

Abstract
Phylogenetic analyses based on concatenated amino acid sequences from orthologous loci from eight genomes of alpha herpesviruses infecting birds provided strong support for the following hypotheses: (1) gallid HV3 is a sister taxon to gallid HV2 but gallid HV1 is not closely related to the other two chicken herpesviruses; (2) meleagrid HV1 is closer to both gallid HV2 and gallid HV3 than is gallid HV1; and (3) within gallid HV2, the virulent GA genome forms an outgroup to both the avirulent CVI988 genome and the highly virulent Md5 and Md11 genomes. Analysis of the pattern of synonymous nucleotide substitution between orthologous genes shared by four complete genomes of gallid HV2 showed strong evidence of past events of homologous recombination that homogenized certain loci between genomes. Eight of these loci represented cases of loci homogenized between the CVI988, on the one hand, and the Md5 and Md11 genomes, on the other hand. Two others represented loci where the GA genome was homogenized with those of Md5 and Md11. The two loci (UL49.5 and RLORF12) that were homogenized among the virulent genomes GA, Md5, and Md11 are candidates for contributing to viral virulence.
A number of alphaherpesviruses (family Herpesviridae, subfamily Alphaherpesvirinae) have been described from birds, including gallid herpesvirus 2 (HV2), the causative agent of Marek’s disease. Gallid HV2 is highly contagious in chickens and induces T cell lymphomas in susceptible birds, causing annual losses to the worldwide poultry industry estimated at US$100 billion (Morrow and Fehler 2004). In the 100 years since Marek’s disease was first described, the disease has substantially increased in severity of symptoms. The first vaccines for gallid HV2 were developed in the late 1960’s and early 1970’s; but new, more virulent strains of the virus have emerged that are resistant to vaccine-induced immune responses (Nair 2005).
The virulence of gallid HV2 is expected to be a polygenic trait, with allelic variants at a number of loci playing a role (Shamblin et al. 2004). Thus, homologous recombination is expected to play an important role in the evolution of virulence, because it can bring together new mutations that originally evolved against different genetic backgrounds. DNA replication in alphaherpesviruses is recombination-dependent, providing a mechanism for homologous recombination between genetically distinct viruses infecting the same cell (Thiry et al. 2004). Sequence analysis has provided evidence of past homologous recombination events in several herpesviruses, including alphaherpesviruses (Bowden et al. 2004; Norberg et al. 2004; Peters et al. 2006).
Evidence of past homologous recombination at protein-coding loci can be obtained by examining the pattern of nucleotide substitution in pairwise comparisons between genomes (Hughes and Friedman 2004, 2005; Hughes and Langley 2006). Particularly useful information can be obtained by examining the distribution of the number of synonymous substitutions per synonymous site (dS) at orthologous protein-coding genes. If dS between two related genomes is unusually high at one locus compared to that at other loci, that is evidence that the sequence at the former locus was acquired through homologous recombination in one or both of the genomes (Hughes and Friedman 2004, 2005; Hughes and Langley 2006). Synonymous sites rather than nonsynonymous sites are used for these analyses because the rate of substitution at the latter is expected to differ markedly among genes as a result of different intensities of purifying selection (Nei 1987).
Here we use homology search to identify putatively orthologous loci in genomes of gallid HV1, gallid HV2, gallid HV3, meleagrid HV1, and psittacid HV1. In the case of gallid HV2, four complete genomes were available analysis; these include the highly virulent Md5 and Md11 (Tulman et al. 2000; Niikura et al. 2006); the virulent GA strain (Eidson and Schmittle 1968; Brunovskis and Velicer 1995; Lee et al. 2000); and the avirulent strain CVI988, which has been used as the basis of a vaccine (Rispens et al. 1972; Spatz et al. 2007). We apply phylogenetic analysis to orthologous sequences in order to understand the evolutionary relationships of these viruses. In addition, we examine the patterns of synonymous nucleotide substitution at orthologous loci shared by four genomes of gallid HV2 in order to identify loci involved in past events of homologous recombination.
