

Abstract
Despite it being a quintessential Phase II detoxification gene, the transcriptional regulation of the rat γ-glutamate cysteine ligase catalytic subunit (GCLC) is controversial. Computer-based sequence analysis identified three putative antioxidant response elements (AREs) at positions −889 to −865 (ARE1), −3170 to −3146 (ARE2) and −3901 to −3877 (ARE3) in the 5’-flanking region of the transcriptional start site. Transfections of individual ARE-luciferase reporter gene constructs into H4IIE cells, a rat hepatoma cell line, identified ARE3 as the functional promoter. Chromatin immunoprecipitation assays using primary rat hepatocytes showed that the transcription factor Nrf2, which is known to regulate ARE-mediated genes, is associated with ARE3. Co-transfection of H4IIE cells with luciferase reporter plasmids containing Gclc ARE3 and an Nrf2 expression plasmid resulted in a 3-fold activation of ARE3-mediated transcription relative to controls. “Loss-of-function” analysis for Nrf2 by small interfering RNA (siRNA) revealed that ARE3-mediated expression was significantly impaired while site-directed mutagenesis of the ARE3-luciferase reporter abolished Nrf2-mediated induction. Treatment with two known Nrf2 inducers, R-(α)-lipoic acid and anetholedithiolethione, showed that the inducible expression of the GCLC gene was also regulated by the ARE3 element. Taken together, these results show that Nrf2 regulates the constitutive expression of rat Gclc through a distal ARE present in its 5’-flanking region. This is the first report showing that rat Gclc is under the transcriptional control of the Nrf2-ARE pathway on a constitutive basis.
Keywords: NF-E2-related factor 2; glutathione, Chromatin immunoprecipitation; detoxification; anetholedithiolethione; R-(α)-lipoic acid; Phase II detoxification
1. Introduction
Glutamate cysteine ligase (GCL) catalyzes the first and rate-limiting step of the de novo synthesis of glutathione (GSH), the most abundant non-protein thiol in the cell (1). GSH plays key roles in detoxifying xenobiotics, peroxides, and electrophiles, while also maintaining the normal intracellular thiol redox status (2–5). The de novo synthesis of GSH from its constituent amino acids involves two ATP-requiring enzymatic steps: the formation of γ-glutamylcysteine from glutamate and cysteine, and subsequent formation of GSH from γ-glutamylcysteine and glycine (6, 7). GCL is the rate-controlling enzyme for GSH synthesis (8). The GCL protein is a heterodimer that can be dissociated under non-denaturing conditions into a catalytic (GCLC, 73 kDa) and a modulatory (GCLM, 29 kDa) subunit, which are encoded by separate genes (9). Although GCLC contains the entire catalytic activity, association with GCLM regulates this activity (8). Since GCL is a major determinant of the overall capacity of GSH synthesis, regulation of GCL subunits has been a topic of extensive research (7). GCL has multiple levels of regulation, which ultimately affect either the catalytic or modifier subunits or both. In addition, the enzyme can be regulated at the kinetic, post-translational and transcriptional levels (7, 10, 11). However, the regulation of GCL at the transcriptional level produces a more persistent effect and thus is important for the maintenance of GSH homeostasis in response to oxidative stress (12).
The distal 5’-flanking region of Gclc has been fully characterized in the human (13) and mouse (14), but not yet in the rat (15). Several DNA cis-elements including those for NF-κB, the AP-1 binding site, and the antioxidant response element (ARE) have been implicated in GCL gene regulation (16). In humans and mice, the ARE (core sequence: GTGACNNNGC) has been identified as the regulatory component responsible for the induction of Gclc both on a constitutive basis and in response to oxidative or electrophilic stress (13, 17–19). Many transcription factors have been reported to bind the ARE, such as Nrf2 family members (Nrf1/2/3), small maf proteins (maf G/K/F), as well as AP-1 transcription factors [Jun (c-Jun, JunB, JunD), and Fos family members (c-Fos, FosB, Fra1, Fra2)] (reviewed by Jaiswal) (20). Among them, ARE-dependent GCLC gene expression is largely dependent upon Nrf2, a member of the ‘Capn’Collar (CNC) family of bZIP proteins (18, 21). Nrf2 is located primarily in the cytosol but upon stimulation, accumulates in the nucleus, where it heterodimerizes with other leucine zipper proteins (e.g. c-Jun and small maf proteins), and binds the ARE to initiate gene transcription (22, 23). Importantly, when overexpressed in cells by transfection, Nrf2 accumulates in the nucleus and activates ARE-mediated transcription (24, 25).
The 5’-flanking region of rat Gclc has only been partially characterized (15, 26–30) where only a 1.8 kb sequence upstream of the start site was thoroughly analyzed. In this region, several binding sites for AP-1 and NF-κB were reported (15). Yang and coworkers (30) suggested that an AP-1 sequence in the proximal promoter region of Gclc is critical to its transcriptional upregulation in response to oxidative stress. In part, this contention is because AREs that are present in the human Gclc promoter, are not found in this 1.8 kb region of the rat Gclc promoter. However, Nrf2-ARE binding has been detected in the rat by transfection of an ARE-containing sequence derived from the human Gclc (31). A 44-bp ARE sequence, which shares a 31 bp similarity with the human Gclc ARE, has recently been designated as the rat Gclc ARE, but no additional sequence information has been disclosed (32). Thus it is possible that functional AREs in the rat GCLC gene are present further upstream of the characterized 5’-flanking regions. Regardless, which cis-acting element is primarily involved in the basal gene expression of rat GCLC is still unknown and an ongoing subject of debate.
It has been well established that both GCLC and GCLM are inducible at the level of transcription by various agents such as quinones (e.g. tert-butyl hydroquinone), dithiolethiones [e.g. anetholedithiolethione (ADT)], isothiocyanates (e.g. sulforaphane), and cyclic disulfides [e.g. R-(α)-lipoic acid (LA)] (33). We have previously demonstrated that intraperitoneal injection (40 mg/kg body weight) of LA to rats increases nuclear Nrf2 levels and subsequent binding to a consensus ARE (34). However, whether Nrf2 inducers increase Gclc transcription directly through any specific ARE region present in the 5’-flanking region of the rat GCLC gene needs to be investigated.
The aim of the present study was to determine whether Nrf2 controlled basal as well as inducible transcriptional regulation of rat Gclc through an ARE-dependent mechanism. Three putative AREs were identified in the promoter region of the rat GCLC gene (designated ARE1, 2 and 3), respectively. Complete characterization of these elements as well as putative AP1 sequences show for the first time that constitutive and inducible expression of rat Gclc is regulated at the ARE3 site in an Nrf2-driven manner.
