

Abstract
Experimental autoimmune uveitis (EAU) in its several variants represents human autoimmune uveitis and has been instrumental in obtaining insights into the basic mechanisms of disease. Studies have uncovered that in addition to CD4+ Th1 cells, uveitis can be induced also by CD8+ T cells. Antibodies may have a secondary role after the blood–retinal barrier has been broken. The role in uveitis of a recently discovered IL-17-producing effector T cell type, Th17, is being intensively studied. Th17 cells elicit EAU, can be found in uveitic eyes along with Th1 cells, and are dominant in some types of EAU. In other types of EAU, Th1 cells have a dominant role. The dominant effector type is at least in part determined by conditions under which initial exposure to self-antigen occurs. These findings shed light on the heterogeneity of human disease and may ultimately help to develop better and more rational treatment strategies for human uveitis.
Keywords: Autoimmune disease, uveitis, Th1, Th17, IL-23, IL-17, IFN-γ
Introduction
Experimental autoimmune uveitis in animals serves as a model of human autoimmune uveitis. Experimental auto-immune uveitis (EAU) can be induced in a variety of rodents and in primates, but the most useful for basic studies is of course the mouse, and consequently, it is the only species that will be discussed here. Although EAU is traditionally induced by immunization with a retinal antigen (Ag) such as interphotoreceptor retinoid-binding protein (IRBP) in complete Freund’s adjuvant (CFA), the disease can also be induced in unimmunized recipients by infusion of activated lymphocytes, cultured from immunized donors [1]. More recently, yet another variant of EAU was described, elicited by infusion of Flt3L-mobilized splenic dendritic cells (DC), matured in vitro with bacterial endotoxin (LPS) and anti-CD40 antibody, and pulsed with Ag [2]. Although no animal model covers the full spectrum of human disease, each of these variants has unique characteristics that may make it appropriate to model specific aspects of human uveitis, and each has contributed to dissecting basic mechanisms of disease.
A special variant of EAU is the transgenic model, where mice are made to express neo-self-Ags in the retina, and disease is induced by immunization with the neo-Ag in CFA or by infusion of activated T cells expressing the specific receptor for this Ag. Another special case is the “humanized” EAU model in human leukocyte antigen (HLA)-transgenic mice. These mice develop EAU with retinal arrestin (retinal soluble Ag, S-Ag) much more readily than their wild-type (WT) parental strains. In both these cases, although the target Ags differ, the mechanisms are similar to EAU induced with IRBP or its fragments.
Although it is not known whether the same retinal Ags that elicit EAU in animals are involved in human uveitis, it is believed that the underlying basic mechanisms are shared. This is supported by demonstration of responses to retinal Ags in human uveitis patients at the level of T cells as well as antibodies and by the fact that therapeutic modalities that have been successful in modulating EAU often were effective clinically. Examples of such therapies are cyclosporin and other macrolides (now clinically approved therapies) as well as oral tolerance and interleukin (IL)-2 receptor targeting [3].
Ag-specific effector T cells as inducers and orchestrators of EAU pathology
EAU is a T cell-dependent disease. Studies in the 1990s with long-term Ag-specific T cell lines that were able to induce EAU upon infusion to otherwise unmanipulated recipients led to identification of the autopathogenic effector T cells as CD4+, interferon (IFN)-γ-producing Th1 type cells [4, 5]. IL-4-producing Th2 type cells were believed to be protective, by virtue of being counter-regulatory to Th1. This simplistic view has since been modified and expanded. Firstly, Th2 cells were also shown to have the ability to induce uveitis, provided that one used immunodeficient hosts [6]. Secondly, CD8+ cells, initially thought to be suppressive rather than pathogenic in EAU, based on their role in eye-derived tolerance and on the fact that their depletion did not affect EAU development, were demonstrated to also be able to induce retinal pathology [7, 8]. However, tissue damage was mild compared to that induced by CD4+ T cells.
Experiments in mice and in rats demonstrated that a surprisingly small number of Ag-specific CD4+ T cells are actually needed to induce EAU [9, 10]. Calculating from the number of cells that can be visualized in the retina of mice or rats adoptively transferred with a known number of (labeled) uveitogenic T cells and the minimal number of such cells required to induce disease, 10–15 Ag-specific pathogenic T cells are needed to jumpstart the EAU process. This calls for tremendous amplification of effector mechanisms, which are provided by recruitment of leukocytes from the circulation. It seems that each type of Ag-nonspecific recruited cells has a role in pathogenesis, as depletion of individual cell types inhibits disease. Thus, depletion of granulocytes (though not of eosinophils alone) prevented development of EAU [11], as did depletion of macrophages [12]. This indicates that both these innate inflammatory effector cells are needed for pathogenesis, and they cannot substitute for each other in the process of inflammation. It is interesting to note that athymic recipients transferred with a uveitogenic T cell line have a reduced ability to develop EAU, suggesting that recruitable non-Ag-specific T cells are also part of the inflammatory amplification and/or effector mechanisms [13].
