

Abstract
Depending on its concentration, nitric oxide (NO) has beneficial or toxic effects. In pathological conditions, NO reacts with superoxide to form peroxynitrite, which nitrates proteins forming nitrotyrosine residues (3NY), leading to loss of protein function, perturbation of signal transduction, and cell death. 3NY immunoreactivity is present in many CNS diseases, particularly Multiple Sclerosis (MS). Here, using the high flux NO donor, spermine NONOate, we report that oligodendrocytes are resistant to NO, while motor neurons are NO sensitive. Motor neuron sensitivity correlates with the NO-dependent formation of 3NY, which is significantly more pronounced in motor neurons as compared to oligodendrocytes, suggesting peroxynitrite as the toxic molecule. The heme-metabolizing enzyme, heme-oxygenase-1 (HO-1), is necessary for oligodendrocyte NO resistance, as demonstrated by loss of resistance after HO1 inhibition. Resistance is reinstated by peroxynitrite scavenging with uric acid further implicating peroxynitrite as responsible for NO sensitivity. Most importantly, differential sensitivity to NO is also present in cultures of primary oligodendrocytes and motor neurons. Finally, motor neurons cocultured with oligodendrocytes, or oligodendrocyte-conditioned media, become resistant to NO toxicity. PRELIMINARY STUDIES SUGGEST OLIGODENDROCYTES RELEASE A SOLUBLE FACTOR THAT PROTECTS MOTOR NEURONS. Our findings challenge the current paradigm that oligodendrocytes are the exclusive target of MS pathology.
Keywords: motor neurons, oligodendrocytes, nitric oxide, peroxynitrite, nitrotyrosine, hemoxygenase 1
Introduction
Nitric oxide is a free radical gas that at normal physiological concentrations is essential for many cellular processes, such as neurotransmission, differentiation, and signal transduction (Packer et al., 2003, Peunova & Enikolopov 1995, Stamler et al., 1997, Stuehr, 1999). These physiological processes are regulated by NO produced at steady state concentrations from ~50nM to ~500nM (Clough et al., 1998, Huk et al., 1998, Pacher et al., 2007). Neurons produce NO at 33 nM concentrations during normal physiological functions (low flux NO) (Leonard et al., 2001).
In excessive amounts, NO is toxic and plays a role in Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), CNS injury, and in multiple sclerosis (MS) (Cassina et al. 2002, Hall et al.,1998, Huk et al., 1998, Ischiropoulos & Beckman,2003, Kawase et al., 1996, Panahian & Maines, 2001, Pacher et al.,2007, Vaziri et al., 2004). In pathological conditions, NO can increase to 10 times the concentrations seen before the insult (Clough et al., 1998, Huk et al., 1998, Pacher et al., 2007). Activated microglial and astrocytes can produce NO at steady state concentrations as high as 1uM (high flux NO) (Hall et al.,1998, Kawase et al., 1996, Pacher et al.,2007, Stuehr,1999,Tominaga et al.,1994).
At high fluxes, NO, released during CNS pathology, is more likely to react with oxygen species to form reactive nitrogen species (RNS), such as peroxynitrite (ONOO−), which damage a variety of macromolecules, including proteins (Beckman, 1996, Cassina et al. 2002, Estevez et al., 1998, 2000, Ischiropoulos & Beckman, 2003, Pacher et al., 2007,Tamir et al., 1993). Peroxynitrite-dependent nitration of tyrosine residues, forming 3-nitrotyrosine (3NY), disrupts protein structure and function, thereby interrupting or altering cell signaling (Bishop et al., 2004, 2006, Cassina et al. 2002,, Ischiropoulos & Beckman, 2003, Estevez et al., ,1998 2000, Pacher et al.,2007). Nitrotyrosine is found in the CNS of patients with spinal injury, ALS, PD, AD, and MS, and is considered a footprint for peroxynitrite mediated damage in the cell (Estevez et al., 1998 2000, Ischiropoulos & Beckman,2003, Jack et al., 2007, Pacher et al.,2007, Prat & Antel et al., 2005). In particular, in MS, progression and severity is tightly associated with levels of RNS in the cerebrospinal fluid (CSF) and blood serum (Giovannoni et al., 1997, 1998).
The accepted hypothesis for MS pathogenesis is that it exclusively affects white matter with the demyelination of axons occurring first, followed by axonal injury, with subsequent damage to neurons (Silber and Sharief, 1999). However, growing evidence indicates that axonal injury is an important and early event in MS which may occur before dysfunction of the oligodendrocytes and myelination (Bitsch et al., 2000, De Stefano et al., 2001, Fergurson et al., 1997, Peterson et al., 2001, Trapp et al., 1999, Werner et al.,2001,). How important, if at all, is initial axonal injury in the MS pathology is still unclear (Bitsch et al., 2000, De Stefano et al., 2001, Fergurson et al., 1997, Peterson et al., 2001, Trapp et al., 1999, Werner et al.,2001).
Because oligodendrocytes and axons, as well as their interactions, are targets for the pathological process seen in MS, and NO is involved in MS pathology, we investigated the nitric oxide sensitivity of both oligodendrocytes and motor neurons alone, and in coculture. For this study, we used more physiologically relevant (lower) NO doses than we did in our previous studies of NO sensitivity in motor neurons (Bishop et al, 1999, 2003, 2004, 2005), and dissected the mechanisms of NO toxicity and resistance in both cell types. Our mechanistic studies of these two cell types, in isolation and together, provide support for the hypothesis that axonal damage plays an early role in MS pathology.
Materials and methods
Vertebrate animals
The use of rats conformed to all institutional and government regulations. Our animal facility has an experienced animal care technician. The protocol, with measures to prevent pain and suffering, was approved by our Institutional Animal Care and Use Committee (assurance number 07-001-R). E15 timed pregnant Sprague Dawley rat females were euthanized with carbon dioxide and sterilized by ethanol wash. The uteri were removed and the embryos harvested for spinal cords.
Primary Motor Neuron Isolation
The spinal cords (cervical and thoracic portions) were isolated from each embryo as per the protocol of Dr. Alvaro Estevez (Bishop et al., 1999, 2004, Estevez et al., 1998, 2000, Schnaar & Schnaffner, 1981). The meninges layers were dissected away and the dorsal roots removed. The ventral part of the spinal cord was dissected away, minced and separated by BSA gradient followed by Optiprep gradient into fractions enriched for motor neurons. This was repeated several times to “enrich” further for motor neurons (Bishop et al., 1999,2004) followed by immunopanning (Estevez et al., 1998, 2000). The motor neurons were plated on flasks coated with a mixture of laminin and poly-D-lysine at a density of 2×106 cells per flask and cultured under 37°C, 5% CO2 in minimal Eagle’s Medium supplemented with D-glucose, L-glutamine and 5% fetal bovine serum. (Bishop et. al., 1999, 2004, Estevez et. al., 1998, 2000).
Primary Oligodendrocyte Isolation
Primary oligodendrocytes were isolated from adult female Sprague Dawley Rats as described (Johnson, et al., 2002). The spinal cord (ventral to sacral) was isolated, placed in cold Liebovitz’s L-15 medium, then transferred to a dish containing 0.25% trypsin with 50 ug/mL DNase I in Hanks. The minced cord was incubated at 37°C for 1 hr in trypsin, and centrifuged at 400–500g for 5 minutes at 4°C. The supernatant was removed and replaced with L-15 medium containing 10% FBS and 50 ug/mL DNase. The cord fragments were triturated in the L-15 medium. The resultant cell suspension was added to a percoll gradient, then centrifuged at 30,000g for 30 minutes at 4°C. The myelin layer was removed, the oligodendrocyte cell fraction was transferred to a tube containing 2% FBS L-15, and then plated in specialized media as described (Johnson et al., 2002).