Methods
The sequences of 8 avian herpesvirus complete genomes were retrieved from the NCBI database: gallid HV1 (NC_006623); gallid HV2 CVI988 (DQ530348); gallid HV2 GA (AF147806); gallid HV2 Md5 (AF243438); gallid HV2 Md11 (AY510475); gallid HV3 (NC_002577); meleagrid HV1 (NC_002641); and psittacid HV1 (NC_005264). Gene families were identified by applying the Blastclust software (Altschul et al.1997) to predicted protein translations with the parameter L (minimum length coverage) set at 0.5 and the parameter S (BLAST score divided by the alignment length) set at 0.3. Putative orthologs were identified as families with exactly one representative per genome. Orthologs were aligned at the amino acid level using Clustal W (Thompson et al. 1994) and the alignment imposed on the DNA sequences.
Because of the long repeat regions of gallid HV2 are known to include duplicated copies of certain genes (Tulman et al. 2000), in identifying orthologs we applied the Blastclust program separately to duplicated regions. We applied the Dotter (Sonhammer and Durbin 1995) and Pip-Maker (Schwartz et al. 2000) programs to the complete genome DNA sequences in order identify quasi-identical repeats within each genome. Genes encoded within the repeats were identified and subtracted from the rest of the gene repertoire for each genome. Search for orthologs using Blastclust was done on two sets of genes: genes encoded outside the duplicated region and genes encoded within the duplicated region. Orthologs from both sets were then combined to perform the rest of the analysis.
The number of synonymous nucleotide substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) were estimated for all pairwise comparisons of orthologs among the four genomes of gallid HV2 by Nei and Gojobori’s (1986), Li’s (1993), and Yang and Nielsen’s (2000) methods. All methods yielded essentially identical results, as expected since dS and dN were low (< 0.1 in all comparisons). Therefore we report on the results of the Nei and Gojobori method, which has the lowest variance of estimation (Nei and Kumar 2000).
Phylogenetic analyses of all eight viral genomes were applied to concatenated protein sequences because synonymous sites were saturated or nearly so in most comparisons between different viral species. We used a variety of methods including the following: (1) maximum parsimony (MP) using branch-and-bound search (Swofford 2003); (2) neighbor-joining (NJ; Saitou and Nei 1986) based on the gamma distance which accounts for rate variation among sites, using the MEGA 3 program (Kumar et al. 2004); and (3) the quartet maximum likelihood method (QML) using the TREE-PUZZLE 5.2 program (Strimmer and von Haeseler 1996;Schmidt et al. 2002) based on the JTT model (Jones et al. 1992) with gamma correction for rate variation among sites. The shape parameter of the gamma distribution was estimated by the TREE-PUZZLE 5.2 program. Phylogenies of the four gallid HV2 genomes were constructed by the NJ method independently on the basis of dS and dN. Reliability of branching patterns in MP and NJ trees were assessed by bootstrapping (Felsenstein 1985). In the case of QML trees, the percent of puzzling steps supporting a branch represents a measure of the relative confidence in the branch.
In order to identify gallid HV2 genes involved in past recombination events, we identified genes with unusually high degrees of synonymous divergence by cluster analysis applied to the matrix of pairwise dS values among the four gallid HV2 genomes (Hughes and Friedman 2004, 2005; Hughes and Langley 2007). We conducted non-hierarchical k-means clustering using McQueen’s algorithm (Johnson and Wichern 1992). This is a method of creating clusters of observed multivariate data points such that variability within clusters is minimized and variability between clusters is maximized. Starting with k = 2, we increased k by steps of one until we identified a cluster of genes with anomalous dS values.
Results
Phylogenetic Analysis
We identified 31 orthologues present in the genomes of gallid HV1; gallid HV2 CVI988; gallid HV2 GA; gallid HV2 Md5; gallid HV2 Md11; gallid HV3; meleagrid HV1; and psittacid HV1. When the location of each gene in gallid HV1 was plotted against the location of the same gene in gallid HV2 Md5, we observed a pattern indicative of two large-scale inversions between these two genomes (Figure 1A). The inverted regions corresponded minimally to positions 19,461-49,439 and 111,204-118,782 of gallid HV2 Md5 (Figure 1A). By contrast, the relationship between positions in gallid HV3 and that in gallid HV2 Md5 was much more linear (Figure 1B). The only except to this linearity was the UL10 gene, encoding glycoprotein M, located at positions 30824-32098 in gallid HV3 but 43848-45122 in gallid HV2 Md5 (Figure 1B). This suggests that a translocation of the UL10 open reading frame has occurred since the common ancestor of gallid HV2 and gallid HV3.
