn mimic design B (Figure 1d). These turn mimics are derived from alkynes that are conveniently accessible from amino acids, so the R1 side-chain can correspond to natural amino acids. Structurally, the “extra” amino group in mimics B relative to A is a minor change, but one that can have a significant impact on the interactions these molecules generate at hot-spots, their solubilities in aqueous media, and their pharmacological properties. This paper describes how a library of fluorescein-labelled bivalent molecules was assembled from these monovalent building blocks B, and tested for binding to cells that are stably transfected with the TrkA or C receptors. Analogs of the most strongly bound compounds were prepared with biotin labels to replace the fluorescein. These were re-tested in binding assays to confirm they still bound strongly, then were screened via cell survival, neuritogenic differentiation, and signal transduction assays. The first two of these assays (survival and neurite outgrowth) monitor effects the compounds have on the cells as a result of these ligands binding the Trk receptors. The third assay type (monitoring intracellular tyrosine phosphorylation) is to elucidate pathways by which these effects occur. One conclusion from the data collected here was anticipated: these modified bivalent compounds derived from mimics B bind the TrkC receptor. However, another finding was not: subsets of these mimics induce divergent activities in ex vivo assays. Specifically, one set of compounds only potentiated survival effect of sub-optimal NT-3, and did not induce neuritogenic outgrowth, while another did the reverse, and there was no overlap between the two sets. Some implications of these unusual findings are discussed.
Results and Discussion
Library Syntheses
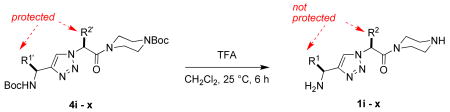
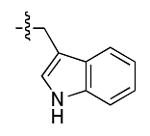
The most compelling feature of the library synthesis is the simplicity with which mimics with natural protein amino acid side-chains can be constructed (Scheme 1). Two fragments were required to synthesize the monovalent building mimics 1: an azide and an alkyne, both conveniently derived from amino acids (via methods shown in the supporting information) with the side chains indicated. In fact, most of the monovalent mimics were purified and stored in the protected forms 4 (see supporting for complete numbering system), then deprotected immediately prior to use. Table 1 indicates the side-chains of the compounds 4, and the deprotection conditions.
Scheme 1.


Preparation of the deprotected monomers 1.
Table 1.
Peptidomimetics 4 and 1.
 | |||
|---|---|---|---|
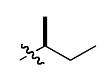
| Compound 4 | R1′ | R2′ | yield (%)a |
| i | 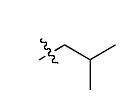 |
 |
64 |
| j |  |
 |
74 |
| k |  |
81 | |
| l | 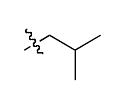 |
 |
98 |
| m |  |
 |
60 |
| n | 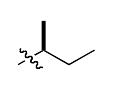 |
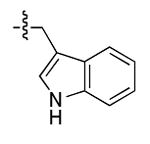 |
68 |
| o |  |
77 | |
| p | 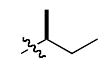 |
 |
78 |
| q | 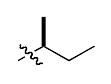 |
 |
50 |
| r | 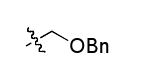 |
 |
40b |
| s | 69b | ||
| t |  |
60b | |
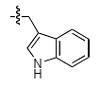
| u |  |
 |
55b |
| v | 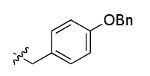 |
 |
84b |
| w | 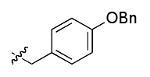 |
 |
62b |
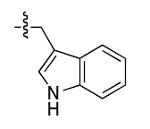
| x | 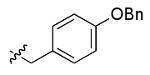 |
 |
68b |
From intermediate 3 (see supporting) after flash chromatography.
After removing the side-chain benzyl protection.
The monomers 1 were assembled into the bivalent molecules 5 (shown below in non-ionized forms) using a modification of our published procedure (see supporting material).(46) The goal of this combinatorial technique is to easily prepare large numbers of bivalent molecules in adequate purities for a first pass biological screen (typically above 85 or 90 %). The simplest prediction would be that monovalent molecules would tend to be antagonists that block the neurotrophins from binding, whereas bivalent compounds could occupy two hot-spots on one receptor molecule giving bivalent antagonists, or on two different receptor molecules giving bivalent agonists. However, the work presented here illustrates that prediction is overly simplistic. Purities assessed via UV detection at 254 nm or via evaporative light scattering (ELS) are different because the techniques have different sensitivity profiles. The crude purities assessed via both methods for the library of molecules 5 were excellent (>90 %) or acceptable (85 – 90 %) except in the cases where serine-containing monovalent molecules were involved, ie fragments r - u. Thus, crude materials were used in the FACS-based screening described below, except for the latter compounds; those were purified by preparative HPLC before screening.
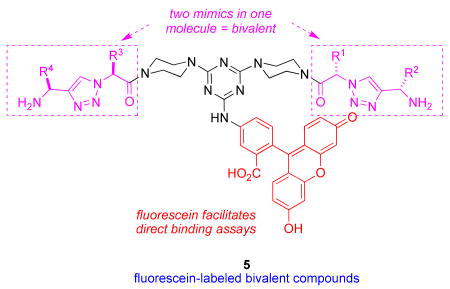
Direct Binding Studies On Fluorescein-labeled Peptidomimetics
Direct FACScan-based binding assays were performed using fluorescein-labeled compounds, testing the binding on NIH-3T3 cells stably transfected to express the neurotrophin receptors: TrkA, TrkC, or p75. These are the receptors to which an NT-3 mimic could potentially bind. NIH-3T3 cells stably transfected to express IGF-1R receptors were used as a control for selectivity.
From a library of 152 fluorescein-labeled-peptidomimetics, we found 10 that bind significantly and selectively to TrkC cells. Of those 10 hits, 5 peptidomimetics also bind with lower affinity to p75 (Figure 2a, for binding data on all fluorescein-labeled compounds see Supplemental Table 1).
Figure 2.