The role of serum antibodies in EAU was for a long time unclear. Immunization with a retinal Ag to induce uveitis invariably results in production of serum antibodies; however, whereas the transfer of immune T cells induces full-blown EAU, the transfer of hyperimmune serum into healthy recipients fails to result in disease. We found that uveitogenic T cells and antibodies synergize to elicit pathology. If antibodies are injected together with a suboptimal number of pathogenic T cells that by itself is insufficient to induce significant EAU, severe pathology can result [14]. Thus, although antibodies are unable to enter into the healthy eye, once the blood–retinal barrier is disrupted by the T cells, antibodies provide a disease-amplifying mechanism.
Most recently, a new Ag-specific autopathogenic effector T cell subset has been recognized, known as Th17, after its characteristic cytokine product, IL-17. This “new kid on the block” has over the past few years taken the scientific stage by storm and has replaced the Th1 in the thinking of many researchers as the major culprit in autoimmune tissue damage. The following paragraphs will be largely devoted to dissecting and placing in context the information that has accumulated thus far on this new effector phenotype, so as to gain perspective on its role in uveitis and in cell-mediated autoimmunity in general.
IL-12 and IL-23 as master regulators of effector T cell responses
Since its discovery in 1989, IL-12, a heterodimeric cytokine composed of the subunits p40 and p35, was known as the signature cytokine produced by the innate immune cells that influences the effector responses of innate and adaptive cell mediated immunity. IL-12 was established as the major cytokine promoting commitment of naïve CD4 cells into the Th1 effector pathway, as well as a potent stimulator of IFN-γ production from natural killer (NK) and CD8 T cells. For over a decade, IL-12, through the Th1 effector response, was also implicated as a major player in the development of various autoimmune diseases.
However, during the late 1990s, studies on the role of IL-12 in a variety of cell-mediated autoimmunity models, including EAU, revealed contradictory results of anti-inflammatory effects. An example is resistance to EAU in the IL-12p40 knockout (KO) mice and in mice treated with anti-IL-12p40 antibodies [15]. Likewise, whereas paradoxically, in vivo administration of recombinant IL-12 inhibited EAU development [16]. These contradictory results, and the realization that related heterodimeric cytokines could share different combinations of their component chains, led to the discovery of IL-23.
IL-23 is a new member of the IL-12 family that shares with IL-12 the p40 subunit and uses the IL-12 receptor subunit IL-12Rβ1, whereas instead of the p35 subunit, it has a p19 subunit and a unique IL-23 receptor subunit.
Studies with IL-23p19 KO mice and anti-p19 antibodies allowed to dissect the separate roles of these two IL-12p40-containing cytokines. These studies revealed that pathogenesis of autoimmune models such as experimental autoimmune encephalomyelitis (EAE) [17, 18] and collagen-induced arthritis (CIA) [19] is dependent on IL-23 and not on IL-12. We recently also demonstrated that a similar situation exists in EAU [20].
The pathogenic role of IL-23 in autoimmune disease models is mediated through a newly discovered subtype of CD4 T helper cells that is distinct from the hitherto known effector phenotypes Th1 or Th2. This new subset of Th cells is characterized by a distinct set of cytokines that include IL-17A, IL-17F, IL-22, IL-6, and tumor necrosis factor (TNF) and by a lineage-specific transcription factor, RORγt (Fig. 1). They were named Th17, after their hallmark effector cytokine. It was suggested that IL-23 has a tissue-specific function in fighting inflammation and in the development of autoimmune diseases. This contention is based mainly on the role of IL-23 in supporting the Th17 lineage and on the finding that IL-17 receptors are expressed on peripheral tissues and not on immune cells [21], as well as on the ability of IL-17 to induce antimicrobial peptides from keratinocytes [22].
Fig. 1.

Effector T cell lineages. Based on information from published literature
In human disease, IL-23 was found to be associated with arthritis [23]. A correlation between IL-23 and ocular disease was identified in uveitis associated with Vogt–Koyanagi–Harada (VKH) disease [24] and in Behcet’s disease [25]. In both of these diseases, patients with active uveitis exhibited higher IL-23 levels in the serum and in activated peripheral blood mononuclear cells (PBMCs) in comparison to patients with inactive uveitis or to normal controls. The causal relationship in humans is difficult to establish. However, in the animal model of EAU, the role of IL-23 is necessary and nonredundant for disease pathogenesis, in that IL-23 KO mice are resistant to EAU, and mice treated with specific anti-IL-23p19 antibodies develop a strongly attenuated disease [20]. The need for IL-23 can be explained at least in part by its involvement in promoting the Th17 effector response; however, this may not be its only role in EAU. Notably, neutralization of IL-23 could prevent disease development only if administered at the innate phase starting at disease induction, whereas neutralizing IL-23 at the effector phase (starting day 7 postimmunization) had no effect on EAU development. This may suggest that IL-23 is essential for the generation of the uveitogenic effector cells mainly during the early phase, possibly even before commitment to the Th17 phenotype, but once the effector T cells were already generated, they were no longer strictly dependent on IL-23.