2. Materials and Methods
2.1 Chemicals and antibodies
Restriction enzymes and T4 DNA ligase for subcloning were from New England BioLabs (Ipswich, MA). All PCR reactions for cloning the full-length Gclc promoter were performed using the LA Taq Kit (TaKaRa Bio, Shiga, Japan). The PCR products were cloned into the TOPO isomerase linked vector, pCR 2.1, using the TOPO TA Cloning Kit from Invitrogen (Carlsbad, CA). All transformations used in cloning the Gclc promoter employed TOP10 One Shot competent cells (Invitrogen, Carlsbad, CA). QIAamp DNA Micro kit was used to isolate genomic DNA from liver tissue (Qiagen, Valencia, CA). The Dual Luciferase Reporter Assay System™ and reporter plasmids, pGL4 minimal promoter vector, pGL4 Basic vector, and phRL-CMV vector were from Promega (Madison, WI). The expression vector for Nrf2 (pcDNA3.1-Nrf2) was a kind gift from Dr. Anil Jaiswal (35). Custom oligonucleotides used in PCR cloning, subcloning, and DNA sequencing were purchased from Invitrogen. Sequence service was provided by the Center for Gene Research and Biocomputing at Oregon State University. Rabbit anti-Nrf2 (H-300) antibody, Nrf2 siRNA and scrambled sequences were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). R-(α)-lipoic acid (LA) was a gift of Asta Medica (Frankfurt, Mainz, Germany). ADT was a kind gift from Dr. Balz Frei (Oregon State University). All chemicals used were the highest purity available and at least analytical grade.
2.2 Animals
Male Fischer 344 rats (age: 3 months) were purchased from the National Institute of Aging colonies. Rats were maintained in separate security barriers operated by Lab Animal Resources, Oregon State University according to the guidelines established in the Guide for the Care and Use of Laboratory Animals (Animal Resources Commission on Life Sciences, National Research Council) and under specific pathogen free (SPF) conditions.
2.3 Sequencing the 5’-flanking region of the rat GCLC gene and identification of putative ARE(s)
To identify the presence and location of putative ARE(s) in the rat Gclc promoter, the 5 kb upstream from the translation start site was downloaded from the NCBI database (www.ncbi.nlm.nih.gov/genome/guide/rat). This sequence was used to search for putative ARE(s) with the help of the MatInspector (36) and the Matrix Family Library software (version 2.4; MatInspector, Genomatix, Munich, Germany) using the ARE primary core sequence (RTGAYNNNGCR) as the probe. The location of the transcription start site for the rat GCLC gene has already been determined (15).
2.4 Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation analysis was conducted using control rabbit IgG, anti-Nrf2 (Santa Cruz; sc-13032) antibodies. PCR primers are described in Table 1. Briefly, proteins and DNA were cross-linked with 1% formaldehyde for 10 minutes at room temperature and cells lysed in SDS-lysis buffer containing protease inhibitors and then sheared to an average length of 500–800 bp by sonication using a Sonic Dismembrator Model F60 (Fisher Scientific, Pittsburgh, PA). Sheared chromatin was immunocleared with salmon sperm DNA/protein A-sepharose and 10% of the precleared chromatin was stored and labeled as “input DNA”. The remaining chromatin was immunoprecipated with IgG (control) or Nrf2 antibodies (2 µg) overnight at 4°C. Immune complexes were adsorbed onto salmon sperm DNA/protein A-sepharose beads. Immunoprecipitates were washed sequentially with wash buffers to reduce background. Protein–DNA complexes were eluted from the antibody with elution buffer (1% SDS, 0.1 M NaHCO3) and formaldehyde cross-links reversed by addition of NaCl (5 M) and heating at 65 °C overnight. DNA was purified using the QIAquick purification kit (Qiagen, Valencia, CA) and PCR performed using primer pairs that spanned each of the ARE and AP1 elements (Table 1). Amplification was detected by agarose gel electrophoresis and visualized by ethidium bromide staining. PCR products were quantified using SYBR Green Mastermix (New England Biolabs) with the Mini Opticon 2 Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Specific enrichment of DNA by anti-Nrf2 antibody was calculated by subtracting the PCR value of normal IgG from that of anti-Nrf2 antibody and by normalizing that value to the PCR input. Triplicate PCR reactions were conducted for each sample, and the experiments were repeated at least thrice. The specificity of the PCR products was confirmed by melting curve analysis and size (agarose gel electrophoresis).
Table 1.
Primers used for ChIP analysis of Gclc Promoter Elements
| Gclc 5’- Element | Primers used for ChIP analysis |
|---|---|
| ARE1 | FP: 5’-TTGCAAAACATTAATCGAACAACTA-3’ RP: 5’-TTACTGGTTATTTACATGCGTTCAT-3’ |
| ARE2 | FP: 5’-TTCCTCCTGACAGTGAGTTGACACTCTGGA-3’ RP: 5’-TTTTTAAGGCAGCTACTGAAAAGTC-3’ |
| ARE3 | FP: 5’-AAGAAGCCAAGAAGGAAGTGTG-3’ RP: 5’-ACGAGAAGAAGCTCTGGGTCTT-3’ |
‘FP’ and ‘RP’ denote forward and reverse primer, respectively.
2.5 Construction of full-length Gclc promoter construct
To validate the use of specific ARE plasmid constructs, the full-length Gclc promoter region incorporating −5868 to −1 bp from the translation start site was cloned into a pGL4 basic vector (Promega). Because of the high GC-rich regions contained in the 5’-flanking region of Gclc, primers designed with sequences containing high GC content failed to produce an amplified product. To overcome this obstacle, the following cloning strategy was empirically devised. QIAamp DNA Micro kit (Qiagen) was used to isolate genomic DNA from F344 rat liver tissue. The Gclc promoter was cloned using the following primers 5’-CAGGACTTTGCCATTTGTCC-3’ and 5’-CGGGAGGCAGATATTCACAT-3’ to amplify a region 310bp downstream and 5,869bp upstream of the translation start site of Gclc. The PCR product was cloned into the TOPO isomerase linked vector, pCR 2.1, using the TOPO TA Cloning Kit (Invitrogen). The primers to amplify the fragment (−5868 to −1 bp) were as follows: 5’-CAGGACTTTGCCATTTGTCC-3’ and 5’-GCCGCGTCCTCCTCCT-3’. The Gclc promoter region was then cloned into the pCR 2.1 vector in the reverse orientation using the TOPO TA Cloning Kit and manufacturers instructions. Finally, the Gclc promoter region was directionally cloned into the pGL4 Basic vector using the Kpn1 and EcoRV restriction sites. The entire insert was sequenced for fidelity. Throughout, PCR reactions were using the TaKaRa LA Taq Kit, and all vectors were characterized by Bgl1 endonuclease restriction digests and sequencing. All transformations used in cloning the Gclc promoter employed TOP10 One Shot competent cells.
2.6 Site-directed mutagenesis of full-length Gclc promoter construct
Site-directed mutagenesis of the ARE3 sequence was performed to determine the relative contribution of the ARE and/or AP1 core sequences on reporter gene activity. A specific mutation in the ARE3 sequence of the full-length Gclc pGL4 construct (FL) was performed using the Quickchange II XL kit (Stratagene). Creation of pGL4 FL mutated ARE construct (FL AREm) used the forward primer: 5’-GCAGAGTGcTGAGATTCGGTGAGGCGAAACGGCGCGCCGGG-3’ and the reverse complement of this primer. Bases underlined in the primer represent the minimal core ARE sequence; bases in lower case represent differences between the Rat Genome 3.4 vs. Fischer 344 genomic sequence; and bases in bold represent mutations introduced by site-directed mutagenesis. Mutagenesis was accomplished using manufacturer’s instructions with the following modifications: PCR amplification was performed using GCl buffer (TaKaRa) designed for amplification of GC-rich fragments. All transformations were performed using XL10-Gold Ultracompetent cells (Stratagene). pGL4 Gclc construct mutants were characterized by restriction endonuclease digestion and sequencing.