Tissue Culture:cell lines
NSC34D
The NSC34D cell line, a further differentiated version of the NSC34 cell line used in our previous studies, was used in our studies detailed here. NSC34 cells are a fusion of primary mouse spinal cord motor neurons with spinal neuroblastoma cells that, upon terminal differentiation with retinoic acid or serum reduction (10% to 2%), become NSC34D. The NSC34Ds provide a homogenous line that have the characteristics of differentiated motor neurons, such as the expression of N Methyl D Aspartate Receptors (NMDAR), but with the very limited ability to divide (Cashman et al., 1992, Eggett et al., 2000, Matsumoto et al.,1995). Cells were grown in 1:1 DMEM without sodium pyruvate/HAMS F12 mixture, 1% NEAA and 2% FBS as described (Eggett et al., 2000).
MO3.13 Cells (human oligodendrocyte model)
The oligodendrocyte M03.13 cell line was used as an oligodendrocyte model. MO3.13 is an immortalized human-human hybrid cell line created from the fusion of a 6-thioguanine resistant mutant of the human rhabdomyosarcoma with adult human oligodendrocytes (McLaurin et al., 1995). These cells differentiate upon serum deprivation (10% to 2%) and have properties of mature primary oligodendrocyte, such as the expression of myelin basic protein (MBP), CNPase, proteolipid protein, and O1 protein (Buntinx et al., 2003, , Li et al., 2002, McLaurin et al. 1995 ). Cells were grown in a humidified 5% CO2 environment and plated in a 1:1 DMEM without sodium pyruvate/HAMS F12 mixture, 1% NEAA and 2% FBS.
Co- Cultures of NSC 34D and MO3.13 cells
Mixed culture plates of oligodendrocytes and motor neurons were plated (at about a 1:3 ratio) and maintained in DMEM,/HAMS F12 mixture supplemented with 2% heat inactivated-FBS.
Tissue Culture Primary Cells
Oligodendrocytes
Cells were plated in 12-well plates with 1 mL O-medium which was comprised of a variety of factors that maintained differentiated oligodendrocytes and selected against fast growing cells as described (Johnson et al.2002). The oligos were maintained at 37°C with 5% CO2 for 6–7 days.
Motor neurons
Cells were incubated in neurobasal media with factors conducive to motor neurons, as described (Estevez et al., 1998), and maintained at 37 °C with 5% CO2 for 2–3 days.
Cell Survival Assay
Cell viability was indicated primarily by Trypan blue exclusion, intact morphology, and neurite outgrowth. Many cells that died, not only rounded up on the plate, but lifted off and became “floaters”, >99% of which did not exclude Trypan blue (Bishop et al.,1999). Lack of Trypan blue exclusion was used in early experiments as the means to detect dead cells. Eventually we relied on cell counts as live cells remained attached to plates and were not rounded (Bishop et al., 1999,2004). Percent cell survival was calculated by dividing post-NO treatment total live cell counts by pre-NO treatment total live cell counts and multiplying by 100%.
Immunocytochemistry
Primary cells were grown in media designated for that cell type. Cells were fixed with 4% paraformaldehyde for 10 minutes, washed with PBS and then permeabilized with a 0.1–0.25% Triton X-100 solution. After being rinsed with PBS, cells were blocked with a 4% BSA solution in 0.1% PBST, rinsed with PBS and then incubated with the appropriate antibody.
Labeling and Detection of Motor Neurons: MAP2 Staining
Microtubule-associated protein 2 (MAP2) is a critical marker for mature CNS neurons (Herzog and Weber, 1978, Brugg and Matus, 1991). Primary motor neurons were incubated overnight at 4°C with rabbit anti-MAP2 antibody (1:500, Chemicon), washed with PBS and incubated for 1 hr with goat-anti-rabbit Alexa Fluor 488 IgG (1:1000; Molecular Probes, Eugene, OR). Fluorescing antibodies were detected using broadband filters (Chroma Series 41028). Oligodendrocytes do not stain green with MAP2 while neurons do stain green.
Labeling and Detection of Oligodendrocytes: MBP Staining
Primary oligodendrocytes were stained with a marker for mature oligodendrocytes, myelin basic protein (MBP) (Baumann and Pham-Dinh, 2001). Primary oligodendrocytes were incubated with mouse monoclonal anti-MBP antibody (1:1000; Covance, Berkeley, CA), rinsed with PBS and incubated for 1 hr with goat-anti-mouse Alexa Fluor 488 IgG2b secondary antibody (1:1000; Molecular Probes, Eugene, OR). Fluorescing antibodies were detected using broadband filters (Chroma Series 41028).
Note that for negative controls, we used the MAP in a plate of oligodendrocytes, and the MBP on the neurons to control for non specific staining. We also used only the secondary Ab on plates to control for non specific staining. In all cases, the frame was black, with no green labeled morphology.
NO Treatment Protocols
Motor neurons and oligodendrocytes were plated at the same cell density, fed the same volume of media at the same temperature and pH and treated with a range of NO doses. For treatment at subtoxic “physiological” NO dose, the compound (z)-1-[2-Aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate] DETA-NONOate with a half life of 16 hours was used (Bishop et al., 1999,2004). Spermine-NONOate, N-[4-[1-(3-Aminopropyl)-2-hydroxy-2-nitrosohydrazino]butyl]-1,3-propanediamine, which has a half-life of ~40 min, was used for the cytotoxic “pathological” NO challenges (Bishop et al., 1999,2004). Both NO donors, particularly DETA-NONOate, release NO with predictable and easily controlled kinetics based on pH and temperature 37°C (Beckman et al,1996, Bishop et al., 1999,2004, Bouton & Demple, 2000, Estevez et al., 1998, 2000, Raoul et al, 2002). The donors were kept on ice at pH 10 to prevent NO release, which was commenced with the addition of the donors to the pH 7.4 37°C media. NO donors release rates have been determined experimentally (Beckman et al, 1996, Bishop et al., 1999,2004, Bouton & Demple, 2000, Estevez et al., 1998, 2000, Raoul et al, 2002) and in our lab (Fig.1A). After treatment for the prescribed amount of time (usually 1 hour) the media was changed repeatedly to eliminate the NO donor. We used spent donor cocktail for the untreated cells as a negative control, and it was found to not release NO, nor kill cells. We then measured damage parameters 24 hours after NO treatment as this was found to be the onset of maximal damage as determined by our previous studies in motor neurons (Bishop et al., 1999, 2004, 2005, 2006).
Figure 1. Differential NO sensitivity of human oligodendrocyte (MO3.13) cells and differentiated motor neuron (NSC34D) cells.

A. The graph is the comparison between the kinetics of NO release from different NO donors:DETA NONOate, spermine NONate and spent donors as a control. B. These are micrographs of NSC34D for cell count and changes in morphology (100× magnification). The first row of panels are a dose curve of DETANONOate: spent donor, a dose that releases 2pm/s, and a 10fold higher dose. The second row of panels is a dose curve of spermine NONOate: spent spermine NONoate, 1/10 the “high NO” dose, and the high NO dose that releases 110pm/s. C. The graph is a quantification of B with % cell survival on the y axis and NO flux on the x axis. % Cell survival is a percentage of a control of untreated cells. The standard error of the mean between experiments (n=4) was calculated. Significance between high flux cell survival of cells exposed to DETANONOate vs spermine NONoate is p<.001, designated with an asterisk. D. The graph is the average of all the experiments (n=3) with a comparison of NO sensitivity of NSC34D vs NSC34. The % cell survival is on the y axis and NO flux is the x axis. % Cell survival is a percentage of a control of untreated cells. The standard error of the mean was determined. There was no significant difference between the experimental groups. E. These panels are micrographs of cells (100× magnification) organized into an MO3.13 row and a NSC34D row. The 60 minute dose of NO is organized into columns. UT is untreated-treated with spent donor, Low NO is 2 pmoles/s NO flux, High NO is 110 pmoles/s NO flux. Cell death was assayed 24h post treatment. F. The graph is the average of all the treatments (n=6) with % cell survival on the y axis and NO flux on the x axis. % Cell survival is a percentage of a control of untreated cells. The standard error of the mean between experiments was calculated, and significance between high flux cell survival of oligodendrocytes vs motor neurons is p<.001, designated by an asterisk. The % cell survival of untreated and High NO-challenged oligodendrocytes does not differ significantly.