In contrast to IL-23, IL-12 can be a suppressive element in the development of autoimmune diseases. In other autoimmune disease models, IL-12p35 KO mice had increased susceptibility to the development of EAE [19, 26] and CIA [19]. This also proved to be true in EAU. The suppressive role of IL-12 could be due to its ability to induce high levels of IFN-γ, which at high systemic levels has a suppressive role in EAU and in many other autoimmune diseases. This was demonstrated by using IFN-γ KO and IFN-γ receptor KO mice, as well as by the treatment of mice with neutralizing anti-IFN-γ antibodies [27–31] and in mice lacking the major IFN-γ transcription factor STAT1 [32]. We reported [16] that in vivo administration of rIL-12 prevented the development of EAU through an IFN-γ-dependent effect. Notably, the inhibitory effect occurred only when IL-12 was given during the induction phase (0 to 4 days postimmunization but not later), when IFN-γ production is mainly by innate cells, including NK and NKT cells, and Th1 cells had not yet been generated. In line with that finding, mice deficient in STAT4 or T-bet transcription factors, which are downstream of IL-12 signaling, were resistant to EAE [32, 33]. These findings suggest that the role of IL-12 in induction of IFN-γ and autoimmune suppression may be separate from its role in generating pathogenic Th1 cells. However, since IL-23 signaling is mediated through several STATs including STAT4 [34], resistance to EAE development in the absence of STAT4 may also be due in part to inhibition of IL-23 signaling.
Th17 vs. Th1 in autoimmunity
Until the discovery of the IL-23/Th17 effector pathway, the IL-12/Th1 effector response was considered as responsible for the pathology seen in EAU and other cell-mediated autoimmune disease models. Th2 was felt to be counter-regulatory, although Th2 cells by themselves are also able to induce pathology in an immunologically compromised host [5, 6]. However, as discussed above, questions arose as to the role of Th1 when both IL-12 and IFN-γ had shown a suppressive role in the development of various autoimmune diseases including EAU. A better explanation was provided by the discovery of IL-23 and as a result the Th17 effector subtype.
Th17 cells: pathogenic effectors in autoimmunity
Th17 cells have been shown to represent a distinct lineage from Th1 and Th2. Although the Th17 effector phenotype was discovered through the discovery of IL-23, it was recently shown that it is not IL-23 that triggers commitment to the Th17 pathway. Rather, the combination of IL-6 with TGF-β (in mice [35, 36]) elicited commitment to the Th17 lineage through the newly discovered transcription factor RORγt. In fact, both the Th1 and Th2 hallmark cytokines, IFN-γ and IL-4, respectively, were found to antagonize Th17 differentiation. The Th17 subtype differs from Th1 and Th2 mainly by the set of cytokines that it expresses, including its hallmark cytokine IL-17A together with IL-6, TNF-α, IL-17F, and IL-22 (Fig. 1) and by the expression of the chemokine receptors and chemokines CCR6 CCL20 and CCL22, CXCL10, and CCL2 [37].
IL-17, initially isolated from a cytotoxic T lymphocyte hybridoma and called CTLA-8, is now known as IL-17A. It is a member of a growing family that currently includes six cytokines, named IL-17A–IL17F. IL-17A is a proinflammatory cytokine, while IL-17E (IL-25) is anti-inflammatory [38]. IL17F appears to be proinflammatory as well [39], although its role and the roles of other IL-17 family members are less well defined.
IL-17A can induce the expression of diverse proinflammatory cytokines and chemokines from a large variety of cells, as expected from the broad tissue expression of IL-17A receptors. In the initial characterizations of IL-17A, it was found to be a potent inducer of IL-6 and IL-8 (CXCL8) by fibroblasts [40, 41]. Subsequent studies have demonstrated proinflammatory effects on a broad range of cellular targets, including epithelial and endothelial cells, fibroblasts, osteoblasts, and monocyte/macrophages. Depending on the target cell population, principal activities of IL-17A include the induction of expression of colony-stimulating factors (e.g., granulocyte colony-stimulating factor and granulocyte–macrophage colony-stimulating factor), CXC chemokines (e.g., CXCL8, CXCL1, and CXCL10), metalloproteinases, and IL-6. Accordingly, IL-17A has potent actions to mobilize, recruit, and activate neutrophils. Indeed, the capacity of T cell-derived IL-17A to expand and recruit the neutrophil pool is a defining feature of these family members and provides a novel mechanism by which T cells coordinate adaptive and innate immunity [41].