2.7 Construction of synthetic ARE and AP1 luciferase reporter vectors
The ARE- and AP1-luciferase reporter plasmids were generated using the pGL4-minimal promoter vector (Promega) containing a minimal TATA promoter upstream of the firefly luciferase gene. The sequences of the inserts used in the different plasmids are summarized in Table 3. Single-stranded oligonucleotides were first annealed to form double-stranded oligonucleotides and then ligated into the pGL4.23[minP] vector following the manufacturer’s instruction (Promega). Each vector was engineered by inserting 3 copies of each of the ARE elements present in the rat Gclc 5’-flanking region. The three different Gclc 25-bp ARE (3X)-driven luciferase reporter constructs [i.e., pGL4.23Gclc-3xARE1-Luc2 (ARE1), pGL4.23Gclc-3xARE2-Luc2 (ARE2), and pGL4.Gclc-3xARE3Luc2 (ARE3)] as well as the ARE mutant (pGL4.23Gclc-3xAREm-Luc2) and the embedded AP1 element in ARE3 (pGL4.23Gclc-3xembeddedAP1-Luc2) were made by insertion of the appropriately hybridized complementary oligonucleotides [75 bp] with 2-bp overhangs into the XhoI/HindIII restriction sites of the pGL4-minimal promoter reporter vector. TOP10 competent cells were transformed with the recombinant DNA after ligation for amplification. After the plasmids were generated, the DNA sequence of the inserts was verified.
Table 3.
Sequence of Inserts in the pGL4 Minimal Promoter Vector
| Plasmid | Insert (5’→3’) |
|---|---|
| pGL-3xARE1 | GATTTACAATGACTAGACACACGTAGATTTACAATGACTAGACACACGTAGATTTACAATGACTAGACACACGTA |
| pGL-3xARE2 | TCCTGACAGTGAGTTGACACTCTGGTCCTGACAGTGAGTTGACACTCTGGTCCTGACAGTGAGTTGACACTCTGG |
| pGL-3xARE3 | GAGTCACGGTGAGGCGGCACGGCGCGAGTCACGGTGAGGCGGCACGGCGCGAGTCACGGTGAGGCGGCACGGCGC |
| pGL-3x embedded AP1 | GTTGAGTCACGGTTGAGTCACGGTTGAGTCACG |
| pGL-3xARE3m | GAGTCACGGGTCGGCGTAACGGCGCGAGTCACGGGTCGGCGTA ACGGCGCGAGTCACGGGTCGGCGTAACGGCGC |
The underlined letters represent those nucleotides that form the minimal functional ARE or AP1 element. The mutated ARE3 core sequence is in bold.
2.8 Cell culture, transfections and luciferase assays
Rat hepatoma-derived H4IIE cells, obtained from American Type Culture Collection (Rockville, MD) were grown in MEM supplemented with 10% FBS, 2 Mm glutamine, 100 U/ml penicillin and 100 U/ml streptomycin and incubated at 37 °C in a 5% CO2 atmosphere. Reporter gene assays were used to determine effects of different ARE and AP1 elements on transcriptional activity of the rat Gclc gene. The pGL4 minimal reporter vector contains a genetically engineered firefly luciferase gene containing a minimal promoter. When promoter regions are cloned into the pGL4 vector upstream of the luciferase gene, there is strong transcriptional activation and expression of luciferase. Mutations in the individual promoter elements can influence transcriptional activity of the luciferase reporter gene that is detected fluorometrically vs. an internal control. Transient transfections were done in cells grown to ~50% confluence using Effectene Transfection reagent (Qiagen, Valencia, CA). The cells were transfected with 0.8 µg of Gclc-Luciferase plasmids. Briefly, the DNA and 4 µl of Enhancer were dissolved in EC buffer from the kit to a total volume of 100 µl. The DNA–enhancer mixture was incubated at room temperature for 5 min. After incubation, 5 µl of Effectene transfection reagent was added to the mixture, mixed, and incubated at room temperature for 10 min to allow transfection–complex formation. Media (200 µl) was added to the mixture and mixed. The solution was then immediately added to the well containing the cells and 1.5 ml of fresh medium. The total amount of plasmid DNA for transfection was adjusted by using empty expression vector (pGL4.23). The control plasmid phRL-CMV encoding Renilla luciferase (0.02 µg) was included in each assay to account for variability in transfection efficiency. Thirty-six hours after transfection, cells were harvested with passive lysis buffer (Promega), and the supernatant was collected by brief centrifugation. Transcriptional activity was determined by the expression of firefly luciferase and was normalized to the renilla luciferase levels by using a Dual Luciferase Reporter Assay Kit™ (Promega) on a Biolumat LB9505 luminometer (Berthold Detection Systems, Pfhorzeim, Germany). The means of at least three independent experiments, each carried out in duplicate, are shown and expressed as the mean ± SEM.
Because the vector construct containing the full length Gclc promoter region was large, transfection protocols were modified vs. the smaller constructs to maximize transfection efficiency. To this end, the following modifications were performed: the transfection reagent used was JetPEI Hepatocyte (Polyplus-Transfection Inc., New York, NY) where the transfection reagent (3.2 µg) was added to 1 µg of the pGL4 plasmid and 0.02 µg the Renilla Luciferase vector in accordance with protocols supplied by the manufacturer. Times of cell harvest following transfection were as indicated above.
2.9 Treatments with ARE inducers
H4IIE cells transfected with luciferase constructs containing empty vector or different ARE elements were treated with LA or ADT to assess Gclc promoter activity. Briefly, a 100 mM LA or ADT stock was prepared in 100% dimethylformamide. Cells were treated with vehicle (0.1% DMF), 50 or 100 µM LA or 30 µM ADT for 24 hours after transfection. Transcriptional activity was measured as outlined in Section 2.5.
2.10 Nrf2 siRNA knockdown
Nrf2 siRNA (50 pM) or scrambled control (50 pM) was transfected to H4IIE cells by Effectene™ (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Briefly, cells (400,000) were seeded in six-well plates and incubated overnight, then transfected with 50 pM siRNA for 12 h using 10 µl Effectene per well. The cells were cultured for another 24 h, then used for experiments. There was almost no visible damage due to the transfection procedure.