FACScan binding assays. a fluorescein-labeled compounds. Cells expressing the indicated receptor were bound with test ligand (20 μM) at 4°C for 20-30 min. Immediately after washing, cells were acquired and analyzed by FACScan/CellQuest. The background mean channel fluorescence (MCF) of NIH-IGF-1R were subtracted to analyze the specific binding to test cells. As positive controls, anti-receptor mAbs were used for each cell line (anti-TrkA mAb 5C3 for NIH-TrkA, anti-TrkC mAb 2B7 for NIH-TrkC, anti-p75 mAb MC192 for NIH-p75 and αIR3 for NIH-IGF-1R; data not shown). Results shown are average MCF ± sem, n=5 independent experiments. b Biotin-labeled compounds. Cells expressing the indicated receptor were first bound with test ligand (50 μM), followed by fluorescein-avidin at 4°C for 20-30 min. After washing, cells were acquired and analyzed by FACScan/CellQuest. The background MCFs of NIH-IGF-1R was subtracted to analyze the specific binding to test cells. Results shown are average fold-increase in MCF versus background ± sem, n=3 independent experiments.
Syntheses of Biotinylated Analogs Of The Fluoresceinated Peptidomimetics Showing Binding
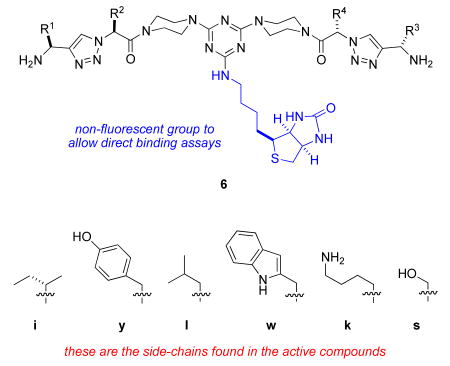
The direct FACS binding assays described above (Figure 2a) showed that 10 of the bivalent mimics 5 bound relatively well to transfected cells expressing the TrkC receptor. Consequently, 10 of these compounds were re-prepared with a biotin label replacing the fluorescein. The purpose of this change was two fold. First, repetition of the FACS binding assays would expose any cases in which the fluorescein label (of compounds 5) contributed significantly to the binding, since the corresponding biotinylated forms 6 would presumably not bind strongly in those cases. In our experience, fluorescein-containing peptides and peptidomimetics are generally less water-soluble and have higher background than their unlabeled or biotin-containing counterparts (unpublished observations). Second, the biotinylated forms are more water-soluble. This is an advantage because data from the cell survival assays that are described below are more reliable if no organic solvents are used to introduce the compounds into the cell medium.
Table 2 shows the structures of the 8 members of series 6 that emerged as the most interesting. That table does not show the structures of 6I (a combination of the amino acid side-chains “YI” and “IE”) and 6J (“YI” and “YE”) because these bound the TrkC transfected cells more weakly than their parent fluoresceinylated compounds 5px and 5vx, respectively.
Table 2.
Summary of the effects of key compounds 6A - H on cells.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| potentiate survival effect of sub-optimal NT-3 | induce neurogenic differentiation | |||||||
| 6 | A | B | C | D | E | F | G | H |
| R1 | i | i | i | y | y | y | y | y |
| R2 | y | y | y | i | i | w | w | w |
| R3 | l | i | i | l | i | l | i | s |
| R4 | w | w | y | w | w | k | k | k |
Direct Binding Studies On Biotin-labeled Peptidomimetics
To verify that the fluorescein moiety was not relevant to the binding, the biotinylated form of the 10 hits were synthesized (6A - J) and tested. For binding assays using biotinylated peptidomimetics, the cells were first incubated with the compounds at 50 μM, followed by fluorescein-avidin. After washing, cells were analyzed by FACS. The results of these binding assays were similar to those obtained with parental fluorescein-labeled peptidomimetics with the following exceptions. First, 6I (analog of 5px) and 6J (analog of 5vx) bind more weakly to TrkC than their parental fluorescein labeled compounds (Figure 2b). For this reason, 6I and 6J were excluded from the ex vivo studies described below. Second, 6A – C have less selectivity for TrkC versus p75, but these compounds were not excluded from further consideration.
Cell Survival Studies
The 8 biotinylated-peptidomimetic hits shown in Table 1 at 20 μM concentration were exposed to the parent cell lines in complete media containing serum. None of the compounds showed significant cytotoxicity in quantitative MTT assays,(17) with the possible exception of 6H that caused a 20% reduction of normal growth in all cells (data not shown).
Figure 3 shows effects of the peptidomimetics on receptor-mediated cell survival using NIH-3T3 cells expressing TrkA, TrkC, or IGF-1R. When cells were placed in serum-free media (SFM) they underwent apoptosis. However, they can be protected by their appropriate growth factors (NIH-TrkA is protected by NGF, NIH-TrkC is protected by NT-3, NIH-IGF-1R is protected by IGF-1). Growth factor protection of cells from apoptosis in serum-free media (SFM) is dose-dependent, and suboptimal doses of growth factor can be used that result quantitatively and consistently in reduced survival (∼30-40% of maximal).
Figure 3.


Cell survival assays. NIH-3T3 cells expressing either a TrkC (NIH-TrkC), b TrkA (NIH-TrkA), or c TrkC and p75 (nnr5-TrkC cells) were cultured in SFM supplemented with the indicated peptidomimetics (20 μM) with or without the appropriate growth factor (NGF for TrkA, NT-3 for TrkC) at optimal or suboptimal concentrations as indicated. Survival was measured in MTT assays, and was calculated relative to optimal neurotrophin (2 nM; 100% survival; 0.2 nM is therefore a suboptimal dose). Results shown are average ± SEM, from at least three independent assays, with each assay n = 4-6.
Five of the “hit” peptidomimetics, 6A - E, potentiate the survival-promoting effect of NT-3, acting as partial agonists. However, these compounds do not afford significant intrinsic trophic activity to cells cultured in SFM, in the absence of neurotrophin. The only exception is that, for the TrkC/p75 expressing cells compounds 6C, 6D, and 6E exhibited some partial agonistic activity in SFM (Figure 3c) in the absence of neurotrophin. The other three hits from the FACS studies, 6F - H, have no significant biological effect (Figure 3a).