Figure 1.

Plot of genomic location (start site) of 31 orthologous genes in (A) gallid HV1 and gallid HV2 Md5; and (B) gallid HV3 and gallid HV2 Md5.
Phylogenetic trees were constructed by a variety of methods based on the concatenated sequences of the proteins encoded by the 31 orthologous genes (Figure 2). When rooted by the mid-point method, the cluster of gallid HV1 and psittacid HV1 formed an outgroup to the other sequences (Figure 2). Assuming this rooting, meleagrid HV1 formed an outgroup to gallid HV3 and gallid HV2; and gallid HV3 formed an outgroup to the four genotypes of gallid HV2 (Figure 2). These patterns received strong support in all phylogenetic methods (Figure 2). This result is consistent with a previous phylogenetic analysis by Thureen and Keeler (2006) based on the DNA polymerase (UL30) protein.
Figure 2.
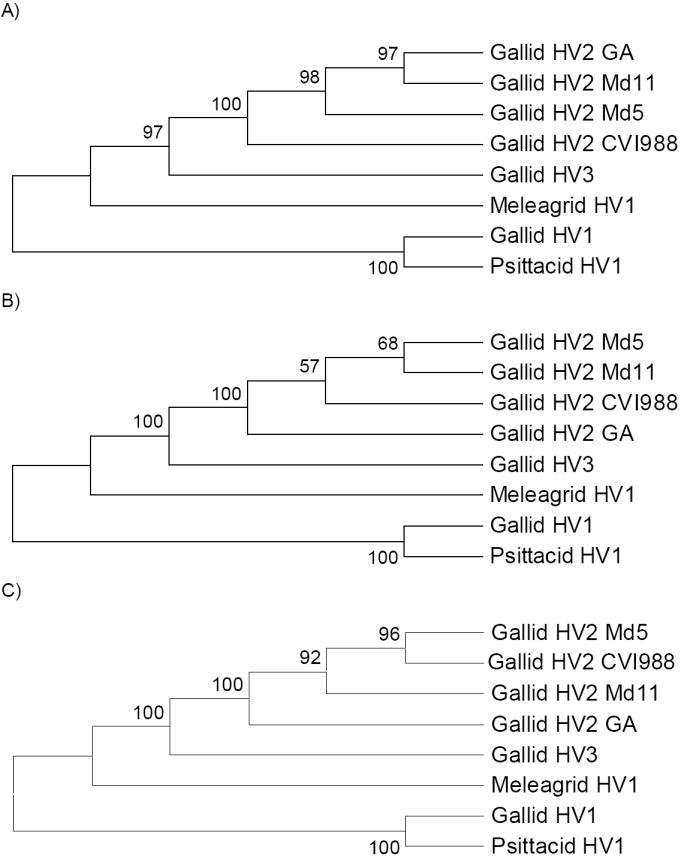
Phylogenetic trees (showing topology only) based on concatenated amino acid sequences of 31 orthologous genes from avian alphaherpesvirus genomes (15,476 aligned amino acid sites); trees are rooted by the mid-point method: (A) NJ tree based on the gamma distance (shape parameter = 1.16); (B) MP tree; and (C) QML tree based on the JTT with gamma (shape parameter = 1.16). Numbers on the branches are percentage of bootstrap samples supporting the branch (A and B) or percentage of puzzling steps supporting the branch (C).