2.11 RNA Isolation and Real-Time PCR
Briefly, total RNA was isolated using RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions, quantified spectrophotometrically (260 nm) and assayed for purity by determining the 260/280 ratio. Total RNA (0.5 mg) was reverse-transcribed with random primers and the resulting cDNA was amplified by real-time PCR using a MJ Research Opticon 2 machine (BioRad, Hercules, CA). To obtain appropriate template concentrations, samples were run concurrently with standard curves generated using plasmid standards. β-actin was used as a housekeeping control for RNA recovery and reverse transcription efficiency. Gene expression was normalized to β-actin mRNA levels and expressed as arbitrary units. The primers used were as follows: Nrf2-F; 5’-TCAGCTACTCCCAGGTTGCCCA-3’, Nrf2-R; 5’-GGCAAGCGACTCATGGTCATCTAC-3’, β-actin-F; 5’-CCTTCCTTCCTGGGTATGGAATCC-3’ Beta-actin-R; 5’-GAGCAAT GATCTTGATCTTCATGGTG-3’, where ‘F’ and ‘R’ denote forward and reverse primers, respectively.
2.12 Statistical Analysis
The data throughout are expressed as the mean ± SEM. Statistical analysis was performed using GraphPad Prism software version 3.03 (GraphPad Software Inc., San Diego, CA). We used a two-tailed Student’s t test to compare the luciferase activity of individual GCLC promoter constructs. A P value less than 0.05 was considered to be significant. One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc analysis was used when multiple comparisons were made to a control.
3. Results
3.1 Identification of a functional ARE in the Gclc promoter
We examined the rat Gclc promoter for potential antioxidant response elements (AREs) by a computer-based analysis. Using the TRANSFAC database and the ARE consensus sequence as a probe (12) revealed that there are three ARE motifs (designated ARE1-3) within 5 kb of the Gclc 5’-flanking region (Figure S1). When compared to the human Gclc promoter sequence, ARE3 is identical to the human Gclc ARE4 while the rat ARE1 and ARE2 contain a single mismatch vs. the human sequence (13, 37–39). Figure 1 represents a schematic orientation of the identified ARE sites in the 5’ flanking region of the rat Gclc promoter. The nucleotide sequence of each ARE is depicted in Table 2. Overall, the identity between the rat and human sequence suggests that the ARE3 site of the rat sequence might be important in Gclc transcription. However, sequencing of the F344 rat Gclc promoter region revealed a single nucleotide change from the rat 3.4 genomic sequence within the ARE3 locus. This mutation resulted in a change from ‘T’ to ‘C’ at position −3905 from the transcriptional start site. Interestingly, this T to C mutation produced an additional ARE element in the reverse orientation to ARE3.
Figure 1. Position of ARE promoters in the 5’-flanking region of rat Gclc.

Representation of ARE sequences in the 5’-flanking region of rat Gclc. Gray boxes denote potential ARE sites. The numbers shown represent DNA bases in the 5’-promoter, as counted from the translational start site.
Table 2.
Putative ARE Elements
| Gclc 5’-Element | Sequence |
|---|---|
| ARE1 | 5’-GATCTTATTGGGAAATGGGATTTACAATGACTAGACACACGTATGTCTAATCT-3’ |
| ARE2 | 5’-GTGCAAATCTGTTTCCTCCTGACAGTGAGTTGACACTCTGGAGACTGCTTCAT-3’ |
| ARE3 | 5’-CGGCCAGCCCGCGCAGAGTGTTGAGTCACGGTGAGGCGGCACGGCGCGCCGGG-3’ |
Bases underlined represent the minimal core ARE sequence
3.2 Nrf2 binds to Gclc ARE3 in an endogenous chromatin configuration
Since Nrf2 is the most potent transcriptional activator among CNC proteins (40–43), Nrf2 may enhance cytoprotective gene expression even if the other transcription factors are present and occupy the ARE. To examine whether the three identified AREs in the 5’-flanking region of Gclc has the ability to bind Nrf2 in vivo, we determined the extent of Nrf2 associated with individual ARE elements in their native chromatin environment by using the chromatin immunoprecipitation assay (ChIP). To control for possible nonspecific interactions and DNA contamination, samples precipitated with rabbit immunoglobulin G were included. Primary rat hepatocytes were cross-linked with formaldehyde, and chromatin was immunoprecipitated using an anti-Nrf2 antibody. PCR analysis revealed Nrf2 binding only to the region between 3.2 and 4 kb upstream of the transcriptional start site (Figure 1 and Figure 2), which harbors the functional ARE3. The recruitment of Nrf2 to these ARE sites was specific because no signal was detected in the immunoglobulin G control samples (data not shown). No recruitment of Nrf2 to the nonspecific intervening region located between ARE1 and ARE2 was observed. The results obtained from DNA that was PCR amplified from chromatin extracts before immunoprecipitation (input) are shown for comparison (Figure 2). This suggests that Nrf2 binds only to ARE3 out of the three putative AREs identified in the 5’-promoter of rat Gclc.
Figure 2. Nrf2 binds only to ARE3 in vivo.
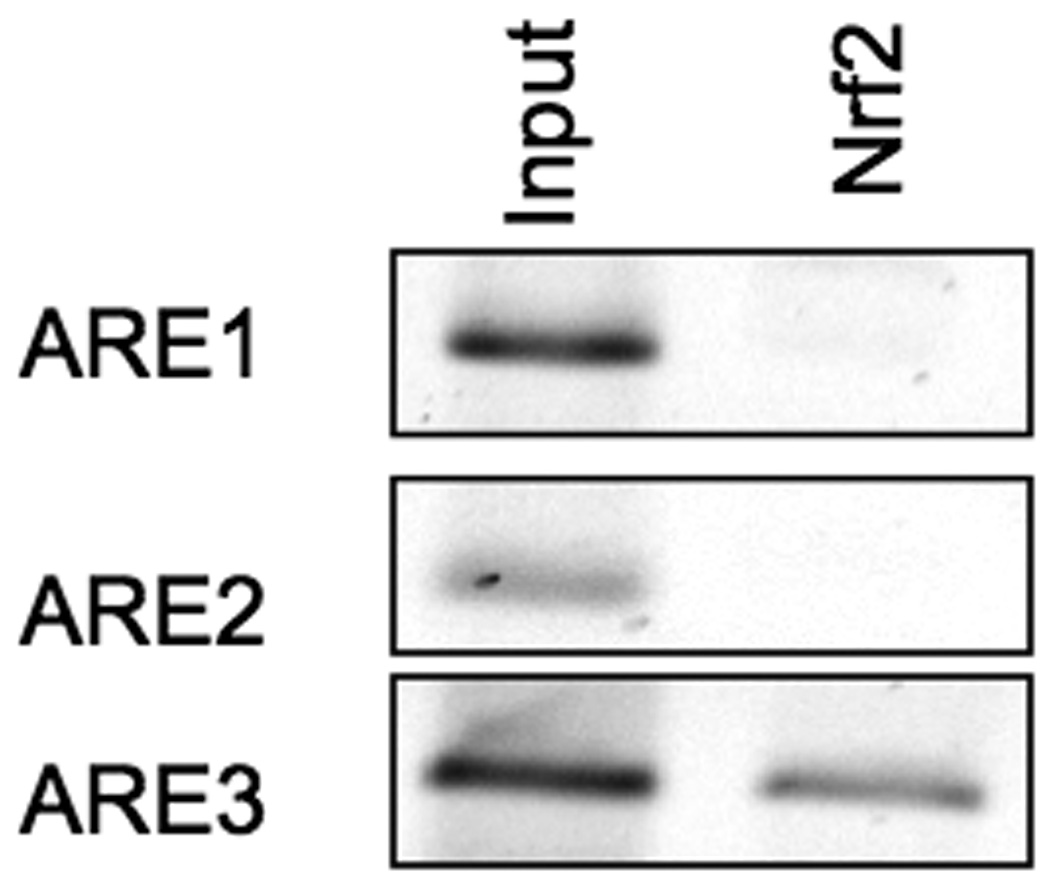
Primary rat hepatocytes were cross-linked with formaldehyde for 10 minutes, and ChIP analysis was performed using antibodies against Nrf2. Normal rabbit IgG was used as a control. Results show Nrf2 only binds to Gclc ARE3 efficiently (N=3).