In receptor-selectivity controls, none of the 8 hit compounds afford trophic protection of TrkA-expressing cells nor do they potentiate the survival-promoting effect of NGF (Figure 3b). Likewise, none alter the survival of IGF-1R-expressing cells nor do they potentiate the survival-promoting effect of IGF-1 (data not shown). These data are consistent with the binding data, and confirm that cell survival is TrkC receptor-dependent.
As further confirmation of selective activity, similar data were obtained when these three cell lines were tested in survival assays using the fluorescein-labeled compounds (data not shown). The fluorescein-labeled compounds promoted survival only for TrkC-expressing cells.
Data from the FACS studies indicated the hit compounds exhibited low but significant binding to cells that overexpress the p75 receptor. Cellular consequences of activation of the neurotrophin Trk and p75 receptors can be interdependent in some cases. Positive data from the cell survival assays described above featured cells that overexpress only the Trk receptors. To gauge the potential impact of p75, survival was tested in cells expressing TrkC and p75 (nnr5-TrkC: nnr5 cells stably expressing TrkC) or control TrkA and p75 (PC12 cells). The parent nnr5 cells are a variant of PC12 that lost TrkA expression. The objective of these experiments was to determine if the functional effects discussed above changed for cells that also express the p75 receptor. The survival data in Figure 3c for these cells are comparable to that obtained with NIH-3T3 cells expressing TrkC or TrkA in the absence of p75 (not shown) indicating that potentiation of trophic survival effects by 6A - E are not affected by p75 co-expression. Further, the compounds that did not promote cell survival in the TrkC expressing cells, 6F - H, do not gain trophic survival promoting activity when p75 is co-expressed.
Neuritogenic Differentiation
Neuritogenic differentiation is a biological endpoint when neuronal cells are engaged by a neurotrophic growth factor. Moreover, it is known that co-expression of p75 regulates TrkC-mediated differentiation.(47) Thus, the peptidomimetics were tested, at 20 μM, in assays of neuritogenic differentiation on cells expressing TrkC and p75 (nnr5-TrkC, Figure 4a). Quantification of these data is shown in Figure 4b.
Figure 4.
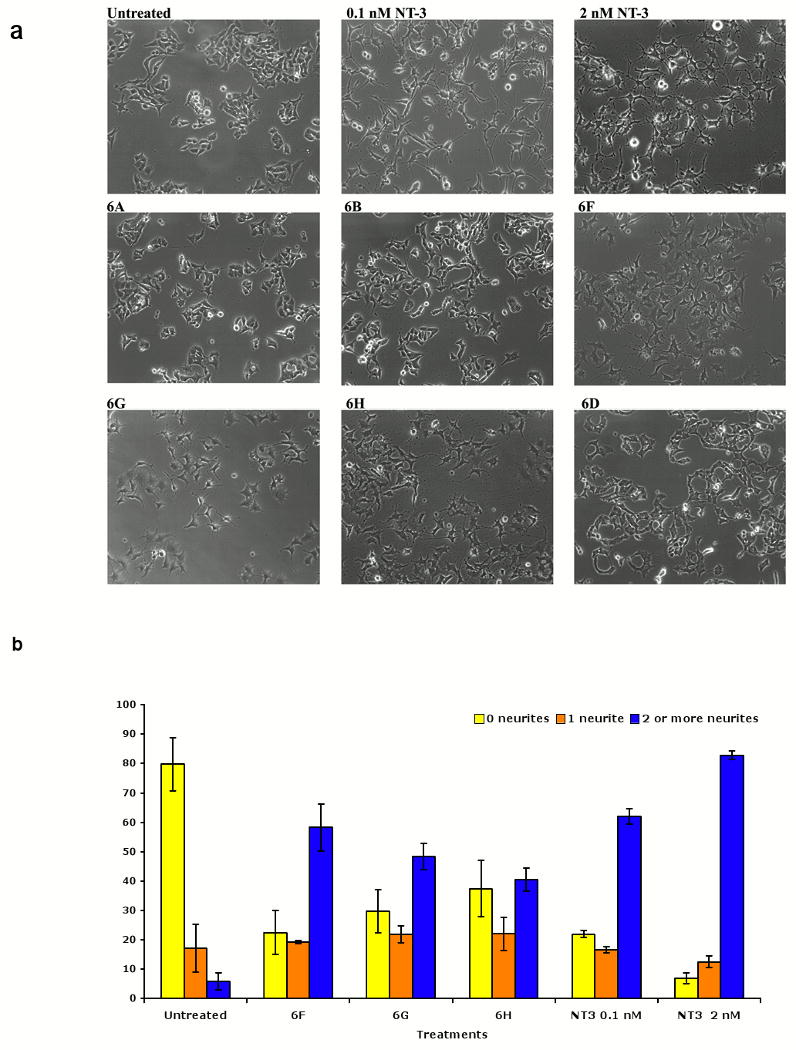
Differential regulation of neuritogenesis by peptidomimetics. Cells expressing TrkC and p75 (nnr5-TrkC) were plated in full medium (untreated cells), ± neurotrophin ± peptidomimetics (20 μM). Fields were photographed 72 h after treatment. Neurite outgrowth was determined as percentage of cells with neurites (defined as length >2 cell bodies). Average ± SEM from three independent experiments, in each experiment measured 5 random pictures/condition were scored. Similar assays using PC12 cells resulted in cellular differentiation by NGF but not by peptidomimetics (data not shown). a Representative pictures from nnr5-TrkC cells. b Quantification of nnr5-TrkC cell differentiation.