However, the phylogenetic methods did not agree with one another with respect to the relationships of gallid HV2 genomes. Both MP and QML placed gallid HV2 GA as an outgroup to the other gallid HV2 genomes, though with quite weak bootstrap support in the case of MP (Figure 2B-C). By contrast, NJ based on the gamma distance placed gallid HV2 CVI988 as an outgroup to the other three gallid HV2 genomes, with significant (98%) bootstrap support (Figure 2A). The NJ tree clustered gallid HV2 Md11 with gallid HV2 GA, likewise with significant (97%) bootstrap support (Figure 2A). By contrast, the MP tree clustered gallid HV2 Md11 with gallid HV2 Md5, although the bootstrap support was relatively low (Figure 2B). The QML tree, on the other hand, gave strong support to yet another pattern, clustering gallid HV2 Md5 and Gallid HV2 CVI988 (Figure 2C).
Recombination in Gallid HV2
We identified 74 orthologs shared by the four genomes of gallid HV2 and estimated the number of synonymous nucleotide substitutions per synonymous site (dS) pairwise between each genome at each of these loci. We then applied k-means clustering to these dS values in order to identify clusters of genes with distinctive patterns of synonymous substitution. With k = 3, three clusters were identified that showed strikingly distinctive patterns (Table 1). Mean dS values differed significantly among the three clusters for all comparisons except that between the highly similar Md5 and Md11 genomes (Table 1). Cluster 1 included 64 genes for which dS values were lowest between Md5 and Md11; intermediate between those two genomes and CVI988; and highest between GA and the other three genomes (Table 1). Cluster 2 consisted of eight genes that showed no synonymous differences among Md5, Md11, and CVI988, but relatively high dS values between these three genomes and GA (Table 1). Cluster 3 included three genes that showed no synonymous differences among Md5, Md11, and GA, but relatively high dS values between the latter three genomes and CVI988 (Table 1).
Table 1.
Mean dS (± S.E.) in clusters of gallid HV2 genes identified by cluster analysis.
| Comparison | Cluster 1 (N = 64) | Cluster 2 (N=8) | Cluster 3 (N = 2) | |
|---|---|---|---|---|
| Md5 | vs. MD11 | 0.00046 ± 0.00023 | 0.00000 ± 0.00000 | 0.00000 ± 0.00000 |
| vs. CVI988 | 0.00205 ± 0.00042 | 0.00000 ± 0.00000 | 0.04425 ± 0.00005a | |
| vs. GA | 0.00468 ± 0.00075 | 0.04855 ± 0.00746 | 0.00000 ± 0.00000a | |
| Md11 | vs. CVI988 | 0.00210 ± 0.00042 | 0.00000 ± 0.00000 | 0.04425 ± 0.00005a |
| vs. GA | 0.00473 ± 0.00075 | 0.04854 ± 0.00746 | 0.00000 ± 0.00000a | |
| CVI988 | vs. GA | 0.00519 ± 0.00076 | 0.04855 ± 0.00746 | 0.04425 ± 0.00005a |
Difference among cluster means significant at P < 0.001 (one-way ANOVA).
Cluster 2 genes thus appeared to correspond to loci at which recombination has acted to homogenize CVI988 with Md5 and Md11, whereas cluster 3 included cases where recombination has homogenized GA with Md5 and Md11. Phylogenetic analyses based on both dS and dN in the concatenated coding sequences of the genes in each cluster supported these interpretations (Figure 3A-F). These phylogenetic trees were rooted by the midpoint method. In the case of Cluster 1, Md5 and Md11 clustered together with highly significant (100%) bootstrap support in trees based on both dS and dN, while GA showed the longest branch and was thus used as outgroup (Figure 3A-B). In the case of Cluster 2, GA again showed the longest branch, while Md5, Md11, and CVI988 were identical at synonymous sites (Figure 3C). In the tree based on dN in Cluster 2 genes, Md11 and CVI988 clustered together with modest (77%) bootstrap support; but distances among Md11, CVI988, and Md5 were very small compared to those between these genomes and GA (Figure 2D). On the other hand, in the case of cluster 3, Md5, Md11, and GA were all identical at both synonymous and nonsynonymous sites, while CVI988 showed a long branch (Figure 3E-F).
Figure 3.