3.3 ARE3 regulates the constitutive expression of rat Gclc
To characterize the contribution of each ARE to the basal transcriptional regulation of Gclc, ARE constructs were created by synthesizing three tandem copies of each ARE element and subcloning them into the pGL4 minimal promoter vector. Following transient transfection into H4IIE cells, each ARE was then analyzed for luciferase activity (Figure 3). The results revealed that only ARE3 at nucleotide position - 3916 was capable of mediating expression over that seen in the empty vector (0.014 ± 0.0012 Relative Luc Activity [RLA] for ARE3 versus 0.0054 ± 0.0014 RLA for empty vector). All other AREs failed to significantly instigate basal expression. Furthermore, an ARE3 mutant in which the core ARE sequence was altered, failed to increase luciferase activity over that seen in the empty vector (Figure 3).
Figure 3. ARE3 regulates constitutive expression of rat Gclc.

H4IIE cells were transiently transfected with 0.8 µg of either of the Gclc-ARE elements or the ARE3 mutant (ARE3m) cloned into the pGL4 luciferase reporter with a minimal promoter together with a CMV-Renilla luciferase transcription control plasmid. Luciferase activity was determined 36 hr after transfection. Results are representative of 3 independent experiments and presented as relative luciferase activities normalized to the Renilla luciferase internal control. ‘a’ denotes statistically significant values compared with empty vector (p≤ 0.001), while ‘b’ denotes statistically significant values compared with ARE3 (p ≤ 0.05).
To corroborate the data presented in Figure 3, a full-length construct of the Gclc 5’-flanking region was made and transfected into H4IIE cells. Despite its size, transfection of this vector (FL) was sufficient to drive robust luciferase activity vs. that observed for the pGL4.10 empty vector (Figure 4). However, a mutated construct (FL ARE3m) designed to abrogate Nrf2 binding only to the ARE3 locus had only minimal activity (−94.4%) relative to the pGL4.10FL native vector. Thus, our results validate the data we obtained from the ChIP assays (Figure 2) showing that ARE3 was the only functional element among the three AREs in the rat Gclc gene.
Figure 4. An intact ARE3 sequence in the 5’ flanking region is necessary for constitutive expression of Gclc.

H4IIE cells were transiently transfected with 1 µg each of the Gclc full length promoter region (FL) or the full length promoter with a point mutation in the sequence of ARE3 (ARE3m) cloned into the pGL4.10 luciferase reporter together with a CMV-Renilla luciferase transcription control plasmid (0.02 µg). The empty vector (pGL4.10) was transfected as a control to check for non-specific transcriptional activity. Luciferase activity was determined in the cytosolic fraction 36 hr after transfection. Results are representative of 5 independent experiments and presented as relative luciferase activities normalized to the Renilla luciferase internal control. ‘a’ denotes statistically significant values compared with empty vector (p≤ 0.001), while ‘b’ denotes statistically significant values compared with the FL vector (p ≤ 0.001).
Based on sequence alignment, the rat Gclc ARE3 is identical with the previously reported minimal “Class III” ARE enhancer sequences: (a/g)TGA(C/T/G)nnnGC(a/g) (1). In contrast, the ARE1 and ARE2 sequences differ from the consensus sequence in that the 3’ GC bases are replaced by AC deoxyribonucleotides. Previous studies reported that the active ARE in the human Gclc (ARE4) also contained an adjacent TPA-responsive element (TRE) that was potentially involved in the induction of the gene (13, 37, 44). Since the ARE consensus sequence resembles the AP-1 binding site and the rat Gclc ARE3 has an embedded AP-1 site at its 5’-end, we transfected an AP1 luciferase construct into H4IIE cells. Results revealed that the AP1 component displays 52% of the basal transcriptional activity of the ARE3 site (data not shown).
3.4 Nrf2-dependent upregulation of Gclc ARE3 transcriptional activity
Nrf2 has been identified as a target transcription factor essential for ARE transactivation. To determine if induction of ARE3-luciferase activity is linked to activation of Nrf2, we examined the effect of constitutively over-expressing Nrf2 on ARE3 transcriptional activity. As detected by western blotting, Nrf2 nuclear levels were enhanced in Nrf2-overexpressing cells by 250% (data not shown). Transfection of an Nrf2 expression plasmid also led to robust induction (96.5% compared to baseline) of Gclc ARE3-driven luciferase activity (Figure 5). On the other hand, there was no induction of the embedded AP1 element over baseline, indicating that Nrf2 neither directly nor indirectly trans-activates Gclc transcription through the AP1 component of ARE3 (Figure S2). As a further control, an ARE3 mutant (GclcARE3 mut) was co-transfected with the Nrf2 expression plasmid in H4IIE cells. This ARE3 mutant abolished the Nrf2 response in reporter gene assays (Figure 5). These results indicate that the identified ARE3 is functional and mediates the activation of Gclc transcription by Nrf2.
Figure 5. Nrf2 activates rat Gclc gene promoter activity through ARE3.

H4IIE cells were transfected with 0.8 µg of either of the three Gclc ARE elements or the ARE mutant, 0.06 µg of pcDNA3.1-Nrf2 (Nrf2) expression plasmid along with 0.02 µg of pHRL-CMV Renilla luciferase plasmid as a control for transfection efficiency. Nrf2 response of each reporter construct is indicated by fold change of activity versus the activity of pcDNA3.1 control plasmid that was co-transfected with it. Nrf2 over expression failed to activate the ARE3 mutant construct. Mean values of relative luciferase activity from at least three independent experiments, each carried out in duplicate, are shown with the ± SEM as detailed in “Materials and Methods”. ‘a’, p ≤0.01 relative to ARE3 alone (Tukey’s post-hoc analysis). Mean difference of the Nrf2 response for the ARE3 mutant group is significant from control group at p ≤ 0.01.
3.5 Nrf2 inducers activate Gclc gene expression through ARE3
To further define whether pharmacological inducers of phase II genes worked through activating the Gclc promoter through the ARE3 locus, we utilized two known inducers of Nrf2, viz. LA and ADT. We transiently transfected H4IIE cells with luciferase reporter constructs containing three copies of each of the Gclc ARE elements and treated them with 100 µM LA or 30 µM ADT for 24 hours. Measurement of luciferase reporter activities comparing unstimulated transfected cells to those after LA (203 ± 18% over vehicle) or ADT (191 ± 17%) treatments show that the activity of only the ARE3 element increases under stimulation by these Nrf2 inducers (Figure 6).