Additional experiments included treatment of the oligodendrocytes with cytotoxic NO challenge, coupled with 20 µM Zinc protoporphyrin (ZnPP-IX), an HO-1 inhibitor (Akins et al., 2004, Yang et al., 2001) or coupled with 10uM uric acid, an efficient peroxynitrite scavenger (Hooper et al.,2000, Pacher et al., 2007). To control for any toxicity of each test agent, some cells were incubated with the test agent alone.
NO Assay
NO release as a function of time was confirmed with a colorimetric NO assay kit that is based on the Greiss Reaction (Active Motive Nitric Oxide Analysis Kit) utilizing nitrate reductase as described (Bishop et al., 1999, 2004). NO concentrations were then calculated based on the read values and a standard curve (repeated in triplicate for each experiment), predicted curves from past studies (Beckman et al, 1996, Bishop et al., 1999,2004, Bouton & Demple, 2000, Estevez et al., 1998, 2000, Raoul et al, 2002), studies in our lab (Fig1A), and reported as a flux (pmoles/s) which is calculated from total NO released during the 1 hour of treatment.
Western Blot/Immunoblot
Protein samples were quantified, using the BIORAD Protein Assay, for equal protein loading of the gels. For immunoblotting, cell extracts were loaded on SDS PAGE, run, transferred onto Nytran blots as described (Bishop et al.,1999, 2004). The blots were washed, blocked with BSA, incubated with rabbit primary anti-3NY (1:1000), a kind gift from Dr. Alvaro Estevez., and developed with a colorimetric secondary antibody.
For positive control, we used peroxynitrite treated albumin and molecular weight markers, both of which exhibited the 3NY formation. For negative controls, we washed the blot with just secondary Ab and found no staining.
Immunoblot Analysis
Blots from western blot analysis were analyzed using the UN-SCAN IT gel densitometry software (Silk Scientific, Orem Utah). Blots were stained with Ponceau to control for even loading and uniform transfer.
Statistical Analysis
Experiments were repeated a minimum of four times. For any data point involving cell counting, a minimum of 200 cells were counted from at least 5 randomly chosen fields. Cell survival was calculated by dividing post-NO treatment total cell counts by pre-NO treatment total cell counts, and multiplying by 100%. The mean of the data points were taken, and the standard error of the mean was calculated. The data was analyzed by a two tailed t test and significance (p value) was calculated. P<.01 was determined to be significant.
Results
Physiologically relevant NO doses
We assayed the release rate of NO from NO donors, to choose pretreatment and challenge doses that were closer to the range of NO concentrations seen by cells during physiological and pathological conditions respectively (Clough et al.,1998, Hall et al.,1998, Kawase et al., 1996, Pacher et al., 2007, Stuehr,1999, Tominaga et al.,1994). For NO donors, we used a final concentration of 1uM DETA-NONOate which donates only NO (Dickhout et al., 2005) at a lower flux rate of 2 pmoles/s ( low dose) (Fig, 1A). For the challenge NO dose, we used 10uM final concentration spermine-NONOate, which donates NO at a higher flux (Cornish et al., 2002) of 110 pmoles/s (high dose) (Fig 1A).
In light of the fact that high flux NO seen in pathological situations produces RNS with resultant 3NY formation (Cornish et al., 2002 Tamir et al., 1993, 1996), and cell death (Estevez et al., 1998 2000, Ischiropoulos & Beckman,2003, Jack et al., 2007, Pacher et al.,2007, Prat & Antel et al., 2005) we wanted to ask if spermine-NONOate, unlike DETA-NONOate, does indeed show greater toxicity. We exposed NSC34D (motor neuron cell line) to a dose response curve of DETA versus spermine and found a significant difference of cell survival at the highest dose (104%+/−10 vs 16%+/−8 (n=4),p<.001) (Fig.1B&C). Thus, NO doses detailed in Figure 1 mimic physiological versus pathological NO-mediated conditions (Pacher et al., 2007), and are valid tools to study cellular NO resistance mechanisms.
For studies with motor neurons and oligodendrocytes, we utilized primary cells and recognized model cell lines. In particular, for our motor neurons, we used NSC34D which is terminally differentiated NSC34,both of which are accepted models of motor neurons (Bishop et al , 1999, 2003, 2004,2005 ,2006, Cashman et al., 1992, Durham et al.1993, Eggett et al., 2000, Matsumoto et al.,1995). More than 99% of NSC34D cells are positive for the motor neuron markers NMDAR, which would presumably make the NSC34D cells more NO sensitive than the NSC34 cells (Bishop et al., 2004, Cashman et al.1992, Egget et al., 2000). However, we have found no increase in NO sensitivity at the high NO doses (110pm/s) in NSC34D as compared to NSC34, (12+/−5 vs 13+/−19 ; n=3)(Fig.1D), thus both motor neuron lines exhibit NO sensitivity which indicates that either NSC34 or NSC34D is an acceptable model of motor neurons for our studies of NO sensitivity (Bishop et al., 1999, 2004, Cashman et al., 1992, Egget et al., 2000).
Comparison of NO sensitivity of oligodendrocytes and motor neurons
NSC34D and MO3.13 (terminally differentiated oligodendrocyte cell line where >90% express myelin basic protein) were exposed to a range of NO fluxes for one hour, and cell death was assayed twenty four hours later. Untreated cells were exposed to spent NO donor as a control. Even at low NO fluxes (2–13pmoles/s), motor neurons were significantly more NO sensitive than were oligodendrocytes (% cell survival of 61 ± 9% vs 82 ± 11%, n=8, p<.001) (Fig.1E&F). This differential susceptibility was more evident at high NO fluxes (110 pmoles/s) where motor neurons survival was minimal and oligodendrocytes were resistant (12 ± 5% vs 115 ± 7%, n=8, p<.001)(Fig.1E&F). Dead cells that remained attached to the plate were rounded with no neurites/processes and did not exclude trypan blue. Of the cells that lifted from the plate >99% did not exclude trypan blue (Bishop et al.,1999, 2004) (Figure 1E). Thus, the MO3.13 cells, unlike NSC34D cells, were completely unaffected by cytotoxic doses of NO, thereby expressing differential sensitivity to a direct NO challenge in the flux range we used (Figure 1E&F).
Differential NO resistance in primary motor neurons and oligodendrocytes
We asked if this differential NO sensitivity can be verified in primary rat oligodendrocyte and motor neuron cultures. The primary motor neurons were stained for motor neuron specific proteins (MAP2), assayed for purity, and were ~80% pure (Fig. 2A). The primary oligodendrocytes were stained for myelin basic protein and determined to be ~65% pure (Fig. 2A). The oligodendrocyte morphology changed profoundly upon fixation and staining, but was still good enough for the purposes of assaying purity, since we were merely counting immunoreactive cells. For the experiments, we did not fix the cells.
Figure 2. Viability of pure primary oligodendrocytes and pure primary motor neurons as a function of NO flux.