The main role of IL-17 in immune defense is against extracellular bacteria and fungi. This role is mediated mainly by recruitment of neutrophils and macrophages to the site of inflammation and by upregulating antibacterial peptides such as the S100 family and β-defensin from fibroblasts [22]. Such responses also have a high potential for causing bystander tissue damage. The connection of IL-17 to autoimmune tissue damage was first identified in EAE and CIA, both autoimmune disease models induced by immunization with a self-Ag in CFA, i.e., in the context of bacterial products [18, 19]. Subsequently, IL-17 was also shown to be involved in colitis, again a disease with a strong microbial involvement. Thus, the role of Th17 cells in autoimmunity may be the consequence of subverting a response designed for antimicrobial defense toward self-reactivity.
Involvement of Th17 cells as uveitogenic effectors was demonstrated in the EAU model by the findings that in vivo neutralization of IL-17 significantly ameliorates disease severity and adoptive transfer of retinal Ag-specific Th17 cells into unimmunized hosts precipitates EAU [20, 42, 43]. Importantly, Th17 cells can induce EAU not only in WT but also in IFN-γ KO mice, showing that Th17 cells can be pathogenic in the absence of IFN-γ and a normal Th1 response. IL-17 is present in extracts of uveitic eyes after immunization of mice with IRBP/CFA [20], and Th17 cells are easily detectable in such eyes by intracellular cytokine staining (Fig. 2). More severe uveitis is associated with a higher proportion of Th17 cells in the ocular inflammatory infiltrate. However, the story is not straightforward because IL-17 KO mice are not resistant to EAU, and Th1 cells can induce EAU in recipients treated with anti-IL-17 antibodies at a dose that prevents induction of disease by active immunization [20]. Thus, EAU does not always require Th17 effector cells, an issue that will be expanded upon in the following sections. This finding raised the possibility that (by being upstream to IL-17) IL-23 may have other roles in the development of autoimmunity, which are separate from its role in promoting the Th17 effector response.
Fig. 2.

Th17 cells as well as Th1 cells infiltrate uveitic eyes. Eyes were obtained at peak of disease from C57BL/6 and B10.RIII mice immunized for EAU. The ocular tissues were dispersed chemically and enzymatically, and the cells were analyzed for phenotype and for intracellular cytokine expression after a brief incubation with PMA/Ionomycin and Brefeldin A [20]
IL-17 and the Th17 effector response may also have a role in human disease, although additional studies are needed to dissect this in more detail. Increased levels of IL-17 were found to correlate with scleritis and with autoimmune uveitis not associated with systemic disease [42], as well as in uveitis associated with VKH and Behcet’s diseases [24, 25]. However, studying the human disease had restricted these findings to the periphery and not the target organ, showing upregulation of IL-17 in the serum and in activated PBMCs. On the other hand, ocular fluids from patients with several different viteoretinal disorders revealed high levels of IL-6 and no detectable levels of IL-17 [44], suggesting that IL-17 is not involved in every eye inflammation or that—unlike in mice—in humans these cells do not home to the eye.
Th1 cells: pathogenic or protective?
IFN-γ is the hallmark Th1 cytokine. Similarly to other cytokines that serve as major regulators of immune responses (e.g., TNF-α and apparently IL-12), it has pleiotropic effects that may be contradictory at different stages or locations of the immune response. Although it is the hallmark of the adaptive Ag-specific CD4+ Th1 and CD8 effector cells, large amounts of IFN-γ are secreted by cell types associated with innate immune responses such as NK, NK T and γδ-T cells. These cells provide the initial IFN-γ responses that are strongly involved in innate antimicrobial host defense and also help shape the adaptive immune response that follows. During the innate immune response, IFN-γ is playing an essential role in activating Ag-presenting cells (APCs) resulting in the upregulation of major histocompatibility complex (MHC)-II and TNF-α for Ag presentation and maturation [45]. However, IFN-γ also impairs the proliferation and survival of newly primed T cells by triggering activation-induced cell death [46]. In our hands, inducing high innate levels of IFN-γ in animals that were concurrently immunized for EAU inhibited disease induction at least in part by causing apoptosis of newly primed effector T cells [16]. Others demonstrated that this process involves stimulation of TIM-3 [47] and GADD45 [48] and leads to Th1 elimination.
Despite the important role of IFN-γ in activating APCs, in autoimmune models, this role of IFN-γ was found to be dispensable, since strong Ag-specific autoreactivity is induced in the absence of IFN-γ signaling (in IFN-γ KO and IFN-γ receptor KO mice). We and others found that in the absence of IFN-γ, Th17 response is dominant, and the lack of suppression by IFN-γ of newly primed T cells and of Th17 differentiation may explain the increased susceptibility to the development of autoimmune diseases in these mice.