Figure 6. Induction of Gclc by ARE inducers is controlled by ARE3.

H4IIE cells were transiently transfected with 0.8 µg of either of the Gclc-ARE elements or the ARE3 mutant (ARE3m) cloned into the pGL4 luciferase reporter with a minimal promoter together with a CMV-Renilla luciferase transcription control plasmid. Eighteen hours after transfection, cells were treated with either 0.01% DMF (vehicle control), 100 µM lipoic acid or 30 µM ADT. Luciferase activity was determined in the cytosolic fraction 24 hr after treatment. Luciferase activity of the vehicle control empty reporter construct was arbitrarily set as 100, and the mean values of relative luciferase activity from at least three independent experiments, are shown with the ± SEM. Results are representative of 3 independent experiments and presented as relative luciferase activities normalized to the Renilla luciferase internal control. ‘a’ denotes statistically significant values compared with empty vector (p≤ 0.001), while ‘b’ denotes statistically significant values compared with ARE3 (p ≤ 0.05).
In order to assess whether the above Nrf2 inducers also upregulated Gclc transcription in the context of the full Gclc 5’-flanking region, ADT and LA (see methods) were added to H4IIE cells containing the FL or the FL ARE3m mutated luciferase reporter constructs. Both ADT and LA supplementation resulted in a 50% and 59% increase in relative luciferase activity, respectively, versus vehicle-treated controls (Figure S4). As anticipated from our results in Figure 4, almost no basal activity was observed using the ARE3 mutant vector. Even though LA and ADT were both able to induce an observable increase in promoter activity in the mutant vector, the level of induction was miniscule when compared to the activities observed in the full-length construct (Figure S4). Thus, using the full length sequence of the Gclc promoter region corroborates our results indicating that ARE3 not only drives basal promoter activity, but also largely controls inducible activity for this gene when known phase II inducing compounds are used as stimulants.
3.6 Effect of Nrf2 siRNA on ARE3-mediated transcription of GCLC
To further confirm the role of Nrf2/ARE in GCLC gene expression, we used small interfering RNAs (siRNAs) to inhibit the endogenous expression of Nrf2 in H4IIE cells. The transfection of H4IIE cells with a siRNA specific for Nrf2 reduced Nrf2 message expression by about 30% (Figure S3A). By contrast, the scrambled siRNA did not significantly affect expression of Nrf2 message or its protein levels as compared to endogenous levels in non-transfected cells (Figure S3A and data not shown). Nrf2 siRNA was also co-transfected with ARE3 to determine its effect on the ARE promoter activity. Briefly, H4IIE cells were first transfected with Nrf2 siRNA. Four hours after the siRNA transfection, the minimal promoter vector or ARE3 promoter-luciferase plasmids were transfected. Luciferase assays were performed 36 h after the second transfection. Cotransfecting the ARE–luciferase construct enabled the measurement of residual transcriptional ARE activity in Nrf2-depleted cells. The silencing of Nrf2 led to a significant abrogation in ARE3-driven luciferase activity (Fig. S3B). These data reveal that Nrf2 is directly involved in ARE3-mediated basal transcription of the rat Gclc gene.
Taken together, these studies indicate that the rat GCLC promoter contains one functional ARE that is responsible for Nrf2-dependent basal transcription.
4. Discussion
GSH is the most abundant non-protein thiol in cells and it plays key roles in multiple biological functions, including conjugation and detoxification of xenobiotics, scavenging free radicals, and maintenance of normal cellular redox status (45). It is well known that GSH levels are maintained via de novo synthesis through GCL gene expression (8, 46, 47). The current work demonstrates that constitutive GCLC gene expression in the rat liver is regulated through the ARE (ARE3). We also present evidence that rat Gclc is under the direct transcriptional control of Nrf2 at least on a basal level, and potentially, by induction of Phase II enzyme inducers These conclusions are supported by the following observations: (a) computer-based searches of the Gclc 5’-flanking region show three putative ARE elements exist in this gene; (b) transfection of H4IIE cells with Gclc ARE and AP1 promoter constructs indicate only one of the AREs (ARE3) and its embedded TRE element have transcriptional activity; (c) chromatin immunoprecipitation assays show that Nrf2 binds only to ARE3 in the Gclc 5’-flanking region, (d) site-directed mutation of the ARE3 element abolishes luciferase activity, and (e) ADT and LA stimulate Gclc promoter activity solely through the ARE3 locus. Collectively, the results of the present study establish that the transcriptional regulation of rat Gclc is governed by the Nrf2-ARE regulon.
The data presented in this study are in keeping with other work in humans and mice, which show that regulation of Gclc expression is mediated in an Nrf2/ARE-dependent manner. However, a direct link for the role of Nrf2-ARE binding in basal Gclc gene regulation has never been established in rats. For this species, deciphering the cis-regulatory element responsible for the basal transcriptional regulation of Gclc, has been controversial. Lu and co-workers reported that basal as well as inducible transcription of rat Gclc is regulated by an AP1 element located 450 bp upstream from the transcriptional start site (15). This study also did not identify any ARE in the rat Gclc promoter region. Subsequent work, including one from the same group, reported that the Nrf2 is essential for rat Gclc transcription (27). In addition, it was reported that Nrf2 regulated the transcription of rat Gclc indirectly through the AP1 site (27). However, there are two significant points that must be considered which may help in resolving the results of these previous studies with our findings. First, only the first 2 kb upstream of the transcriptional start site for Gclc was previously analyzed, so the potential role of distal elements was not assessed (15, 27, 30). In fact, the active ARE3 site lies outside this 2 kb region and therefore, has not been previously investigated. Secondly, the rat Gclc promoter lacking the distal ARE elements (i.e. ARE3) was analyzed in fibroblasts of Nrf2 null mice, hence an accurate mechanism was not obtained (27). Nevertheless, the mechanism of transcriptional regulation of rat Gclc has been an unsolved problem.
During the preparation of this manuscript, but after the publication of a thesis on which it is based (http://en.scientificcommons.org/22769607 issue date June 29, 2007), another publication ahead of print also independently corroborated the importance of ARE3 in regulating the basal as well as the electrophile-mediated transcription of Gclc. However, it is highly surprising that this study did not identify the two AREs downstream of ARE3 and more proximal to the transcriptional start site of Gclc. In fact, the core sequences of ARE1 & 2 (located −891 and −3172 kb upstream of the transcriptional start site) confirm with the classical ARE core promoter. Our data show that Nrf2 does not bind to either ARE1 or 2 (Figure 2) and furthermore, these two response elements repress the transcriptional activity of the pGL4 minimal promoter. It is tempting to speculate that a mixture of repressive bZip heterodimers (small maf proteins, Bach1 & 2, Fra1) in the absence of Nrf2 might contribute to the negative transcriptional regulation through these elements.