NJ trees of four gallid HV2 genomes based on concatenated coding sequences of orthologous genes: (A) dS in Cluster 1; (B) dN in Cluster 1 (Table 1); (C) dS in Cluster 2; (D) dN in Cluster 2; (E) dS in Cluster 3; (F) dN in Cluster 3; (G) dS in orthologs shared with gallid HV3, rooted with gallid HV3 and excluding members of Cluster 2 and Cluster 3 (topology only shown). Numbers of aligned codons were as follows: A-B: 34,488: C-D: 3,408; E-F: 162; and G: 32,305.
In order to test further the appropriateness of rooting trees of non-recombined genes from gallid HV2 using GA as an outgroup (Figure 3A-B), we assembled a data set of orthologs shared by gallid HV3 and the four gallid HV2 genomes. This data set included 8 of the 10 members of clusters 2 and 3 involved in putative recombination events; excluding these genes, there were 55 remaining genes. An NJ tree based on dS in the concatenated sequence of these 55 genes was constructed using gallid HV3 as the outgroup (Figure 3G). In this tree, the hypothesis that GA forms an outgroup to the other three gallid HV2 genomes was supported by a significant (96% bootstrap support) internal branch (Figure 3G).
Cluster 2 included two sets of loci physically adjacent in the gallid HV2 genome: (1) UL6 and UL7; and (2) UL16, UL17, and UL18 (Table 2). The fact that these loci are linked suggests that a single recombination event may have acted to homogenize UL6 and UL7 between CVI988 and a genome closely related to Md5 or Md11. Similarly, a single recombination event may have acted to homogenize UL16, UL77, and UL18 between CVI988 and a genome closely related to Md5 or Md11.
Table 2.
Members of clusters 2 and 3.
| Cluster | Gene symbol (Md11; AY510475) | Protein function |
|---|---|---|
| Cluster 2 | LORF1 | Hypothetical |
| UL6 | Capsid protein | |
| UL7 | Capsid protein | |
| UL12 | Deoxyribonuclease | |
| UL16 | Tegument protein | |
| UL17 | Tegument protein | |
| UL18 | Capsid protein | |
| UL32 | Nuclear phosphoprotein | |
| Cluster 3 | UL49.5 | Envelope protein |
| RLORF12 | Binds growth-related translationally controlled tumor protein |
Discussion
Phylogenetic analyses based on concatenated amino acid sequences from 31 orthologous loci from eight genomes of alpha herpesviruses infecting birds provided strong support for the hypothesis that gallid HV3 is a sister taxon to gallid HV2 (Figure 2). By contrast, gallid HV1 was not found to be closely related to the other two chicken herpesviruses (Figure 2). This conclusion was further supported by evidence for large-scale genomic inversions between gallid HV2 and gallid HV1 but not between gallid HV2 and gallid HV3 (Figure 1). In addition, the phylogenetic analyses supported the hypothesis that the turkey virus meleagrid HV1 was closer to both gallid HV2 and gallid HV3 than was gallid HV1 (Figure 2). Thus, the herpesviruses infecting the chicken (Gallidae) did not form a monophyletic group apart from the virus infecting turkey (Meleagridae), indicating that the phylogeny of these viruses is not congruent with that of their hosts and therefore supporting the hypothesis of past host switches in the evolution of these viruses.
Analysis of the pattern of synonymous nucleotide substitution between orthologous genes shared by four complete genomes of gallid HV2 showed strong evidence of past events of homologous recombination that homogenized certain loci between genomes. Eight of these loci represented cases where the CVI988 genome closely resembled Md5 and Md11. Since these eight loci included two groups of adjacent loci, one including two loci and the other three loci, the homogenization of the eight loci need only have required five separate recombinational events. In addition, there were two loci at which GA was identical to Md5 and Md11.