NT-3 is known to induce dose-dependent cellular differentiation in cells expressing TrkC and p75 (nnr5-TrkC), optimally at 2 nM and sub-optimally at 0.1 nM. Mimetics 6F - H also were shown to induce significant differentiation of these cells, to a degree comparable to 0.1 nM NT3. These compounds also potentiate the differentiation induced by 0.1 nM NT3 in what appears to be an additive manner (data not shown). In cellular selectivity controls, none of the peptidomimetics that induce the differentiation of nnr5-TrkC cells promote the differentiation of PC12 cells, nor do they potentiate the differentiation induced by 0.2 nM NGF (data not shown).
It is notable that the compounds that promote cellular differentiation do not promote survival (e.g. see Figure 3). Hence, these compounds promote one neurotrophic activity (neurogenic differentiation) but not the other (trophic survival).
Conversely, the mimetics that promote survival (6A - E) do not induce significant differentiation of nnr5-TrkC cells and do not significantly potentiate the differentiation promoted by 0.1 nM NT-3 (data not shown). Hence, these compounds promote one neurotrophic activity (trophic survival) but not the other (neurogenic differentiation).
Signal Transduction and Receptor Activation
The fact that peptidomimetics 6F - H afford neurogenesis but not survival merited further study of the biochemical signals arising from these ligands. Consequently, biochemical assays were done by western blot analyses of the tyrosine phosphorylation and activation state of TrkC, Akt, and MAPK (Figure 5). Thus, PC12 (expressing TrkA and p75) or nnr5-TrkC (expressing TrkC and p75) cells were exposed to the peptidomimetics at 20 μM (and, in controls, to NTFs at 2 nM) for 5 - 20 min and detergent lysates were studied by Western blotting. After 20 min, 6F - H induce significant tyrosine phosphorylation of TrkC, and Akt in nnr5-TrkC cells (Figure 5a, see panel d for quantification). Tyrosine phosphorylation of these proteins is a measure of activation. In control PC12 cells, there is no activation of TrkA, Akt, or MAPK by these compounds at any time point (Figure 5b, 20 min time point shown for p-Akt). These data confirm that the signals activated by these ligands require expression of TrkC. The kinetics of activation are slower than NT-3. At 5 min of ligand exposure, these compounds do not afford significant tyrosine phosphorylation in nnr5-TrkC cells (eg Figure 5c). In addition, there was no significant activation of MAPK by these compounds at any time point in the +TrkC/+p75 cells (data not shown). These biochemical data correlate well with the biological data, as it is known that in the cells tested AKT activation is important for differentiation, whereas MAPK activation is mainly associated with survival.
Figure 5.
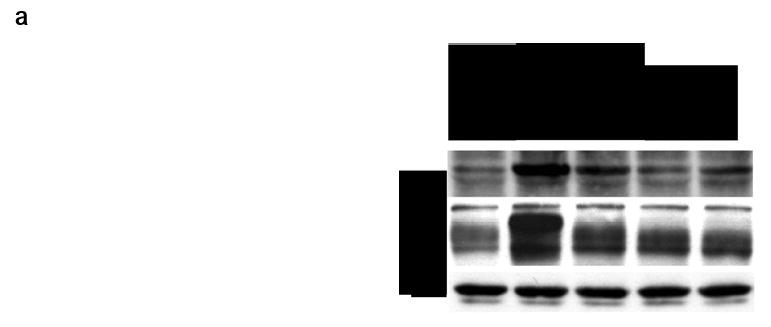
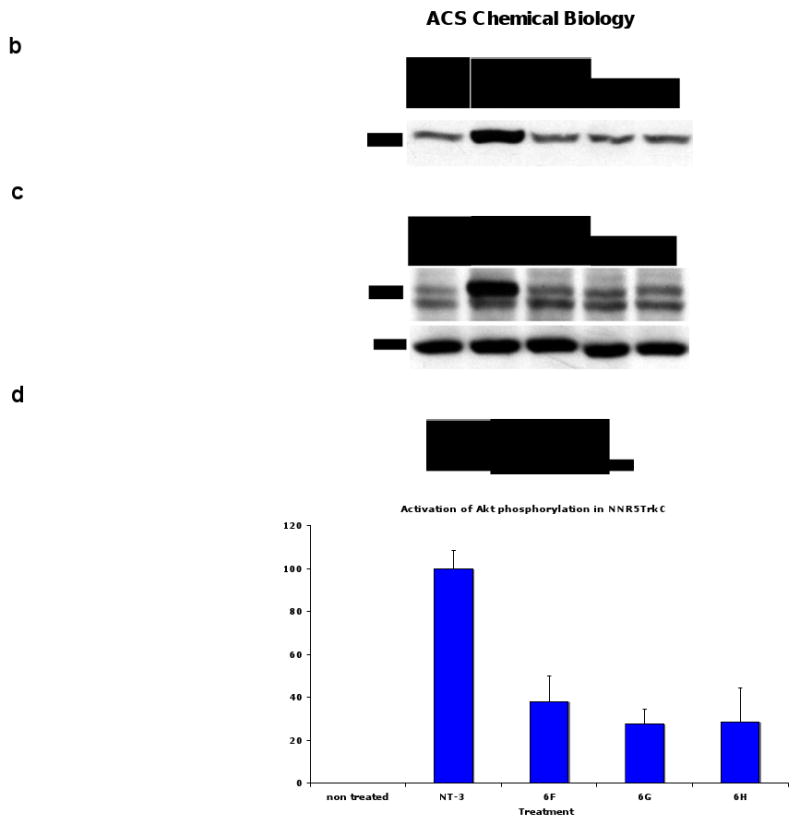
Signal transduction assays. a Effect of peptidomimetics on TrkC and Akt phosphorylation in nnr5-TrkC. Cells were exposed to the indicated peptidomimetics (20 μM) or NT-3 (2 nM) for 20 min. Detergent lysates were analyzed by Western blotting with anti-pTyr mAb 4G10 or anti-phospho-Akt. After stripping, membranes were re-blotted with anti-actin to quantify protein loading. Data representative of 3-4 independent experiments. b Lack of Akt activation by peptidomimetics in PC12. c Similar assays as in panel a, except that ligand stimulation was done for 5 min. d Quantification of data for Akt phosphorylation by densitometry relative to 2 nM NT-3 and represented as average ± SD (n = 4 independent experiments).