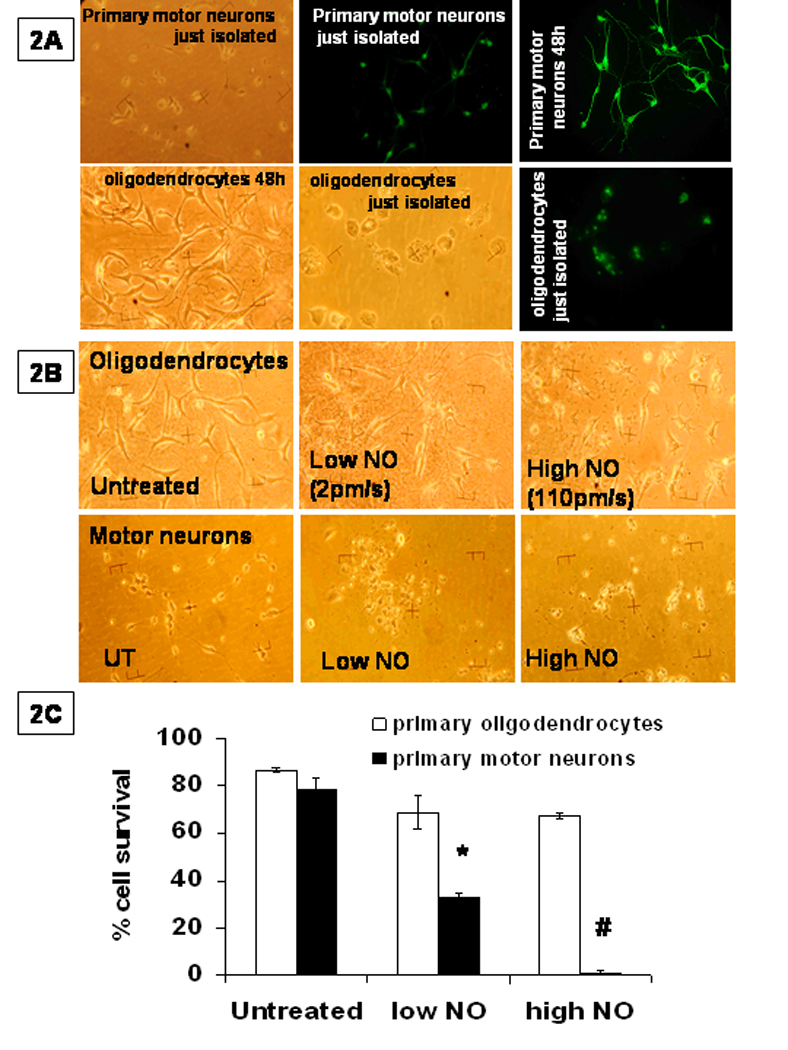
A. The first row is micrographs of primary motor neurons with the first panel phase contrast, the second and third panel fluorescence images of cells labelled for the neuronal marker MAP2. For the lower row, which contains primary oligodendrocytes, the first panel is phase contrast, while the second panel is fixed cells under phase contrast, with the third panel fluorescence images of cells labelled with oligo maker, myelin basic protein MBP. The % purity = (number of MBP positive cells / total cells)*100. Non fixed cells were used for all experiments. B.) These are photomicrographs of primary motor neurons and primary oligodendrocytes treated with low dose NO and then high dose of NO. Note, most of the fields of motor neurons treated with high NO fluxes were empty. We chose a field with neurons to show the decay of morphology. C.) Cell survival was quantified; the values represent the mean ± SEM of n=4 performed in triplicate. The significance (P<.001) was determined between cell survival after high flux NO challenge of oligodendrocytes vs motor neurons and designated by an #.
We found that primary oligodendrocytes were resistant to treatment with the low dose of NO (2–13pm/s DETA NONoate) (69± 7%, n=4), while the primary motor neurons were significantly (p<.001) more sensitive (33 ± 2%, n=4) (Figure 2B,C). When the treatment dose was 110pm/s NO (administered by spermine-NONOate), we found that primary oligodendrocytes were still resistant (67 ± 1 %, n=4), while motor neurons were quite sensitive (1 ± 1%, n=4,p<.001). For the micrograph in Figure 2B, we chose one of the few fields of HNO motor neurons that contained cells, rather than debris, to illustrate the decay in morphology. At the highest flux of NO there was a change in oligodendrocyte morphology, suggesting some NO sensitivity, but not nearly as much as seen in the primary motor neurons (Figure 2B,C).
HO1-dependent mechanism for differential NO resistance on the two cell types
We have found in previous studies that, in motor neurons, HO1 is important for induced adaptive resistance-a phenomenon where pretreatment with low dose of NO lends NO resistance to normally quite NO sensitive motor neurons (Bishop et al., 1998, 2004, 2006, Fung et al., 1999, Kitamura et al., 2003). Here we use lower NO doses that are within the physiological range (2pmoles/s) for the pretreatment dose to induce resistance to a more physiologically relevant challenge dose (110pmoles/s) and found that yes, IAR can still be demonstrated in motor neurons (IAR 72%+/−5 vs High NO alone 9+/−4, n=4, p<.001) (Fig. 3A&B). We utilized a more specific HO1 inhibitor, zinc protoporphyrin (ZnPPIX) (Akins et al., 2004, Yang et al., 2001) and found that IAR in motor neurons is abrogated by the inhibition of HO1 activity (72+/−5 vs 30+/−14, n=4, p<.001) (Fig.3A&B).
Figure 3. NO resistance is turned off by HO1 inhibitor and involves peroxynitrite.