The immune system evolved to fight pathogens. Th1 and Th17 effector lymphocytes are directed at eradication of intracellular and extracellular pathogens, respectively. During the development of autoimmune disease, autoreactive Th1 and Th17 cells are activated out of their original context, and both of these cell types are found together in the target organ. However, whereas the pathogenic role of the Th17 effectors is supported by inhibition of autoimmune tissue damage as a result of IL-17 neutralization [18, 20], neutralization of IFN-γ has a disease-enhancing effect [27, 28]. Thus, with the discovery of the Th17 cells, the role of Th1 as a pathogenic effector phenotype is being questioned. Despite decades of evidence that polarized Th1 cell lines induce autoimmune pathology when injected into naive recipients, some researchers are now suggesting that the role of the Th1 cells is to suppress and clear inflammation caused by Th17 cells [42].
Th1 and Th17 effector cells induce retinal pathology independently of each other
The pathogenicity of Th17 vs Th1 cells and the question whether Th1 cells are pathogenic or protective have been addressed by using polarized T cell lines having a Th1 or a Th17 phenotype and infused into recipients lacking the reciprocal hallmark cytokine, either through antibody neutralization or by genetic deficiency [20]. Polarized Th17 cells derived from IRBP-immunized IFN-γ KO mice (and thus incapable of producing IFN-γ) injected into either WT or IFN-γ KO recipients induced EAU. Conversely, a polarized Th1 cell line that is unable to produce IL-17, injected into naive recipients treated with saturating doses of neutralizing IL-17 antibody, induced severe uveitis (Fig. 3). It is interesting to note that whereas the Th17 line typically recruited a mostly granulocytic inflammatory infiltrate into the eye, the Th1 line recruited a predominantly mononuclear infiltrate. Notably, the severity of tissue pathology induced by Th1 cells was no less than that induced by Th17 cells (Fig. 3). It is important to underscore that, in contrast to other studies with polarized T cells or T cell lines, which did not stringently exclude participation of the reciprocal cytokine in pathology, this study has demonstrated that Th1 and Th17 cells are both pathogenic effectors that induce tissue damage independently of each other [20].
Fig. 3.
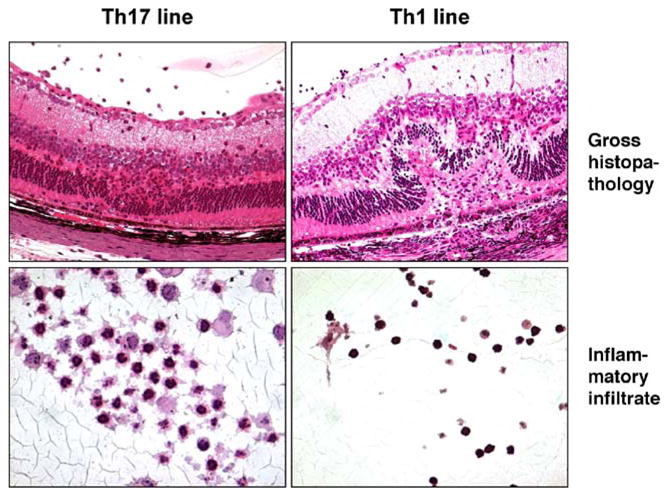
EAU can be induced by Th17 or by Th1 cells. B10.RIII mice received adoptive transfer of IRBP-specific cells from a polarized Th1 cell line unable to produce IL-17 or of cells from IFN-γ KO mice polarized toward the Th17 [20]. Eyes were collected at the peak of disease, processed for histopathology, and stained with hematoxylin and eosin. Note the neutrophilic infiltrate associated with Th17-induced EAU, consistent with its characteristic to recruit granulocytes
One can argue that T cell lines are not reflective of autoimmune disease that arises endogenously as a result of some environmental trigger. However, independent lines of evidence suggest that at least in the EAU model, endogenous disease can also be either Th17 or Th1-driven, although independence from the reciprocal effector phenotype cannot be as sharply defined. In the “classical” model of EAU induced by immunization with IRBP in CFA, there is a strong IL-17 response, and neutralization of IL-17 (but not of IFN-γ) aborts induction of disease, even though the Ag-specific IFN-γ response is relatively undiminished [20]. If the adaptive IFN-γ response is a correlate of Th1, this would suggest that the Th1 response is insufficient in the CFA-induced model to induce disease. Conversely, in the recently developed alternative EAU model, induced with Flt3L-mobilized, in vitro-matured, and Ag-pulsed DC (DC-EAU), there is a strong IFN-γ response, and uveitogenic DC from WT mice are unable to induce EAU in IFN-γ KO recipients, despite development of an undiminished Ag-specific IL-17 response in these mice [2]. To the extent that an adaptive IL-17 response is a correlate of Th17, this would suggest that in the DC-induced model, a Th1 effector is necessary, and a Th17 effector response by itself is insufficient to induce disease.