Conclusions
Table 2 shows a summary of the structures and activities. Peptidomimetics 6A – E that afford cell survival effects in TrkC expressing cells all contain an isoleucine (8 of 10 monovalent units) or leucine (2 of 10) side-chain in conjunction with tyrosine (6 of 10 monovalent units) or tryptophan (4 of 10). However, the peptidomimetics that induced neuritogenic differentiation 6F – H all contained the monovalent unit with Tyr and Trp side-chains, in conjunction with another: x-Lys where x = leucine, isoleucine or serine.
The fact that some peptidomimetics can afford trophic survival but completely lack neuritogenic differentiation, while others induce neuritogenic differentiation but lack survival promoting action shows it is possible to uncouple these signals by using synthetic ligand. Previously, this uncoupling has only been achievable by mutagenesis of the intracellular domain of receptors, eg a deletion of five-amino acids in the intracellular juxtamembrane region of TrkA abolishes differentiation without affecting survival.(48)
In general, it is quite unusual for agonistic ligand binding at the ectodomain to uncouple signals that are mediated by intracellular adaptor proteins. In principle, this would require the ligand to cause a partial conformational change in the receptor intracellular domain such that only some (but not all) adaptor proteins can be activated. However, two additional scenarios would also explain the biology of peptidomimetics as biased agonists. First, prolonged MAPK signaling leading to differentiation can take place by interaction of the ARMS protein with Trks.(49) Potentially a TrkC-ARMS complex may be preferentially activated by the peptidomimetics that afford differentiation. Second, sustained versus transient signal transduction, and also the kinetics of ligand-induced receptor internalization can affect the functional outcome towards differentiation or survival.(50) It is possible that the peptidomimetics that afford survival may have sustained kinetics of activation, and kinetic studies are in progress.
Biased agonists are of great biological interest, but very few have been documented for receptor tyrosine kinases. Aberrant expression of trkC mRNA and dysfunctional NT-3 responsiveness have been correlated with neurodegenerative diseases such as Alzheimer's disease (AD),(51) motor neuron diseases such as amyotropic lateral sclerosis (ALS).(52) Thus, survival-promoting peptidomimetics may be particularly interesting as therapeutics when trophic support is desired without inducing new neuritic growth and new connections. Neuritogenesis-promoting peptidomimetics may be interesting to study the mechanisms of axonal growth inhibition.
Finally, we note that the side-chains supported on the turn mimics used in this study were chosen by considering ones that are frequently found in hot-spots for protein-protein interactions in general (eg Trp, Ile, Lys), and not for correspondence with the turns of the neurotrophins. It is therefore surprising that so many active compounds were identified. Current studies are devoted to an analogous library where the side chains do correspond with those of the neurotrophins; it will be intriguing to see the cellular activities that these compounds induced. After that comparison, the next steps will be to investigate related unnatural amino acids, then move the most promising compounds on to in vivo studies in models for specific neurodegenerative diseases.
Methods
Cell Lines—NIH3T3 fibroblasts transfected with either human trkA cDNA (NIH-TrkA), human trkC cDNA (NIH-TrkC), or human IGF-1R cDNA (NIH-IGF-1R) were drug-selected (0.5 mg/ml G418) and stable expressing subclones were isolated that express relatively high levels of their receptor (∼200,000–500,000 receptors/cell). Cells are grown under drug selection and are routinely screened for receptor expression by FACScan using monoclonal antibodies directed to the receptor extracellular domains.
For the differentiation assays we used PC12 cells (express TrkA and p75 neurotrophin receptors and respond to NGF) and nnr5-TrkC cells (a variant of PC12 that lost TrkA, and into which human trkC cDNA was stably transfected, respond to NT-3).(53,54)
Antibodies—mouse anti-TrkA mAb 5C3(55), mouse anti-TrkC mAb 2B7 (56) and mouse anti-IGF-1R mAb alphaIR3 were purified using protein G-Sepharose (Pharmacia, Baie d'Urfe, Quebec, Canada).
FACScan analyses—Cells (2×105) in 100 ml of FACScan binding buffer (phosphate-buffered saline, 0.5% BSA, and 0.1% NaN3) were immunostained as described.(17) Saturating mAbs, or non-binding mouse IgGs (background control) were added to cells for 20 min at 4 °C, excess primary antibody was washed off, and cells were immunostained with fluoresceinated goat anti-mouse IgG (fluorescein-G-a-M) secondary antibody. As additional cellular controls NIH3T3 cells not expressing NGF receptors were used (e.g. NIH-IGF-1R or wild type NIH3T3 cells). Cells were acquired on a FACScan, and bellshaped histograms were analyzed using the LYSIS II program.
FACScan analyses of peptidomimetics— Peptidomimetics labeled with fluorescein were used in FACScan binding studies, as described above. The assay was slightly modified to extend the incubation of the “primary” reagent to 40 minutes, followed by two washes to remove unbound material. No secondary reagent was added, as the compounds are directly labeled. In the cases where peptidomimetics were labeled with biotin, the secondary reagent was fluorescein-avidin and the study progressed as described above. For binding assays using biotinylated peptidomimetics, the cells were first incubated with the compounds at 50 μM, followed by fluorescein-avidin. After washing, cells were fixed with 1% paraformaldehyde and analyzed by FACS.
Proliferation and Survival Assays—Cells (5,000–10,000 cells/well) were added to 96-well plates (Becton Dickinson, Lincoln Park, NJ) and cultured either in media containing 5% fetal bovine serum or in serum free media with 0.1% BSA (SFM). Serial dilutions of neurotrophins or peptidomimetics were then added. A suboptimal dose of neurotrophins (0.1 – 0.2 nM, affording ∼25% of survival) was used to test the effect of combination of neurotrophin with peptidomimetics. The proliferative/survival profile of the cells was quantitated using the tetrazolium salt reagent (3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma) and optical density (OD) readings as described.(57)
Neuritogenic differentiation assays—were performed as described.(53,54) Briefly, PC12 cells or nnr5-TrkC cells were plated at low density in complete media and allowed to adhere to the dish. Then the indicated treatment (NGF, NT-3 or peptidomimetics) was added for 48-72 h. The morphology of the cells was scored from pictures taken randomly and in a blinded fashion (4 pictures/well, 4 independent wells per condition).