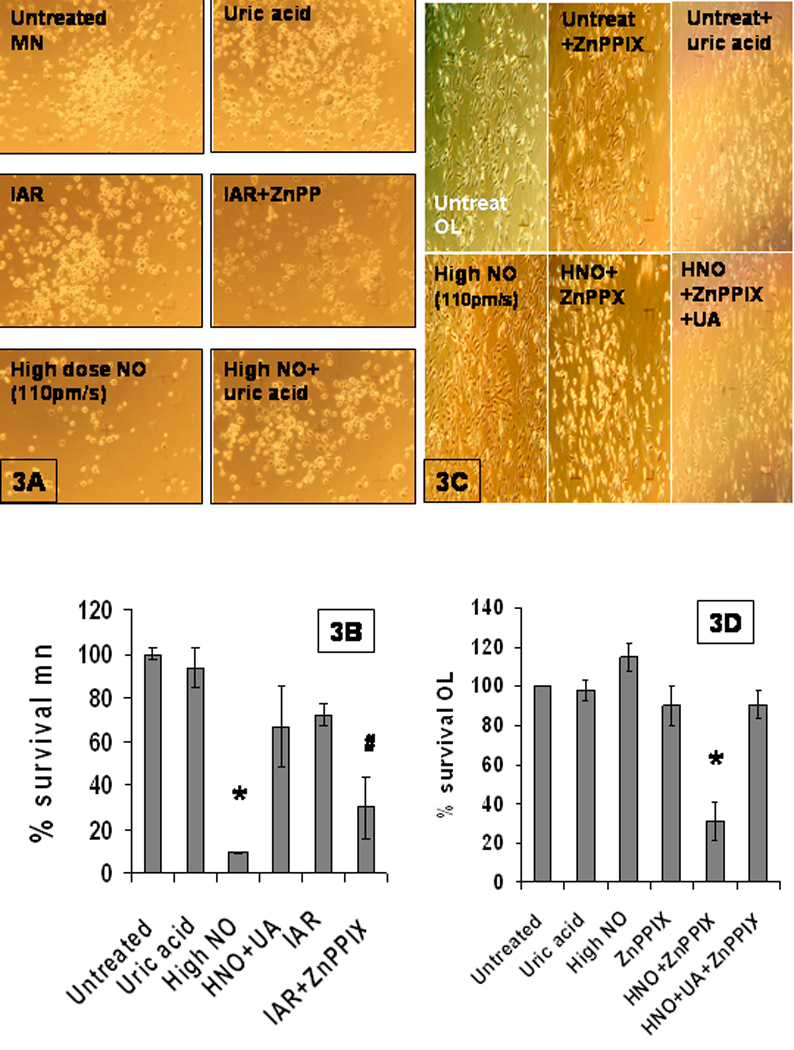
A.) Photomicrographs of motor neurons, all at 100× magnification, are organized in panels: UT--cells treated with spent NO donor, UA--cells treated with 10uM uric acid alone; IAR (induced adaptive resistance)--cells pretreated with low dose NO (2pm/s) followed 2h later by high dose NO (~100pm/s); high doseNO (~100pm/s) , IAR+ZnPPIX--IAR in the presence of 20uM ZnPPIX, high dose NO+UA. B.) The cell survival data is organized along the same labeling as seen in A. The cell survival data was quantified, SEM determined, and p<.001 was determined to be a significant difference between high NO and high NO with uric acid designated by an asterisk. The # is the p<.001 significant difference between IAR and IAR+ZnPPIX. C.) Photomicrographs of oligodendrocytes, all at 100× magnification, are organized in panels: UT-- cells treated with spent NO donor, ZnPPIX -- cells treated with the 20uM HO1 inhibitor alone; UA-- cells treated with 10uM uric acid alone; the HighNO panel -- (~100pm/s NO); HNO + ZnPPIX --cells challenged with high dose NO (110pm/s) in the presence of the HO1 inhibitor; and finally, the HNO + ZnPPIX + UA-- cells challenged with high dose NO (~110pm/s) in the presence of the HO1 inhibitor + uric acid. D.) The cell survival data is organized along the same labeling as seen in C. The cell survival data was quantified, and SEM determined. There was significant difference (p<.001) between HNO, HNO + ZnPPIX, and HNO + ZnPPIX +uric acid, designated by an asterisk
With our previous studies in motor neurons (Bishop et al., 1998, 2004, 2006) and the above study in mind, we asked if the constitutive resistance exhibited by the oligodendrocytes was dependent on HO1. We found that yes, NO resistance to cytotoxic challenge exhibited by the oligodendrocytes is abrogated by the addition of 20uM ZnPPIX (compare 115% +/− 7(n=9) vs 31%+/−10 (n=6) p<.001) (Fig..3C&D). We controlled for possible toxicity of the ZnPPIX by incubating cells with the HO1 inhibitor alone, and found little toxicity (Fig. 3C&D). This profound ZnPPIX-mediated decrease in NO resistance oligodendrocytes indicates that HO1 activity is needed for the oligodendrocyte constitutive NO resistance.
Involvement of peroxynitrite in NO toxicity and NO resistance
In light of the many studies which indicate that much of NO toxicity seen in MS, ALS or spinal injury, is due to protein nitration with subsequent formation of nitrotyrosine residues (3NY) by the RNS, peroxynitrite, (Esteves, 1998, Ischiropoulos & Beckman, 2003, Jack et al., 2007,Pacher et al., 2007, , Prat & Antel 2005) we asked whether the NO toxicity we see is due to peroxynitrite. For our studies we utilized uric acid, which is a specific a peroxynitrite scavenger (Hooper et al., 2000) to determine if it ameliorated NO sensitivity in the motor neurons. Motor neuron NO sensitivity was abrogated by the addition of 10uM uric acid (9%+/.4 vs 67%+/-19, n=4,p<.001) indicating that a significant portion of the NO toxicity seen in motor neurons is due to peroxynitrite(Fig 3A&B).
We asked if the oligodendrocyte resistance was, in fact, to peroxyntrite, by investigating if the NO-sensitive HO1-inhibited oligodendrocytes could be rescued with addition of uric acid. When these cells were incubated with the 10uM uric acid before challenge, NO-mediated cell death was prevented (31%+/−10 (n=6) vs 91%+/−7 (n=4)p<.003)(Fig. 3C&D). In fact, incubation of the HO1-inhibited oligodendrocytes with uric acid before cytotoxic NO treatment restored them to the % cell survival of oligodendrocyctes that were incubated with the HO1 inhibitor alone (91%+/−7 (n=4) vs 90%+/− 10 (n=6)), with no significant difference, indicating that the cell saving effect of uric acid was >99% (Fig 3C&D) Thus we can conclude that >99% of the toxicity seen in NO-challenged HO1-inhibited oligodendrocytes is due to peroxynitrite.
Involvement of 3NY formation in NO sensitivity
In addition to the pharmacological evidence, we wanted to determine, in motor neurons, if the cytotoxic NO doses we used for our experiments produced peroxynitrite as indicated by the intracellular formation of 3NY. Motor neurons were treated, lysed, and analyzed by Western Blot and probed for 3NY formation, with nitrated albumin as a positive control and untreated lysate as a negative control (Figure 4). We used the densitometry software on the whole lane, rather than on individual bands, as a source of comparison (Figure 4). In motor neurons challenged with high dose NO alone we see an increase in 3NY formation, and in IAR we see a mitigation of 3NY formation at these physiologically relevant doses (46% above untreated versus 31% above untreated). When IAR is abrogated by the addition of the HO1 inhibitor ZnPPIX we see a concomitant increase in 3NY formation (34% 3NY increase vs 89% 3NY increase) linking HO1 activity to 3NY inhibition. In motor neurons challenged with high dose NO we see more 3NY formation which is abrogated by the addition of the peroxynitrite scavenger, uric acid (46% increase of 3NY levels above untreated control vs 10% increase of 3NY formation above untreated controls, n=3).
Figure 4. MO3.13 NO resistance is turned off by HO1 inhibitor and involves peroxynitrite.

A).Western blot from motor neuron cell lysates. 1. MW=molecular weight, 2. UA—10uM uric acid, 3. HNO---high dose NO (110pm/s) 4. HNO+UA—high dose NO + uric acid, 5.IAR---low dose NO (2pm/s) followed 2h later by high dose NO (110pm/s) 6. IAR+ZnPPIX---IAR protocol in the presence of HO1 inhibitor (20uM). B.) Quantification of data from A and other western blots. Xfold increase is from 3NY total from untreated (treated with spent NO donors). C).1. MW=molecular weight, 2. 3NY=nitrated albumin, 3.=Untreated M03.13,4.=low dose NO MO3.13, 5.=high NO MO3.13, 6=high NO MO3.13 7.=high NO NSC34D, 8.=high NO NSC34D, 9.=low NO NSC34D, 10.=untreated NSC34D. D.) Quantification of data from C and other western blots. Xfold increase is from 3NY total from untreated (treated with spent NO donors).
We asked if the differential sensitivity seen in oligodendrocytes versus motor neurons is reflected in differences in 3NY formation in response to cytotoxic NO challenge, hence further suggesting that the NO resistance seen in oligodendrocytes is to peroxynitrite rather than NO per se (Figure 4C&D). Motor neuron lysates were loaded on one half of the gel and the oligodendrocyte lysates were loaded on the other half. In motor neurons, high NO doses(110pm/s) do yield significantly increased 3NY formation (~8 fold) (n=4) above UT, indicating that NO challenged results in intracellular 3NY formation, thereby implicating intracellular peroxynitrite formation (Figure 4C&D). In fact, there is an increase of 3NY formation of at least ~4 fold (n=4) as a result of high dose NO challenge in motor neurons, as compared to that in oligodendrocytes (Figure 4C&D). In some particular bands there is a 10 fold increase of 3NY in NO challenged motor neurons as compared to NO challenged oligodendrocytes. Clearly, Figure 4C and other westerns indicate that the NO challenge yields peroxynitrite, and that oligodendrocytes somehow mitigate the peroxynitrite mediated 3NY formation in response to NO challenge.