In previous reports investigating transcriptional regulation of rat Gclc, DNA binding assays were performed to assess transcription factors bound to the consensus sequences because the complete sequence of the promoter had not been cloned. However, the DNA binding assay is limited by its inability to disclose the native state of competition between two reactive elements at one time. In this report, we obtained more direct evidence using chromatin immunoprecipitation, which involves immunoprecipitating chromatin bound by Nrf2 and amplifying the TRE- or ARE-containing regions individually by PCR. ChIP assays conclusively showed that only ARE3 (located between −3916 and −3877 bases from the transcriptional start site) displayed Nrf2 binding in the in vivo state.
A significant finding of the present work is that constitutive transcription of rat Gclc results directly from binding of Nrf2-ARE3 binding. The association of Nrf2 with rat ARE3 is consistent with Nrf2 regulation of GCLC in human cell lines (18). The distal ARE/EpRE sites located in the human Gclc promoter region, between −3802 and −2752 bp, are required both for constitutive and induced expression of the human GCLC gene (13). Recently, it was reported that like other AREs, the most distal ARE in the human Gclc promoter region consists of a consensus ARE sequence containing an embedded TRE element (38). The presence of a 5’ AP1-like element is known to enhance NQO1 gene ARE-mediated gene expression (20, 48). However, the function of these regulatory sequences and the interaction between them has not been fully elucidated. In this context, it is noteworthy that the rat Gclc ARE3 responsive to Nrf2 contains a functional binding site for the AP1 family of transcription factors similar to the functional Gclc ARE in humans. The 5’-TGAC-3’ tetranucleotide within the ARE3 core sequence resembles the half-site recognized by members of the AP-1 family (consensus: 5’-TGACTCA-3’). This also matches with results from Yang and co-workers where blocking AP-1 with dominant negative c-Jun lowered the basal expression of Gclc (26). Thus, our results thus provide a molecular mechanism for the basal transcriptional regulation of rat Gclc and provide the first direct evidence that Nrf2 targets rat Gclc as part of maintaining constitutive glutathione synthesis.
The ARE sequence regulates the constitutive as well as inducible transcription of Gclc in the human as well as the mouse. Therefore, we asked whether LA and ADT, two thiol-containing phase II inducers regulated rat Gclc transcription strictly through the ARE3. Data presented in Figure 6 show that both these inducers increase luciferase activity by driving transcription exclusively through the ARE3 element and not ARE1 or ARE2.
GSH levels can be severely limiting in cases of acute xenobiotic challenges. So, far strategies to increase GSH have focused on boosting its rate-limiting substrate cysteine (administration of N-acetylcysteine) rather than increasing GCL levels. The evidence provided in this study opens the potential for augmenting GSH levels through increased transcription of its synthetic enzymes. This will be beneficial for toxicological studies in the rat model.
4.1 Conclusion
In summary, we found that basal and inducible transcription of rat Gclc occurs via the Nrf2/ARE3 complex since mutation of a previously uncharacterized ARE abrogates Gclc promoter activity. Moreover, ChIP assays detected appreciable Nrf2 binding to ARE3. Finally, a role for Nrf2 in the induction of Gclc promoter activity is also demonstrated by the ability of Nrf2 siRNA to abolish the activation of Gclc promoter activity via ARE3.
Supplementary Material
Putative ARE sites in the promoter region of rat Gclc
Sequence of the 5’-flanking region of Gclc, which is conserved between human, mouse and rat. The putative ARE elements are conserved between the three species are underlined.
{kind=link}
Nrf2 controls Gclc transcription through the ARE site but not the embedded AP1 component of the ARE3 element
H4IIE cells were transfected with 0.8 µg of the Gclc ARE3 element or the AP1 component only of the ARE3 element, 0.06 µg of pcDNA3.1-Nrf2 (Nrf2) expression plasmid along with 0.02 µg of pHRL-CMV Renilla luciferase plasmid as a control for transfection efficiency. Nrf2 response of each reporter construct is indicated by fold change of activity versus the activity of pHRL-CMV control plasmid that was co-transfected with it. Nrf2 over expression failed to activate the construct containing only the embedded AP1 component of the ARE3 element. The mean values of relative luciferase activity from at least three independent experiments was calculated. ‘a’, p ≤0.01 relative to ARE3 alone.
{kind=link}
Effects of Nrf2 siRNA on ARE3-dependent Gclc transcription in H4IIE cells.
H4IIE cells were transfected with 50 pM control siRNA or 50 pM Nrf2 siRNA as described under Materials and Methods, and then transfected with pARE3-rGclc-Luc2. (A) 24 hours after siRNA transfection, Nrf2 RNA levels were detected by RT-PCR. (B) Nrf2 knockdown inhibits luciferase activity of ARE3, but not the empty vector. a, p ≤ 0.05 versus ARE3 control. Luciferase activities after treatment with control siRNA is shown as a negative control. Results are shown as mean ± S.E.M. of three independent experiments.
{kind=link}
Nrf2 inducers only activate an intact ARE site present in the luciferase construct containing the full length 5’ flanking region of the Gclc gene
H4IIE cells were transiently transfected with 1 µg each of the Gclc full length promoter region (FL) or the full-length promoter with alterations in the sequence of ARE3 (FL ARE3m) together with a CMV-Renilla (0.02 µg). The pGL4 basic vector was transfected as a control to check for non-specific transcriptional activity. Twenty hours after transfection, cells were treated with either 0.001% DMF (vehicle control), 50 µM LA or 30 µM ADT. Luciferase activity was determined 16 hr after treatment. Results are representative of 4 independent experiments and presented as relative luciferase activities normalized to the Renilla luciferase internal control. ‘a’ denotes statistically significant values compared with vehicle treatment of the FL promoter (p≤ 0.001).