Western Blots—Blots were performed as described.(57) Briefly, cells were detergent-solubilized, and protein concentrations were determined. Lysates were fractionated by SDS-polyacrylamide gel electrophoresis under reducing conditions, transferred to PDVF membranes (Xymotech Biosystems, Montreal, Quebec, Canada), and immunoblotted with anti-phosphotyrosine (a-pY) antibody 4G10 (Upstate Biotechnology, Lake Placid, NY), anti-phosphoMAPK, or anti-phosphoAKT as indicated. Blots were visualized using the enhanced chemiluminescence (ECL) system (NEN Life Science Products), and data were standardized to total protein loaded, as determined by re-probing membranes with total MAPK, total AKT, and from Coomassie blue staining of the gels.
Supplementary Material
Acknowledgments
We thank Dr Jing Liu for help with the manuscript preparation, and The National Institutes of Health (MH070040, GM076261), The Canadian Institutes of Health Research, and the Robert A. Welch Foundation (A-1121). TAMU/LBMS-Applications Laboratory headed by Dr Shane Tichy provided mass spectrometric support. The NMR instrumentation in the Biomolecular NMR Laboratory at Texas A&M University was supported by a grant from the National Science Foundation (DBI-9970232) and the Texas A&M University System.
References
- 1.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–21. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–91. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 3.Barbacid M. The Trk Family of Neurotrophin Receptors. J Neurobiol. 1994;25:1386–403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 4.Segal RA. Selectivity in Neurotrophin Signaling: Theme and Variations. Annu Rev Neurosci. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Moheban DB, Conway BR, Bhattacharyya A, Segal RA. Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J Neurosci. 2000;20:5671–8. doi: 10.1523/JNEUROSCI.20-15-05671.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaccaro MC, B Lee H, Pattarawarapan M, Xia Z, Caron A, L'Heureux PJ, Bengio Y, Burgess K, Saragovi HU. Selective Small Molecule Peptidomimetic of TrkC and TrkA Receptors Afford Discrete or Complete Neurotrophic Activities. Chemistry & Biol. 2005;12:1015–28. doi: 10.1016/j.chembiol.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Mahadeo D, Kaplan L, Chao MV, Hempstead BL. High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. Implications for multi-subunit polypeptide receptors. J Biol Chem. 1994;269:6884–91. [PubMed] [Google Scholar]
- 8.Carter BD, Dechant G, Frade JM, Kaltschmidt C, Barde YA. Neurotrophins and their p75 receptor. Cold Spring Harbor Symposia on Quantitative Biology. 1996;61:407–15. [PubMed] [Google Scholar]
- 9.Dechant G, Tsoulfas P, Parada LF, Barde YA. The Neurotrophin Receptor p75 Binds Neurotrophin-3 on Sympathetic Neurons with High Affinity and Specificity. J Neurosci. 1997;17:5281–7. doi: 10.1523/JNEUROSCI.17-14-05281.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barker PA. p75NTR: A study in contrasts. Cell Death Differentiation. 1998;5:346–56. doi: 10.1038/sj.cdd.4400375. [DOI] [PubMed] [Google Scholar]
- 11.Rabizadeh S, Ye X, Wang JJ, Bredesen DE. Neurotrophin dependence mediated by p75NTR: contrast between rescue by BDNF and NGF. Cell Death Differentiation. 1999;6:1222–7. doi: 10.1038/sj.cdd.4400614. [DOI] [PubMed] [Google Scholar]
- 12.Zaccaro M, Ivanisevic L, Perez P, Meakin S, Saragovi H. p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J Biol Chem. 2001;276:31023–9. doi: 10.1074/jbc.M104630200. [DOI] [PubMed] [Google Scholar]
- 13.Saragovi HU, Zaccaro MC. Small Molecule Peptidomimetic Ligands of Neurotrophin Receptors, Identifying Binding Sites, Activation Sites and Regulartory Sites. Curr Pharm Des. 2002;8:99–110. doi: 10.2174/1381612023393215. [DOI] [PubMed] [Google Scholar]
- 14.LeSauteur L, Wei L, Gibbs B, Saragovi HU. Small Peptide Mimics of Nerve Growth Factor Bind TrkA Receptors and Affect Biological Responses. J Biol Chem. 1995;270:6564–9. doi: 10.1074/jbc.270.12.6564. [DOI] [PubMed] [Google Scholar]
- 15.LeSauteur L, Cheung NKV, Lisbona R, Saragovi HU. Small Molecule Nerve Growth Factor Analogs Image Receptors in vivo. Nature Biotech. 1996;14:1120–2. doi: 10.1038/nbt0996-1120. [DOI] [PubMed] [Google Scholar]
- 16.Maliartchouk S, Feng Y, Ivanisevic L, Debeir T, Cuello AC, Burgess K, Saragovi HU. A Designed Peptidomimetic Agonistic Ligand of TrkA Nerve Growth Factor Receptors. Mol Pharmacol. 2000;57:385–91. [PubMed] [Google Scholar]
- 17.Maliartchouk S, Debeir T, Beglova N, Cuello A, Gehring K, Saragovi H. Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J Biol Chem. 2000;275:9946–56. doi: 10.1074/jbc.275.14.9946. [DOI] [PubMed] [Google Scholar]