What are the factors that will determine which effector type will be dominant? In the CFA-induced EAU model, the Ag and mycobacterial components from CFA reach the local lymph nodes and are processed and presented by diverse Ag-presenting cells (APCs). One can thus envision engagement of multiple Toll-like receptors and APC types, resulting in strong and varied innate receptor stimulation. In the DC-EAU model, Ag is taken up in vitro by DC matured in the presence of LPS and anti-CD40, resulting a much more homogeneous APC population and a more limited innate receptor engagement. It is interesting to note that the autoimmune disease models where Th17 dominance has initially been demonstrated, EAE [17, 18] and CIA [19], are both induced in the context of CFA. Thus, we hypothesize that the quality/quantity of Toll-like receptor stimulation and type/diversity of APCs may drive preferentially toward Th17 or toward Th1 dominance.
This also bears on the question of Th17 vs Th1 in human disease, where the inducing events surrounding initial exposure to self-Ag are unknown. Importantly, CFA-EAU and DC-EAU, although induced by the exact same Ag in genetically identical individuals, differ from each other in many respects, making them clinically distinct disease entities. Specifically, differences are seen not only in the immunological response profile but also in pathological and clinical disease manifestations including severity, course, cellular composition of the inflammatory infiltrate, and the appearance of the fundus. If conditions during initial exposure to self-Ag affect development of uveitis in humans in a similar fashion, this may help explain the heterogeneous nature of human uveitis, often in the face of responses to the same retinal Ag(s).
Th1/Th17 synergism and stability of the Th17 phenotype
Prior to the discovery of Th17 as a distinct lineage, IL-17 was already recognized as a potent proinflammatory cytokine associated with tissue damage mainly in skin lesions [49]. At that time, IL-17 was still studied in the context of the Th1 response. Thus, several studies examined the combined effect of IL-17 together with IFN-γ and TNF-α on the expression of chemokines and chemokine receptors in keratinocytes, as a possible mechanism of enhanced recruitment of leukocytes in inflammation. It is interesting to note that the effect of IL-17 alone on the induction of inflammation-associated molecules was minor; however, the combination of IL-17 with TNF-α and even more so with IFN-γ revealed a strong synergistic effect on expression of ICAM-1, IL-8, MCP-1, RANTES, CD40, HLA-DR, and MHC-I [50].
A recent study that characterized CD4 T cells in the gut from patients with Crohn’s disease demonstrated a large proportion of cells that coexpress IFN-γ and IL-17 may be in line with these earlier findings. It is interesting to note that these double-producer cells expressed both IL-12Rβ2 and IL-23R and coexpressed the lineage-specific transcription factors T-bet and RORγt. Similar to cells that expressed IL-17 alone, these cells were resistant to suppression of proliferation by regulatory T cells [51]. These findings may suggest that these CD4 cells that express both IL-17 and IFN-γ can respond both to IL-12 and IL-23 and may support the presence of synergism between IL-17 and IFN-γ in the induction of tissue damage.
In the eye, the double-producing T cells expressing both IL-17 and IFN-γ in their cytoplasm abound (Fig. 2). The significance of these cells in EAU and whether they represent a stable population or a transitional phenotype is currently unclear. For example, some of them may be Th17 cells that are transitioning to a different stage of differentiation. Several independent lines of evidence suggest that the Th17 phenotype may not be a stable one. Although stringent examination of this requires use of IL-17 reporter mice, polarized IL-17-producing cells appear to lose production of IL-17 and/or acquire the ability to produce IFN-γ, upon prolonged culture (Ana Hansen and Rachel Caspi, unpublished data), or after transfer into mice (Guangpu Shi and Igal Gery, personal communication). The biological implications of this apparent plasticity remain to be dissected.
Conclusions
Human uveitis is a clinically heterogeneous disease. Due to objective limitations, the basic mechanisms in human disease are difficult to dissect. The EAU model has been extremely useful in obtaining insights into the mechanisms that might drive uveitis in humans. Until a few years ago, EAU was considered to be mediated by Th1 cells of the CD4 lineage. Antibodies, which can enter the ocular compartment after the blood–retinal barrier has been broken by the T cells, also come into play as an effector mechanism, as do recruited inflammatory leukocytes. More recent data revealed that CD8 lymphocytes, usually thought of as suppressive, can also be pathogenic effectors, although their importance in EAU elicited in an immunologically normal host remains to be determined. With the discovery of Th17 cells, a new effector phenotype was added to the lineup. Although Th17 were originally defined as CD4+ T cells, CD8+ T cells are also induced to produce IL-17 under similar conditions as CD4. The relative dominance of these different effector mechanisms and how they mesh with each other, affect the manifestations of disease. These findings shed light on the variability of human uveitic disease and may ultimately help to develop better and more rational treatment strategies for human uveitis through the targeting of specific molecules involved in critical checkpoints of the disease process.