The phylogenies based on concatenated amino acid sequences at 31 orthologous loci acid sequences at 31 orthologous loci did a poor job of resolving the relationships of the four gallid HV2 genomes. This may have occurred partly as a result of the confounding effects of recombination on the phylogenetic analysis, since three of the loci implicated in recombination events (UL6, UL12, and UL18; Table 2) were among the 31 loci used in these analyses. However, another factor may simply have been the relatively small numbers of amino acid differences among the four gallid HV2 genomes and the consequent large stochastic error. On the other hand, a phylogenetic tree of gallid HV2 genomes rooted with gallid HV3 and excluding genes implicated in recombination events provided strong support for the hypothesis that the virulent GA genome forms an outgroup to the both the avirulent CVI988 genome and the highly virulent genomes Md5 and Md11 (Figure 3G).
The results of these analyses suggest that, in reconstructing the evolutionary relationships among a set of genomes encompassing a wide range of evolutionary distances, it may be helpful to approach the problem in a stepwise fashion, using amino acid sequences to construct the phylogeny of more distantly related taxa and DNA sequences to resolve the relationships of closely related taxa, as done here (compare Figures 2 and 3G).
Because the virulent genome GA clustered apart from the two highly virulent genomes Md5 and Md11 in the phylogenetic tree (Figure 3G), recombination among these genomes may have been involved in the transmission of virulence between these two lineages of gallid HV2. As a consequence, the two loci (UL49.5 and RLORF12) that were homogenized among GA, Md5, and Md11 (Table 2) appear to be candidates for contributing to virulence. Consistent with this hypothesis is evidence that the envelope protein encoded by the UL49.5 gene of gallid HV2 is essential for cell-to-cell spread in vitro (Tischer et al. 2002), suggesting an important role in the infection process. Moreover, in the completely sequenced genome of CVI988 (DQ530348) there is a premature stop codon in the RLORF12 reading frame, and deletions in this gene have been reported in other avirulent strains of gallid HV2 (Spatz and Silva 2006).
Given the avirulent nature of CVI988, recombination events involving this genome are unlikely to have involved virulence factors. Nonetheless, the loci involved recombination events among CVI988, Md5, and Md11 included two loci encoding tegument (UL16 and UL17) proteins (Table 2); and there is evidence of host antibody responses directed against tegument proteins in a number of herpesviruses (Lazzarotto et al. 2001; van Drunen Little-van den Hurk 1995). Thus these recombination events also genotypes may have played a role in the evolution of viral interactions with the host.
Acknowledgments
This research was supported by grant GM43940 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden R, Sakaoka H, Donnelly P, Ward R. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect Genet Evol. 2004;4:115–123. doi: 10.1016/j.meegid.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Brunovskis P, Velicer LF. The Marek’s disease virus unique short region: alphaherpesvirus-homologous, fowlpox virus-homologous and MDV-specific genes. Virology. 1995;206:324–338. doi: 10.1016/s0042-6822(95)80048-4. [DOI] [PubMed] [Google Scholar]
- Eidson CS, Schmittle SC. Studies on acute Marek’s Disease I. Characteristics of isolate GA in chickens. Avian Dis. 1968;12:467–476. [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Patterns of sequence divergence in 5’ intergenic spacers and linked coding regions in 10 species of pathogenic Bacteria reveal distinct recombinational histories. Genetics. 2004;168:1795–1803. doi: 10.1534/genetics.104.032979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Nucleotide substitution and recombination at orthologous loci in Staphylococcus aureus. J Bacteriol. 2005;187:2698–2704. doi: 10.1128/JB.187.8.2698-2704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Langley KJ. Nucleotide usage, synonymous substitution pattern, and past recombination in genomes of Streptococcus pyogenes. Infect Genet Evol. 2006;7:188–196. doi: 10.1016/j.meegid.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Johnson R, Wichern D. Applied Multivariate Statistical Methods. 3. Prentice-Hall; Englewood Cliffs NJ: 1992. [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lazzarotto T, Varani S, Gabrielli L, Pignatelli S, Landini MP. The tegument protein ppUL25 of human cytomegalovirus (CMV) is a major target antigen for the anti-CMV antibody response. J Gen Virol. 2001;82:335–338. doi: 10.1099/0022-1317-82-2-335. [DOI] [PubMed] [Google Scholar]