- 18.Longo FM, Manthorpe M, Xie YM, Varon S. Synthetic NGF Peptide Derivatives Prevent Neuronal Death via a p75 Receptor-dependent Mechanism. J Neurosci Res. 1997;48:1–17. doi: 10.1002/(sici)1097-4547(19970401)48:1<1::aid-jnr1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 19.Xie Y, Tisi MA, Yeo TT, Longo FM. Nerve Growth Factors(NGF) Loop 4 Dimeric mimetics Activate ERK and AKT Promote NGF-like Neurotropic Effects. J Biol Chem. 2000;275:29868–74. doi: 10.1074/jbc.M005071200. [DOI] [PubMed] [Google Scholar]
- 20.Xie Y, Longo FM. Neurotrophin small-molecule mimetics. Prog Brain Res. 2000;128:333–47. doi: 10.1016/s0079-6123(00)28030-8. [DOI] [PubMed] [Google Scholar]
- 21.O'Leary PD, Hughes RA. Design of Potent Peptide Mimetics of Brain-derived Neurotrophic Factor. J Biol Chem. 2003;278:25738–44. doi: 10.1074/jbc.M303209200. [DOI] [PubMed] [Google Scholar]
- 22.Beglova N, LeSauteur L, Ekiel I, Saragovi HU, Gehring K. Solution Structure and Internal Motion of a Bioactive Peptide Derived from Nerve Growth Factor. J Biol Chem. 1998;273:23652–8. doi: 10.1074/jbc.273.37.23652. [DOI] [PubMed] [Google Scholar]
- 23.Beglova N, Maliartchouk S, Ekiel I, Zaccaro MC, Saragovi HU, Gehring K. Design and Solution Structure of Functional Peptide Mimetics of Nerve Growth Factor. J Med Chem. 2000;43:3530–40. doi: 10.1021/jm990441x. [DOI] [PubMed] [Google Scholar]
- 24.Burgess K. Solid-Phase Syntheses of β-Turn Analogues To Mimic or Disrupt Protein-Protein Interactions. Acc Chem Res. 2001;34:826–35. doi: 10.1021/ar9901523. [DOI] [PubMed] [Google Scholar]
- 25.Feng Y, Pattarawarapan M, Wang Z, Burgess K. Solid-phase SN2 Macrocyclization Reactions to Form β-Turn Mimics. Org Lett. 1999;1:121–4. doi: 10.1021/ol990597r. [DOI] [PubMed] [Google Scholar]
- 26.Feng Y, Burgess K. Solid Phase SNAr Macrocyclizations to Give Turn-extended-turn Peptidomimetics. Chem Eur J. 1999;5:3261–72. [Google Scholar]
- 27.Pattarawarapan M, Zaccaro MC, Saragovi U, Burgess K. New Templates for Synthesis of Ring-fused C10 β-Turn Peptidomimetics Leading To The First Reported Small Molecule Mimic of Neurotrophin-3. J Med Chem. 2002;45:4387–90. doi: 10.1021/jm0255421. [DOI] [PubMed] [Google Scholar]
- 28.Zaccaro MC, Lee BH, Pattarawarapan M, Xia Z, Caron A, L' Heureux PJ, Bengio Y, Burgess K, Saragovi HU. Selective Small Molecule Peptidomimetic Ligands of TrkA and of TrkC Receptors Afford Discrete or Complete Neurotrophic Activities. Chem Biol. 2005;12:1015–28. doi: 10.1016/j.chembiol.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 29.Bruno MA, Clarke PBS, Seltzer A, Quirion R, Burgess K, Cuello AC, Saragovi HU. Long-Lasting Rescue of Age-Associated Deficits in Cognition and the CNS Cholinergic Phenotype by a Partial Agonist Peptidomimetic Ligand of TrkA. J Neurosci. 2004;24:8009–18. doi: 10.1523/JNEUROSCI.1508-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pattarawarapan M, Reyes S, Xia Z, Zaccaro MC, Saragovi HU, Burgess K. Selective Formation of Homo- and Heterobivalent Peptidomimetics. J Med Chem. 2003;46:3565–7. doi: 10.1021/jm034103e. [DOI] [PubMed] [Google Scholar]
- 31.Reyes SJ, Burgess K. Heterovalent Selectivity and the Combinatorial Advantage. Chem Soc Rev. 2006;35:416–23. doi: 10.1039/b516721n. [DOI] [PubMed] [Google Scholar]
- 32.Price R, Milne SA, Sharkey J, Matsuoka N. Advances in Small Molecules Promoting Neurotrophic Function. Pharmacology & Therapeutics. 2007;115:292–306. doi: 10.1016/j.pharmthera.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Lewis MA, Hunihan L, Franco D, Robertson B, Palmer J, St Laurent DR, Balasubramanian BN, Li Y, Westphal RS. Identification and characterization of compounds that potentiate NT-3-mediated Trk receptor activity. Molecular Pharmacology. 2006;69:1396–404. doi: 10.1124/mol.105.020255. [DOI] [PubMed] [Google Scholar]
- 34.Wilkie N, Wingrove PB, Bilsland JG, Young L, Harper SJ, Hefti F, Ellis S, Pollack SJ. The non-peptidyl fungal metabolite L-783,281 activates TRK neurotrophin receptors. J Neur. 2001;78:1135–45. doi: 10.1046/j.1471-4159.2001.00504.x. [DOI] [PubMed] [Google Scholar]
- 35.Jang SW, Okada M, Sayeed I, Xiao G, Stein D, Jin P, Ye K. Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death. Proc Natl Acad Sci U S A. 2007;104:16329–34. doi: 10.1073/pnas.0706662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin B, Pirrung MC, Deng L, Li Z, Liu Y, Webster NJG. Neuroprotection by small molecule activators of the nerve growth factor receptor. J Pharmacol Exp Ther. 2007;322:59–69. doi: 10.1124/jpet.106.118034. [DOI] [PubMed] [Google Scholar]
- 37.Fournier J, Keane PE, Ferrara P, Soubrie P. SR 57746A: an orally active non-peptide compound with neurotrophic and neuroprotective effects. CNS Drug Rev. 1997;3:148–67. [Google Scholar]
- 38.Massa SM, Xie Y, Yang T, Harrington AW, Kim ML, Yoon SO, Kraemer R, Moore LA, Hempstead BL, Longo FM. Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J Neurosci. 2006;26:5288–300. doi: 10.1523/JNEUROSCI.3547-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Longo FM, Massa SM. Small molecule modulation of p75 neurotrophin receptor functions. CNS Neurol Disord: Drug Targets. 2008;7:63–70. doi: 10.2174/187152708783885093. [DOI] [PubMed] [Google Scholar]