Mitigation of motor neuron NO sensitivity upon coculture with oligodendrocytes
Since oligodendrocytes are NO-resistant at the fluxes we used, and motor neurons are quite NO sensitive we asked if the oligodendrocytes can bestow their NO resistance upon motor neurons, or vise versa. We cocultured the two cell types and the ratio at which both cell types exhibited optimal health was ~66% motor neurons and ~33% oligodendrocytes (Figure 5A). It was found that in coculture the oligodendrocytes myelinate motor neuron axons as indicated morphologically by Nodes of Ranvier (Figure 5A). Although this is chiefly a morphological detail, it further legitimizes the coculture as an effective model to study the interplay between motor neurons and oligodendrocytes. We observed the cultures in phase contrast, and then stained the cultures with a fluorescent antibody specific to neurons, MAP2. More than 99% of the neurons in the culture expressed the neuron specific antibody, while none of the oligodendrocytes expressed the neuron specific antibody. Thus the green cells seen in the cocultures in Figure 5 were neurons. For the oligodendrocyte cultures, since they exhibited no green cells, we checked the phase contrast to make sure we were at an NO dose (50pmoles/s) where there was no oligodendrocyte loss.
Figure 5. Cocultures of motor neurons and oligodendrocytes.

A. A micrograph of the cocultured motor neurons and oligodendrocytes, with oligodendrocytes myelinating motor neuron axons as indicated morphologically by Nodes of Ranvier, which are indicated by white arrows. These are all at 320× magnification. B. Micrographs of cells (mag 320×), all labelled with MAP-2 (a neuron specific protein). The green cells were exclusively motor neurons. The top row is untreated: oligodendrocytes, motor neurons and cocultures. The bottom row is high dose NO (50pm/s which is half of toxic dose in other experiments):oligodendrocytes, motor neurons, cocultures C. This is a quantification of the data from the micrographs in A. Standard error of the mean (n=4) was calculated. Comparison of the two groups: NO challenged motor neurons vs NO challenged motor neurons cocultured with oligodendrocytes, was made and found to be significant (P<.001) as indicated by asterisk. D. A micrograph of motor neurons untreated, challenged with high NO (110pm/s), untreated in the presence of oligo conditioned media (OLCM), challenged with high NO in the presence of OLCM, challenged in the presence of OLCM treated with the HO1 inhibitor (20uM ZnPPIX), or challenged in the presence of OLCM incubated with 10% trypsin.. E. This is a quantification of D and is the average of at least (n=4) experiments. SEM was calculated and the difference in % cell survival between NO challenged neurons with and without OLCM was significant (p<.001), designated by an asterisk. The difference between NO challenged neurons with OLCM versus NO challenged neurons with ZnPPIX-treated OLCM was significant, designated by an #. The difference between NO challenged cells with OLCM and NO challenged cells with OLCM treated with 10% trypsin was significant, designated by a +
We exposed pure motor neurons, pure oligodendrocytes ,and cocultures to NO, and asked if motor neurons cocultured with oligodendrocytes were more resistant to NO than were motor neurons alone. Motor neurons in coculture were significantly more resistant (109%+/−5 (n=4) vs 53% +/− 2, n=4, p<.001) (Figure 5 B &C). Clearly the oligodendrocytes bestow upon the neurons their native resistance. We then asked if the oligodendrocytes merely shield the motor neurons from NO or if they secrete a factor that protects neurons. We incubated one set of motor neurons with media conditioned by oligodendrocytes and another set of neurons with neuron conditioned media. We challenged both sets of motor neurons with NO and found that there was significant protection exerted by the oligodendrocyte conditioned media as indicated by the increase in % cell survival (41%+/−0.1 vs 80%+/−0.4, n=4, p<.001) (Fig 5D&E). This protection was attenuated by incubation of the oligo CM with the HO1 inhibitor, ZnPPIX,(64%+/−5) and augmented by the incubation of the oligo CM with the protease, trypsin (96%+/−6) indicating that the secreted factor may be HO1, or a protein with HO1 like activity.
Discussion
With this study, we have established that motor neurons are more sensitive to NO than are oligodendrocytes, that this differential NO resistance seen in oligodendrocytes is due to resistance to peroxynitrite rather than to NO per se, and that the NO sensitivity seen in motor neurons is, again, to peroxynitrite rather than NO. In addition, we found that the resistance seen in oligodendrocytes is HO1 dependent. Most importantly, we have found that oligodendrocytes can imbue motor neurons with NO resistance when cocultured with neurons and that oligodendrocytes secrete an unknown neuron protective factor that possibly could be HO1 itself or a protein with HO1 activity. These results add to the current findings of prominent researchers in the MS field and suggest directions for further study-one of which is isolation and characterization of the secreted factor.
NO is a proven common denominator in spinal injury, ALS and MS (Beckman et al., 1996; Giovannoni et al., 1997, 1998, Ishiropolous & Beckman et al., 2001). In ALS there is substantial motor neuron death with concomitant accumulation of NO and other reactive nitrogenous species (RNS) with such reliability that these NO metabolites (3NY proteins) are markers for the disease (Ischiropolous & Beckman et al., 2003, Reiter et al.,2002). These same RNS have been detected in patients with MS where metabolites of NO, nitrate and nitrite, and free 3NY and 3NY proteins are found in the CSF of MS individuals, the concentrations of which are correlated with the severity and duration of the disease (Acar et al., 2003; Bizzoero et al., 2005, Giovannoni et al., 1997, 1998, Liu et al.,2005). Therefore our studies of NO sensitivity in oligodendrocytes and motor neurons, are apropos to MS, and offer further directions for research.
Although MS has been considered a disease predominantly affecting white matter brain tissue with demyelination first, followed by axonal injury and damage to neurons (Silber and Sharief, 1999), we may have to re-examine this current paradigm of MS etiology in light of our findings of increased NO sensitivity of motor neurons as compared to oligodendrocytes. In one study, amyloid precursor protein, a sensitive marker of axonal damage, was found within acute MS brain lesions. In fact, several investigators have data that has led them to the conclusion that axonal damage induces MS, with demyelination occurring after (Bitsch et al., 2000, De Stefano et al., 2001, Fergurson et al., 1997).
Several important studies address NO sensitivity of oligodendrocytes (Prat & Antel et al.,2005), or motor neurons (Estevez et al.,1998, Jack et al., 2007, Ischoropolous & Beckman et al..,2003) individually. Our study of NO sensitivity in both cell types, in direct comparison and in coculture, is an important step towards elucidating which cell type starts the cascade of demyelination and cell death in MS. Our observations from our direct comparison of motor neurons having significantly more NO sensitivity than oligodendrocytes, and the fact that oligodendrocytes lend NO resistance to motor neurons in coculture, lead one to imagine a scenario, in MS, where the excess NO released overwhelms the protective effects of oligodendrocytes, or even turns oligodendrocytes against neurons, causing neuronal death with subsequent demyelination. In normal CNS functions, the interplay between the axons of neurons and oligodendrocytes is critical for maturation of oligodendrocytes. Disturbance of this cellular relationship possibly leads to MS pathology (Silber and Sharief, 1999).
Finally, our finding of an HO1-mediated mechanism for oligodendrocyte NO-resistance, which can be bestowed upon motor neurons in coculture, offers a possible therapeutic target for mitigation of axonal injury seen in MS.
Acknowledgments
We gratefully acknowledge the expertise of Dr. Neil R. Cashman and his kind gift of the NSC34 cells and the expertise of Dr. Bruce Demple. I would also like to acknowledge the support of NASA and Dr. Robert R. Richmond (NASA Biology Directorate), the Louis B. Stokes Alliance for Minority Participation (LSAMP), and NIH R15 AREA grant.