References
- 1.Agarwal RK, Caspi RR. Rodent models of experimental autoimmune uveitis. Methods Mol Med. 2004;102:395–420. doi: 10.1385/1-59259-805-6:395. [DOI] [PubMed] [Google Scholar]
- 2.Tang J, Zhu W, Silver PB, Su SB, Chan CC, Caspi RR. Autoimmune uveitis elicited with antigen-pulsed dendritic cells has a distinct clinical signature and is driven by unique effector mechanisms: initial encounter with autoantigen defines disease phenotype. J Immunol. 2007;178:5578–5587. doi: 10.4049/jimmunol.178.9.5578. [DOI] [PubMed] [Google Scholar]
- 3.Nussenblatt RB. Bench to bedside: new approaches to the immunotherapy of uveitic disease. Int Rev Immunol. 2002;21:273–289. doi: 10.1080/08830180212067. [DOI] [PubMed] [Google Scholar]
- 4.Caspi RR, Silver PB, Chan CC, Sun B, Agarwal RK, Wells J, Oddo S, Fujino Y, Najafian F, Wilder RL. Genetic susceptibility to experimental autoimmune uveoretinitis in the rat is associated with an elevated Th1 response. J Immunol. 1996;157:2668–2675. [PubMed] [Google Scholar]
- 5.Caspi RR. Th1 and Th2 responses in pathogenesis and regulation of experimental autoimmune uveoretinitis. Int Rev Immunol. 2002;21:197–208. doi: 10.1080/08830180212063. [DOI] [PubMed] [Google Scholar]
- 6.Kim SJ, Zhang M, Vistica BP, Chan CC, Shen DF, Wawrousek EF, Gery I. Induction of ocular inflammation by T-helper lymphocytes type 2. Invest Ophthalmol Vis Sci. 2002;43:758–765. [PubMed] [Google Scholar]
- 7.McPherson SW, Yang J, Chan CC, Dou C, Gregerson DS. Resting CD8 T cells recognize beta-galactosidase expressed in the immune-privileged retina and mediate autoimmune disease when activated. Immunology. 2003;110:386–396. doi: 10.1046/j.1365-2567.2003.01750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song L, Le J, Ye F, Shao H, Kaplan HJ, Sun D. Sequence 168 to 177 of interphotoreceptor retinoid-binding protein (IRBP) is an antigenic epitope for autoreactive CD8T cells in the B10RIII mouse. J Neuroimmunol. 2008;193:68–76. doi: 10.1016/j.jneuroim.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prendergast RA, Iliff CE, Coskuncan NM, Caspi RR, Sartani G, Tarrant TK, Lutty GA, McLeod DS. T cell traffic and the inflammatory response in experimental autoimmune uveoretinitis. Invest Ophthalmol Vis Sci. 1998;39:754–762. [PubMed] [Google Scholar]
- 10.Caspi RR. Ocular autoimmunity: the price of privilege? Immunol Rev. 2006;213:23–35. doi: 10.1111/j.1600-065X.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- 11.Su SB, Grajewski RS, Luger D, Agarwal RK, Silver PB, Tang J, Tuo J, Chan CC, Caspi RR. Altered chemokine profile associated with exacerbated autoimmune pathology under conditions of genetic interferon-gamma deficiency. Invest Ophthalmol Vis Sci. 2007;48:4616–4625. doi: 10.1167/iovs.07-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forrester JV, Huitinga I, Lumsden L, Dijkstra CD. Marrow-derived activated macrophages are required during the effector phase of experimental autoimmune uveoretinitis in rats. Curr Eye Res. 1998;17:426–437. doi: 10.1080/02713689808951224. [DOI] [PubMed] [Google Scholar]
- 13.Caspi RR, Chan CC, Fujino Y, Najafian F, Grover S, Hansen CT, Wilder RL. Recruitment of antigen-nonspecific cells plays a pivotal role in the pathogenesis of a T cell-mediated organ-specific autoimmune disease, experimental autoimmune uveoretinitis. J Neuroimmunol. 1993;47:177–188. doi: 10.1016/0165-5728(93)90028-w. [DOI] [PubMed] [Google Scholar]
- 14.Pennesi G, Mattapallil MJ, Sun SH, Avichezer D, Silver PB, Karabekian Z, David CS, Hargrave PA, McDowell JH, Smith WC, et al. A humanized model of experimental autoimmune uveitis in HLA class II transgenic mice. J Clin Invest. 2003;111:1171–1180. doi: 10.1172/JCI15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarrant TK, Silver PB, Chan CC, Wiggert B, Caspi RR. Endogenous IL-12 is required for induction and expression of experimental autoimmune uveitis. J Immunol. 1998;161:122–127. [PubMed] [Google Scholar]
- 16.Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, Caspi RR. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis. J Exp Med. 1999;189:219–230. doi: 10.1084/jem.189.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 18.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan C-C, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: context of initial Ag exposure determines dominant effector category. J Exp Med. 2008 doi: 10.1084/jem.20071258. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spriggs MK. Interleukin-17 and its receptor. J Clin Immunol. 1997;17:366–369. doi: 10.1023/a:1027360106635. [DOI] [PubMed] [Google Scholar]