{kind=link}
{kind=link}
Acknowledgements
We would like to thank Dr. Chrissa Kioussi for guidance in performing ChIP assays and to Ms. Judy Butler technical assistance. We would also like to thank Drs. Brian M. Dixon and Alexander Michels for designing Nrf2 primers for qPCR and luciferase assays, respectively. This work was supported by a grant from the NIH (NCCAM P01AT002034-06) and a graduate student fellowship (SS) from the Linus Pauling Institute, OSU. Support facilities were also from the Environmental Health Sciences Center, OSU (NIEHS ES00240).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 2.Ghezzi P, Bonetto V, Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 3.Meisterand A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 4.Schaferand FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 5.Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 6.Dickinsonand DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 7.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. Faseb J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 8.Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys. 2004;423:116–125. doi: 10.1016/j.abb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Huang CS, Anderson ME, Meister A. Amino acid sequence and function of the light subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993;268:20578–20583. [PubMed] [Google Scholar]
- 10.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- 11.Meister A. The fall and rise of cellular glutathione levels: enzyme-based approaches. Curr Top Cell Regul. 1985;26:383–394. doi: 10.1016/b978-0-12-152826-3.50036-x. [DOI] [PubMed] [Google Scholar]
- 12.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272:7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 14.Bea F, Hudson FN, Chait A, Kavanagh TJ, Rosenfeld ME. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ Res. 2003;92:386–393. doi: 10.1161/01.RES.0000059561.65545.16. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Wang J, Huang ZZ, Ou X, Lu SC. Cloning and characterization of the 5'-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wildand AC, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: insights into transcriptional control of antioxidant defenses. Free Radic Res. 2000;32:281–301. doi: 10.1080/10715760000300291. [DOI] [PubMed] [Google Scholar]
- 17.Moinovaand HR, Mulcahy RT. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun. 1999;261:661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 18.Wild AC, Moinova HR, Mulcahy RT. Regulation of gammaglutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 19.Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 21.Chanand JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 22.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15:4184–4193. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, Takagi Y, Yamamoto M. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells. 2002;7:807–820. doi: 10.1046/j.1365-2443.2002.00561.x. [DOI] [PubMed] [Google Scholar]
- 25.Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25:10895–10906. doi: 10.1128/MCB.25.24.10895-10906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Magilnick N, Ou X, Lu SC. Tumour necrosis factor alpha induces co-ordinated activation of rat GSH synthetic enzymes via nuclear factor kappaB and activator protein-1. Biochem J. 2005;391:399–408. doi: 10.1042/BJ20050795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Magilnick N, Lee C, Kalmaz D, Ou X, Chan JY, Lu SC. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee TD, Yang H, Whang J, Lu SC. Cloning and characterization of the human glutathione synthetase 5'-flanking region. Biochem J. 2005;390:521–528. doi: 10.1042/BJ20050439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu SC, Huang ZZ, Yang H, Tsukamoto H. Effect of thioacetamide on the hepatic expression of gamma-glutamylcysteine synthetase subunits in the Rat. Toxicol Appl Pharmacol. 1999;159:161–168. doi: 10.1006/taap.1999.8729. [DOI] [PubMed] [Google Scholar]
- 30.Yang H, Zeng Y, Lee TD, Yang Y, Ou X, Chen L, Haque M, Rippe R, Lu SC. Role of AP-1 in the coordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J Biol Chem. 2002;277:35232–35239. doi: 10.1074/jbc.M203812200. [DOI] [PubMed] [Google Scholar]
- 31.Lu SC, Huang ZZ, Yang JM, Tsukamoto H. Effect of ethanol and high-fat feeding on hepatic gamma-glutamylcysteine synthetase subunit expression in the rat. Hepatology. 1999;30:209–214. doi: 10.1002/hep.510300134. [DOI] [PubMed] [Google Scholar]
- 32.Jeyapauland J, Jaiswal AK. Nrf2 and c-Jun regulation of antioxidant response element (ARE)-mediated expression and induction of gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Pharmacol. 2000;59:1433–1439. doi: 10.1016/s0006-2952(00)00256-2. [DOI] [PubMed] [Google Scholar]
- 33.Soltaninassab SR, Sekhar KR, Meredith MJ, Freeman ML. Multi-faceted regulation of gamma-glutamylcysteine synthetase. J Cell Physiol. 2000;182:163–170. doi: 10.1002/(SICI)1097-4652(200002)182:2<163::AID-JCP4>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 34.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhakshinamoorthy S, Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 36.Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulcahyand RT, Gipp JJ. Identification of a putative antioxidant response element in the 5'-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Biophys Res Commun. 1995;209:227–233. doi: 10.1006/bbrc.1995.1493. [DOI] [PubMed] [Google Scholar]
- 38.Wild AC, Gipp JJ, Mulcahy T. Overlapping antioxidant response element and PMA response element sequences mediate basal and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase catalytic subunit gene. Biochem J. 1998;332(Pt 2):373–381. doi: 10.1042/bj3320373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Go YM, Gipp JJ, Mulcahy RT, Jones DP. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J Biol Chem. 2004;279:5837–5845. doi: 10.1074/jbc.M307547200. [DOI] [PubMed] [Google Scholar]
- 40.Toki T, Itoh J, Kitazawa J, Arai K, Hatakeyama K, Akasaka J, Igarashi K, Nomura N, Yokoyama M, Yamamoto M, Ito E. Human small Maf proteins form heterodimers with CNC family transcription factors and recognize the NFE2 motif. Oncogene. 1997;14:1901–1910. doi: 10.1038/sj.onc.1201024. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, Hayashi N, Yamamoto M. Molecular cloning and functional characterization of a new Cap'n' collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443–6452. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 42.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–868. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–286. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 44.Moinovaand HR, Mulcahy RT. An electrophile responsive element (EpRE) regulates beta-naphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem. 1998;273:14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- 45.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Liu H, Wang H, Shenvi S, Hagen TM, Liu RM. Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci. 2004;1019:346–349. doi: 10.1196/annals.1297.059. [DOI] [PubMed] [Google Scholar]
- 47.Rahman I. Regulation of glutathione in inflammation and chronic lung diseases. Mutat Res. 2005;579:58–80. doi: 10.1016/j.mrfmmm.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 48.Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2001;20:3906–3917. doi: 10.1038/sj.onc.1204506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Putative ARE sites in the promoter region of rat Gclc
Sequence of the 5’-flanking region of Gclc, which is conserved between human, mouse and rat. The putative ARE elements are conserved between the three species are underlined.
Nrf2 controls Gclc transcription through the ARE site but not the embedded AP1 component of the ARE3 element
H4IIE cells were transfected with 0.8 µg of the Gclc ARE3 element or the AP1 component only of the ARE3 element, 0.06 µg of pcDNA3.1-Nrf2 (Nrf2) expression plasmid along with 0.02 µg of pHRL-CMV Renilla luciferase plasmid as a control for transfection efficiency. Nrf2 response of each reporter construct is indicated by fold change of activity versus the activity of pHRL-CMV control plasmid that was co-transfected with it. Nrf2 over expression failed to activate the construct containing only the embedded AP1 component of the ARE3 element. The mean values of relative luciferase activity from at least three independent experiments was calculated. ‘a’, p ≤0.01 relative to ARE3 alone.
Effects of Nrf2 siRNA on ARE3-dependent Gclc transcription in H4IIE cells.
H4IIE cells were transfected with 50 pM control siRNA or 50 pM Nrf2 siRNA as described under Materials and Methods, and then transfected with pARE3-rGclc-Luc2. (A) 24 hours after siRNA transfection, Nrf2 RNA levels were detected by RT-PCR. (B) Nrf2 knockdown inhibits luciferase activity of ARE3, but not the empty vector. a, p ≤ 0.05 versus ARE3 control. Luciferase activities after treatment with control siRNA is shown as a negative control. Results are shown as mean ± S.E.M. of three independent experiments.
Nrf2 inducers only activate an intact ARE site present in the luciferase construct containing the full length 5’ flanking region of the Gclc gene
H4IIE cells were transiently transfected with 1 µg each of the Gclc full length promoter region (FL) or the full-length promoter with alterations in the sequence of ARE3 (FL ARE3m) together with a CMV-Renilla (0.02 µg). The pGL4 basic vector was transfected as a control to check for non-specific transcriptional activity. Twenty hours after transfection, cells were treated with either 0.001% DMF (vehicle control), 50 µM LA or 30 µM ADT. Luciferase activity was determined 16 hr after treatment. Results are representative of 4 independent experiments and presented as relative luciferase activities normalized to the Renilla luciferase internal control. ‘a’ denotes statistically significant values compared with vehicle treatment of the FL promoter (p≤ 0.001).