Footnotes
Note: Responses to the first round of reviewers’ critiques are denoted by italics in the manuscript text and new figures. Deletion of supportive data not relevant to the paper is still noted the ms. My second round of revisions in response to the reviewers are denoted by underlined italics in the figure legend(Fig 5C,D,E), the methods, the results, and discussion. MY THIRD ROUND OF REVISONS (ONE) IS DENOTED IN CAPITAL LETTERS AND I HAVE TRIMMED MY REFERENCES. THANKYOU.
References
- Acar G, Idiman F, Idiman E, Kirkali G, Cakmakci H, Ozakbas S. Nitric oxide as an activity marker in multiple sclerosis. J Neurol. 2003;250(5):588–592. doi: 10.1007/s00415-003-1041-0. [DOI] [PubMed] [Google Scholar]
- Akins R, Jr, McLaughlin T, Boyce R, Gilmour L, Gratton K. Exogenous Metalloporphyrins Alter the Organization and Function of Cultured Neonatal Rat Heart Cells Via Modulation of Heme Oxygenase Activity. Journal of Cellular Physiology. 2004;201:26–34. doi: 10.1002/jcp.20040. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Estevez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- Bishop A, Anderson J. NO signaling in the CNS:from the physiological to the pathological. In: Laranjinha João., editor. Toxicology(Special Issue) Nitric Oxide, Cell Signalling and Death. 2005. [Google Scholar]
- Bishop A, Cashman NR. Induced adaptive resistance to oxidative stress in the CNS: Discussion of possible mechanisms and their therapeutic potential. Current Drug Metabolism. 2003;4(2):171–184. doi: 10.2174/1389200033489514. [DOI] [PubMed] [Google Scholar]
- Bishop A, Fung-Yet S, Perrella MJ, Lee AM, Cashman NR, Demple B. Decreased resistance to nitric oxide in motor neurons of HO-1 null mice. BBRC. 2004;325:3–9. [Google Scholar]
- Bishop A, Marquis JC, Cashman NR, Demple B. Adaptive resistance to nitric oxide in motor neurons. Free Radical Biology & Medicine. 1999;26(7–8):978–986. doi: 10.1016/s0891-5849(98)00284-6. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123(Pt 6):1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Bizzozero OA, DeJesus G, Bixler HA, Pastuszyn A. Evidence of nitrosative damage in the brain white matter of patients with multiple sclerosis. Neurochem Res. 2005;30(1):139–149. doi: 10.1007/s11064-004-9695-2. [DOI] [PubMed] [Google Scholar]
- Buntinx M, Vanderlocht J, Hellings N, Vandenabeele F, Lambrichts I, Raus J, Ameloot M, Stinissen P, Steels P. Characterization of three human oligodendroglial cell lines as a model to study oligodendrocyte injury: Morphology and oligodendrocyte-specific gene expression. J Neurocytol. 2003;32:25–38. doi: 10.1023/a:1027324230923. [DOI] [PubMed] [Google Scholar]
- Businaro R, Fabrizi C, Caronti B, Calderaro C, Fumagalli L, Lauro GM. Myelin basic protein induces heme oxygenase-1 in human astroglial cells. Glia. 2002;37:83–88. doi: 10.1002/glia.10018. [DOI] [PubMed] [Google Scholar]
- Bouton C, Demple B. Nitric oxide-inducible expression of heme oxygenase-1 in human cells. Translation-independent stabilization of the mRNA and evidence for direct action of nitric oxide. J Biol Chem. 2000;275:32688–32693. doi: 10.1074/jbc.275.42.32688. [DOI] [PubMed] [Google Scholar]
- Brugg B, Matus A. Phosphorylation determines the binding of microtubule-associated protein 2 (MAP2) to microtubles in living cells. J Cell Biol. 1991;114:735–743. doi: 10.1083/jcb.114.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Devel. Dynamics. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Cassina P, Peluffo H, Pehar M, Martinez-Palma L, Ressia A, Beckman JS, Estevez AG, Barbeito L. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J.Neurosci.Res. 2002;67:21–29. doi: 10.1002/jnr.10107. [DOI] [PubMed] [Google Scholar]
- Clough GF, Bennett AR, Church MK. Measurement of nitric oxide concentration in human skin in vivo using dermal microdialysis. Exp Physiol. 1998;83(3):431–434. doi: 10.1113/expphysiol.1998.sp004126. [DOI] [PubMed] [Google Scholar]
- Cornish AS, Jijon H, Yachimec C, Madsen KL. Peroxynitrite enhances the ability of salmonella Dublin to invade T84 monolayers. SHOCK. 2002;18(1):93–96. doi: 10.1097/00024382-200207000-00017. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, Matthews PM, Arnold DL. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;58:65–70. doi: 10.1001/archneur.58.1.65. [DOI] [PubMed] [Google Scholar]
- Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhoták S, Austin RC. Vascular Endothelium: Implications in Atherogenesis Arterioscler. Thromb. Vasc. Biol. 2005;25:2623–2629. doi: 10.1161/01.ATV.0000189159.96900.d9. [DOI] [PubMed] [Google Scholar]
- Durham HD, Dahrouge S, Cashman NR. Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. Neurotoxicol. 1993;14:387–395. [PubMed] [Google Scholar]
- Eggett CJ, Crosier S, Manning P, Cookson MR, Menzies FM, McNeil CJ, Shaw PJ. Development and characterisation of a glutamate-sensitive motor neuron cell line. J. Neurochemistry. 2000;74:1895–1902. doi: 10.1046/j.1471-4159.2000.0741895.x. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Spear N, Manuel SM, Radi R, Henderson CE, Barbieto L, Beckman JS. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J. Neurosci. 1998;18(3):923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estevez AG, Sampson JB, Zhuang Y-X, Spear N, Richardson GJ, Crow JP, Tarpey MM, Barbeito L, Beckman JS. Liposome-delivered superoxide dismutase prevents nitric oxidedependent motor neuron death induced by trophic factor withdrawal. Free Radic. Biol. Med. 2000;28:437–446. doi: 10.1016/s0891-5849(99)00261-0. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Richmon JD, Sato M, Sharp FR, Panter SS, Noble LJ. Induction of heme oxygenase-1 (HO-1) in glia after traumatic brain injury. Brain Res. 1996;736(1–2):68–75. doi: 10.1016/0006-8993(96)00680-4. [DOI] [PubMed] [Google Scholar]
- Giovannoni G, Heales SJ, Land JM, Thompson EJ. The potential role of nitric oxide in multiple sclerosis. Mult Scler. 1998;4(3):212–216. doi: 10.1177/135245859800400323. [DOI] [PubMed] [Google Scholar]
- Giovannoni G, Heales SJ, Silver NC, O'Riordan J, Miller RF, Land JM, Clark JB, Thompson EJ. Raised serum nitrate and nitrite levels in patients with multiple sclerosis. J Neurol Sci. 1997;145(1):77–81. doi: 10.1016/s0022-510x(96)00246-8. [DOI] [PubMed] [Google Scholar]
- Gong P, Stewart D, Hu B, Li N, Cook J, Nel A, Alam J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Delta(12,14)-prostaglandin J(2) is mediated by the stress response elements and transcription factor Nrf2. Antioxid Redox Signal. 2002;4:249–257. doi: 10.1089/152308602753666307. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. GLIA. 1998;23(3):249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Herzog W, Weber K. Fractionation of brain microtubule-associated proteins (isolation of two different which stimulate tubulin polymerization in vitro) Eur J Biochem. 1978;98:1–8. doi: 10.1111/j.1432-1033.1978.tb12716.x. [DOI] [PubMed] [Google Scholar]
- Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H, Spitsin SV. Uric Acid, A Peroxynitrite Scavenger, Inhibits CNS Inflammation, Blood-CNS Barrier Permeability Changes, And Tissue Damage. A Mouse Model Of MS FASEB J. 2000;14(5):691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- Huk I, Brovkovych V, Nanobash Vili J, Weigel G, Neumayer Ch, Partyka L, Patton S, Malinski T. Bioflavonoid quercetin scavenges superoxide and increases nitric oxide concentration in ischaemia-reperfusion injury: an experimental study. British Journal of Surgery. 1998;Vol 85(8):1080–1085. doi: 10.1046/j.1365-2168.1998.00787.x. [DOI] [PubMed] [Google Scholar]
- Inglese M. Multiple Sclerosis: New Insights and Trends. Am J Neuroradiol. 2006;27:954–957. [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J. Clin. Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C, Antel J, Brück W, Kuhlmann T. Contrasting potential of nitric oxide and peroxynitrite to mediate oligodendrocyte injury in multiple sclerosis. Glia. 2007;55(9):926–934. doi: 10.1002/glia.20514. [DOI] [PubMed] [Google Scholar]
- Johnson MI, Bunge RP, Wood PM. Protocols for Neural Cell Culture. 3rd edition. New Jersey: Human Press; 2004. Primary Cell Cultures for the Study for Myelination; pp. 95–115. [Google Scholar]
- Juckett M, Zheng Y, Yuan H, Pastor T, Antholine W, Weber M, Vercellotti G. Heme and the endothelium. Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J Biol Chem. 1998;273:23388–23397. doi: 10.1074/jbc.273.36.23388. [DOI] [PubMed] [Google Scholar]
- Kawase M, Kinouchi H, Kato I, Akabane A, Kondo T, Arai S, Fujimura M, Okamoto H, Yoshimoto T. Inducible nitric oxide synthase following hypoxia in rat cultured glial cell. Brain Research. 1996;738:319–322. doi: 10.1016/s0006-8993(96)00924-9. [DOI] [PubMed] [Google Scholar]
- Kinobe R, Ji Y, Nakatsu K. Peroxynitrite-mediated inactivation of heme oxygenases. BMC Pharmacology. 2004;4:26. doi: 10.1186/1471-2210-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Ishida Y, Takata K, Mizutani H, Kakimura J, Inden M, Nakata J, Taniguchi T, Tsukahara T, Akaike A, Shimohama S. Hyperbilirubinemia protects against focal ischemia in rats. J. Neurosci. Res. 2003;71:544–550. doi: 10.1002/jnr.10514. [DOI] [PubMed] [Google Scholar]
- Leonard CS, Michaelis EK, Mitchell KM. Activity-Dependent Nitric Oxide Concentration Dynamics in the Laterodorsal Tegmental Nucleus In Vitro. J Neurophysiol •. 2001;VOL 86:2159–2172. doi: 10.1152/jn.2001.86.5.2159. [DOI] [PubMed] [Google Scholar]
- Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol. 2001;158:2057–2066. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annual Review of Pharmacology and Toxicology. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Yoshino H, Yuki N, Hara Y, Cashman NR, Handa S, Miyatake T. Ganglioside characterization of a cell line displaying motor neuron-like phenotype: GM2 as a possible major ganglioside in motor neurons. J. Neurol. Sci. 1995;131:111–118. doi: 10.1016/0022-510x(95)00101-7. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Trudel GC, Shaw IT, Antel JP, Cashman NR. A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J Neurobiol. 1995 Feb;26(2):283–293. doi: 10.1002/neu.480260212. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer MA, Stasiv Y, Benraiss A, Chmieinicki E, Grinberg A, Westphal H, Goldman SA, Enikolopov G. Nitric oxide negatively regulates mammalian adult neurogenesis. PNAS. 2003;100(16):9566–9571. doi: 10.1073/pnas.1633579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahian N, Maines M. Site of injury-directed induction of heme oxygenase-1 and -2 in experimental spinal cord injury: differential functions in neuronal defense mechanisms? Journal of Neurochemistry. 2001;76:539–554. doi: 10.1046/j.1471-4159.2001.00023.x. 2001. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Peunova N, Enikolopov G. Nitric oxide triggers a switch to growth arrest during differentiation of neuronal cells. Nature. 1995;375:68–73. doi: 10.1038/375068a0. [DOI] [PubMed] [Google Scholar]
- Prat A, Antel J. Pathogenesis of multiple sclerosis. Demyelinating diseases Current Opinion in Neurology. 2005;18(3):225–230. doi: 10.1097/01.wco.0000169737.99040.31. [DOI] [PubMed] [Google Scholar]
- Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 2002 Sep 12;35(6):1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Reiter TA, Pang B, Dedon PC, Demple B. Resistance to nitric oxide-induced necrosis in heme oxygenase-1 overexpressing pulmonary epithelial cells associated with decreased lipid peroxidation. J Biol Chem. 2006:36603–36612. doi: 10.1074/jbc.M602634200. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Dai W, Gan XD, Ali S, Fu J, Back SA, Sanchez RM, Segal MM, Follett PL, Jensen FE, Volpe JJ. Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. Journal of Neuroscience Research. 2002;71(2):237–245. doi: 10.1002/jnr.10472. [DOI] [PubMed] [Google Scholar]
- Schipper H. Heme Oxygenase-1: Transducer of Pathological Brain Iron Sequestration under Oxidative Stress. Ann. N.Y. Acad. Sci. 2004;1012:84–93. doi: 10.1196/annals.1306.007. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Cisse S, Stopa E. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann. Neurol. 1995;37:758–768. doi: 10.1002/ana.410370609. [DOI] [PubMed] [Google Scholar]
- Silber E, Sharief MK. Axonal degeneration in the pathogenesis of multiple sclerosis. J Neurol Sci. 1999;170:11–18. doi: 10.1016/s0022-510x(99)00178-1. [DOI] [PubMed] [Google Scholar]
- Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiasch E, Bach FH. Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide. Antioxid Redox Signal. 2002;4:321–329. doi: 10.1089/152308602753666370. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Jia L, Eu JP, Mcmahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ. Mammalian nitric oxide synthases. Biochim Biophys Acta. 1999;1411(22–3):217–230. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- Tamir S, Burney S, Tannenbaum SR. DNA damage by nitric oxide. Chem. Res. Toxicol. 1996;9:821–827. doi: 10.1021/tx9600311. [DOI] [PubMed] [Google Scholar]
- Tamir S, Lewis RS, de Rojas Walker T, Deen WM, Wishnok JS, Tannenbaum SR. The influence of delivery rate on the chemistry and biological effects of nitric oxide. Chem. Res. Toxicol. 1993;6:895–899. doi: 10.1021/tx00036a021. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Sato S, Ohnishi T, Ohnishi ST. Electron paramagnetic resonance (EPR) detection of nitric oxide produced during forebrain ischemia of the rat. J Cereb Blood Flow Metab. 1994;14(5):715–722. doi: 10.1038/jcbfm.1994.92. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis:relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Lee YS, Lin CY, Lin VW, Sindhu RK. NAD(P)H oxidase, superoxide dismutase, catalase, glutathione peroxidase and nitric oxide synthase expression in subacute spinal cord injury. Brain Res. 2004;995(1):76–83. doi: 10.1016/j.brainres.2003.09.056. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- Yang G, Nguyen X, Ou J, Rekulapelli P, Stevenson DK, Dennery PA. Unique effects of zinc protoporphyrin on HO-1 induction and apoptosis. Blood. 2001;97:1306–1313. doi: 10.1182/blood.v97.5.1306. [DOI] [PubMed] [Google Scholar]