- 22.Kao CY, Chen Y, Thai P, Wachi S, Huang F, Kim C, Harper RW, Wu R. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J Immunol. 2004;173:3482–3491. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- 23.Yago T, Nanke Y, Kawamoto M, Furuya T, Kobashigawa T, Kamatani N, Kotake S. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther. 2007;9:R96. doi: 10.1186/ar2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi W, Yang P, Li B, Wu C, Jin H, Zhu X, Chen L, Zhou H, Huang X, Kijlstra A. IL-23 promotes CD4+ T cells to produce IL-17 in Vogt–Koyanagi–Harada disease. J Allergy Clin Immunol. 2007;119:1218–1224. doi: 10.1016/j.jaci.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Chi W, Zhu X, Yang P, Liu X, Lin X, Zhou H, Huang X, Kijlstra A. IL-23 and IL-17 are upregulated in Behcet’s patients with active uveitis. Invest Ophthalmol Vis Sci. 2008 doi: 10.1167/iovs.07-1390. (in press) [DOI] [PubMed] [Google Scholar]
- 26.Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- 27.Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, Carton H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol. 1988;140:1506–1510. [PubMed] [Google Scholar]
- 28.Caspi RR, Chan CC, Grubbs BG, Silver PB, Wiggert B, Parsa CF, Bahmanyar S, Billiau A, Heremans H. Endogenous systemic IFN-gamma has a protective role against ocular autoimmunity in mice. J Immunol. 1994;152:890–899. [PubMed] [Google Scholar]
- 29.Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol. 1996;26:2393–2398. doi: 10.1002/eji.1830261019. [DOI] [PubMed] [Google Scholar]
- 30.Jones LS, Rizzo LV, Agarwal RK, Tarrant TK, Chan CC, Wiggert B, Caspi RR. IFN-gamma-deficient mice develop experimental autoimmune uveitis in the context of a deviant effector response. J Immunol. 1997;158:5997–6005. [PubMed] [Google Scholar]
- 31.Zhu Y, Ljunggren HG, Mix E, Li HL, van der Meide P, Elhassan AM, Winblad B, Zhu J. Suppression of autoimmune neuritis in IFN-gamma receptor-deficient mice. Exp Neurol. 2001;169:472–478. doi: 10.1006/exnr.2001.7662. [DOI] [PubMed] [Google Scholar]
- 32.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyton RJ, Davies S, Marden C, Fantino C, Reynolds C, Portugal K, Dewchand H, Altmann DM. Stat4-null non-obese diabetic mice: protection from diabetes and experimental allergic encephalomyelitis, but with concomitant epitope spread. Int Immunol. 2005;17:1157–1165. doi: 10.1093/intimm/dxh293. [DOI] [PubMed] [Google Scholar]
- 34.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 35.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 36.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 37.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 38.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seiderer J, Elben I, Diegelmann J, Glas J, Stallhofer J, Tillack C, Pfennig S, Jurgens M, Schmechel S, Konrad A, et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn’s disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 2007 doi: 10.1002/ibd.20339. (in press) [DOI] [PubMed] [Google Scholar]
- 40.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 41.Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- 42.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 43.Peng Y, Han G, Shao H, Wang Y, Kaplan HJ, Sun D. Characterization of IL-17+ interphotoreceptor retinoid-binding protein-specific T cells in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2007;48:4153–4161. doi: 10.1167/iovs.07-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banerjee S, Savant V, Scott RA, Curnow SJ, Wallace GR, Murray PI. Multiplex bead analysis of vitreous humor of patients with vitreoretinal disorders. Invest Ophthalmol Vis Sci. 2007;48:2203–2207. doi: 10.1167/iovs.06-1358. [DOI] [PubMed] [Google Scholar]
- 45.Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med. 2005;202:203–207. doi: 10.1084/jem.20050810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts AI, Devadas S, Zhang X, Zhang L, Keegan A, Greeneltch K, Solomon J, Wei L, Das J, Sun E, et al. The role of activation-induced cell death in the differentiation of T-helper-cell subsets. Immunol Res. 2003;28:285–293. doi: 10.1385/IR:28:3:285. [DOI] [PubMed] [Google Scholar]
- 47.Khademi M, Illes Z, Gielen AW, Marta M, Takazawa N, Baecher-Allan C, Brundin L, Hannerz J, Martin C, Harris RA, et al. T Cell Ig- and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. J Immunol. 2004;172:7169–7176. doi: 10.4049/jimmunol.172.11.7169. [DOI] [PubMed] [Google Scholar]
- 48.Lu B, Yu H, Chow C, Li B, Zheng W, Davis RJ, Flavell RA. GADD45gamma mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity. 2001;14:583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 49.van Beelen AJ, Teunissen MB, Kapsenberg ML, de Jong EC. Interleukin-17 in inflammatory skin disorders. Curr Opin Allergy Clin Immunol. 2007;7:374–381. doi: 10.1097/ACI.0b013e3282ef869e. [DOI] [PubMed] [Google Scholar]
- 50.Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. J Immunol. 1999;162:494–502. [PubMed] [Google Scholar]
- 51.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]