- Lee LF, Wu P, Sui D, Ren D, Kamil J, Kung HJ, Witter RL. The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc Natl Acad Sci USA. 2000;97:6091–6096. doi: 10.1073/pnas.97.11.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH. Unbiased estimates of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- Morrow C, Fehler F. Marek’s disease a worldwide problem. In: Davison TF, Nair V, editors. Marek’s Disease: An Evolving Problem. Elsevier Academic Press; London: 2004. pp. 49–61. [Google Scholar]
- Nair V. Evolution of Marek’s disease – a paradigm for incessant race between the pathogen and the host. Vet J. 2005;170:175–183. doi: 10.1016/j.tvjl.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford University Press; New York: 2000. [Google Scholar]
- Niikura M, Dodgson J, Cheng H. Direct evidence of host genome acquisition by the alphaherpesvirus Marek’s disease virus. Arch Virol. 2006;151:537–549. doi: 10.1007/s00705-005-0633-7. [DOI] [PubMed] [Google Scholar]
- Norberg P, Bergström T, Rekabdar E, Lindh M, Liljeqvist J-Ǻ. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J Virol. 2004;78:10755–10764. doi: 10.1128/JVI.78.19.10755-10764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Tyler SD, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J Virol. 2006;80:9850–9860. doi: 10.1128/JVI.00715-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rispens BH, van Vloten H, Mastenbroek N, Maas H, Schat KA. Control of Marek’s disease in the Netherlands. I Isolation of an avirulent Marek’s disease virus (strain CVI 988) and its use in laboratory vaccination trials. Avian Dis. 1972;16:108–125. [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W. PipMaker – a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamblin CE, Greene N, Arumugaswami V, Dienglewicz RL, Parcells MS. Comparative analysis of Marek’s disease virus (MDV) glycoprotein-, lytic antigen pp38- and transformation antigen Meq-encoding genes: association of meq mutations with MDVs of high virulence. Vet Microbiol. 2004;102:147–167. doi: 10.1016/j.vetmic.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Sonhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Petherbridge L, Zhao Y, Nair V. Comparative full-length sequence analysis of oncogenic and vaccine (Rispens) strains of Marek’s disease virus. J Gen Virol. 2007;88:1080–1096. doi: 10.1099/vir.0.82600-0. [DOI] [PubMed] [Google Scholar]
- Spatz SJ, Silva RF. Polymorphisms in the repeat long regions of oncogenic and attenuated pathotypes of Marek’s disease virus 1. Virus Genes. 2006 doi: 10.1007/s11262-006-0024-5. in press. [DOI] [PubMed] [Google Scholar]
- Strimmer K, von Haeseler A. Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996;13:964–969. [Google Scholar]
- Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods) Sinauer; Sunderland MA: 2002. [Google Scholar]
- Thiry E, Meurens F, Muylkens B, McVoy M, Gogev S, Thuiry J, Vanderplasschen A, Epstein A, Keil G, Schynts F. Recombination in alphaherpesviruses. Rev Med Virol. 2005;15:89–103. doi: 10.1002/rmv.451. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improvement of the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thureen DR, Keeler CL., Jr Psittacid herpesvirus 1 and infectious laryngotracheitis virus: comparative genome sequence analyses of two avian alphaherpesviruses. J Virol. 2006;80:7863–7872. doi: 10.1128/JVI.00134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer BK, Schumacher D, Messerle M, Wagner M, Osterrieder N. The products of the UL10 (gM) and the UL49.5 genes of Marek’s disease virus serotype 1 are essential for virus growth in cultured cells. J Gen Virol. 2002;83:997–1003. doi: 10.1099/0022-1317-83-5-997. [DOI] [PubMed] [Google Scholar]
- Tulman ER, Afonso CL, Lu Z, Zak L, Rock DL, Kutish GF. The genome of a very virulent Marek’s disease virus. J Virol. 2000;74:7980–7988. doi: 10.1128/jvi.74.17.7980-7988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Drunen-Little-van den Hurk S, Garzon S, van den Hurk JV, Babiuk LA, Tijssen P. The role of the major tegument protein VP8 of bovine herpesvirus-1 in infection and immunity. Virology. 1995;206:413–425. doi: 10.1016/s0042-6822(95)80057-3. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]