- 40.Longo FM, Yang T, Knowles JK, Xie Y, Moore LA, Stephen MM. Small molecule neurotrophin receptor ligands: novel strategies for targeting Alzheimer's disease mechanisms. Curr Alzheimer Res. 2007;4:503–6. doi: 10.2174/156720507783018316. [DOI] [PubMed] [Google Scholar]
- 41.Pehar M, Cassina P, Vargas MR, Xie Y, Beckman JS, Massa SM, Longo FM, Barbeito L. Modulation of p75-dependent motor neuron death by a small non-peptidyl mimetic of the neurotrophin loop 1 domain. The European journal of neuroscience. 2006;24:1575–80. doi: 10.1111/j.1460-9568.2006.05040.x. [DOI] [PubMed] [Google Scholar]
- 42.Steiner JP, Connolly MA, Valentine HL, Hamilton GS, Dawson TM, Hester L, Snyder SH. Neurotrophic Actions of Nonimmunosuppressive Analogues of Immunosuppressive Drugs FK506, Rapamycin and Cyclosporin A. Nature Med. 1997;3:421–8. doi: 10.1038/nm0497-421. [DOI] [PubMed] [Google Scholar]
- 43.Steiner JP, Hamilton GS, Ross DT, Valentine HL, Guo H, Connolly MA, Liang S, Ramsey C, Li JHJ, Huang W, Howorth P, Soni R, Fuller M, Sauer H, Nowotnik AC, Suzkak PD. Neurotrophic immunophilin ligands stimulate structural and functional recovery in neurodegenerative animal models. Proc Natl Acad Sci USA. 1997;94:2019–24. doi: 10.1073/pnas.94.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. Journal of Neuroscience. 1998;18:104–11. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jordan GR, McCulloch J, Shahid M, Hill DR, Henry B, Horsburgh K. Regionally selective and dose-dependent effects of the ampakines Org 26576 and Org 24448 on local cerebral glucose utilisation in the mouse as assessed by 14C-2-deoxyglucose autoradiography. Neuropharmacology. 2005;49:254–64. doi: 10.1016/j.neuropharm.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 46.Angell Y, Chen D, Brahimi F, Saragovi HU, Burgess K. A Combinatorial Method for Solution-Phase Synthesis of Labeled Bivalent β-Turn Mimics. J Am Chem Soc. 2008;130:556–65. doi: 10.1021/ja074717z. [DOI] [PubMed] [Google Scholar]
- 47.Ivanisevic L, Banerjee K, Saragovi HU. Differential cross-regulation of TrkA and TrkC tyrosine kinase receptors with P75. Oncogene. 2003;22:5677–85. doi: 10.1038/sj.onc.1206864. [DOI] [PubMed] [Google Scholar]
- 48.Meakin S, MacDonald JI. A novel juxtamembrane deletion in rat TrkA blocks differentiative but not mitogenic cell signaling in response to nerve growth factor. J Neurochem. 1998;71:1875–88. doi: 10.1046/j.1471-4159.1998.71051875.x. [DOI] [PubMed] [Google Scholar]
- 49.Arevalo JC, Conde B, Hempstead BI, Chao MV, Martin-Zanca D, Perez P. TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol & Cell Biol. 2000;20:5908–16. doi: 10.1128/mcb.20.16.5908-5916.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arevalo JC, Conde B, Hempstead BI, Chao MV, Martin-Zanca D, Perez P. A novel mutation within the extracellular domain of TrkA causes constitutive receptor activation. Oncogene. 2001;20:1229–34. doi: 10.1038/sj.onc.1204215. [DOI] [PubMed] [Google Scholar]
- 51.Saragovi HU, Zheng W, Maliartchouk S, DiGugliemo GM, Mawal YR, Kamen A, Woo SB, Cuello AC, Debeir T, Neet KE. A TrkA-selective, fast internalizing nerve growth factor-antibody complex induces trophic but not neuritogenic signals. J Biol Chem. 1998;273:34933–40. doi: 10.1074/jbc.273.52.34933. [DOI] [PubMed] [Google Scholar]
- 52.Saragovi HU, Gehring K. Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol Sci. 2000;21:93–8. doi: 10.1016/s0165-6147(99)01444-3. [DOI] [PubMed] [Google Scholar]
- 53.Ivanisevic L, Banerjee K, Saragovi HU. Differential cross-regulation of TrkA and TrkC tyrosine kinase receptors with P75. Oncogene. 2004 doi: 10.1038/sj.onc.1206864. in the press. [DOI] [PubMed] [Google Scholar]
- 54.Ivanisevic L, Zheng W, Woo SB, Neet KE, Saragovi HU. TrkA Receptor “Hot Spots” for Binding of NT-3 as a Heterologous Ligand. J Biol Chem. 2007;282:16754–63. doi: 10.1074/jbc.M701996200. [DOI] [PubMed] [Google Scholar]
- 55.LeSauteur L, Malartchouk S, LeJeune H, Quirion R, Saragovi HU. Potent and Specific p140-TrkA Agonists Derived from an Anti-receptor Monoclonal Antibody. J Neurosci. 1996;16:1308–16. doi: 10.1523/JNEUROSCI.16-04-01308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Esteban PF, Yoon HY, Becker J, Dorsey SG, Caprari P, Palko ME, Coppola V, Saragovi HU, Randazzo PA, Tessarollo L. A kinase-deficient TrkC receptor isoform activates Arf6-Rac1 signaling through the scaffold protein tamalin. J Cell Biol. 2006;173:291–9. doi: 10.1083/jcb.200512013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maliartchouk S, Saragovi HU. Optimal NGF Trophic Signals Mediated by Synergy of TrkA and p75 Receptor-Specific Ligands. J Neurosci. 1997;17:6031–7. doi: 10.1523/JNEUROSCI.17-16-06031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.