

Abstract
Obesity is associated with increased cancer incidence and mortality. We have previously found that obesity in children is associated with a 50% increased recurrence of acute lymphoblastic leukemia (ALL) in high-risk patients. We have therefore developed novel in vivo and in vitro preclinical models to study the mechanism(s) of this association. Obesity increased relapse after monotherapy with vincristine (p=0.03) in obese mice injected with syngeneic ALL cells. This occurred even though the drug was dosed proportionally to body weight, equalizing blood and tissue drug levels. In co-culture, 3T3-L1 adipocytes significantly impaired the anti-leukemia efficacy of vincristine, as well as 3 other chemotherapies (p<0.05). Interestingly, this protection was independent of cell-cell contact, and it extended to human leukemia cell lines as well. Adipocytes prevented chemotherapy-induced apoptosis, and this was associated with increased expression of the two pro-survival signals Bcl-2 and Pim-2. These findings highlight the role of the adipocyte in fostering leukemia chemotherapy resistance, and may help explain the increased leukemia relapse rate in obese children and adults. Given the growing prevalence of obesity worldwide, these effects are likely to have increasing importance to cancer treatment.
Keywords: lymphoblastic leukemia, obesity, tumor microenvironment, DIO mouse model, adipocytes
Introduction
Obesity is associated with an increased risk of numerous types of cancer in adults1–7. In addition, obese cancer patients have poorer outcomes than their leaner counterparts8–10. Calle et al. estimated, that given the high prevalence of obesity in the United States and the strength of its association with cancer, 14% of all cancer deaths in men and 20% in women are attributable to obesity11.
Leukemia is the most common childhood cancer, affecting ~2000 children per year in the United States12. We have previously shown that obese adults diagnosed with ALL have a 50% higher likelihood of relapse than lean adults13. Three recent studies have examined the relationships between obesity and ALL relapse in children. Two of these reported that obesity tended to increase14 or had no detectable effect on relapse risk15. However, these relatively small studies each had fewer than 25 obese subjects over 10. In a large cohort of 5420 children, including 262 obese subjects ≥ 10 years old, we found that obesity at the time of ALL diagnosis independently increased relapse rates by ~50% in children ≥ 10 years of age16.
The mechanisms underlying the association between obesity and ALL relapse are likely multifactorial. Adipocytes could alter chemotherapy pharmacokinetics and/or induce drug resistance by secretion of adipokines. Other growth factors secreted in the context of obesity could enhance leukemia cell survival. Adipose tissue stromal cells promote solid tumor growth, and thus might affect leukemia cells as well17. To investigate the roles of obesity in ALL relapse, we have established new in vivo and in vitro models. We focused our studies on vincristine, as it is used nearly universally as a first-line agent in the treatment of childhood leukemia, and vincristine resistance in vitro18 and in mouse xenografts19 is strongly prognostic of relapse. Here we report how adipocytes impair the leukemia response to vincristine in vivo and of several drugs in vitro.
Materials and Methods
Diet Induced Obesity (DIO) Model
Male C57Bl/6 mice (Jackson Laboratories, Bar Harbor, ME) were weaned onto a high-fat diet (60% of calories from fat, Research Diets D12492, New Brunswick, NJ) or a control diet (10% of calories from fat, D12450) until transplantations at ~20 weeks of age. All experiments were approved by the CHLA Institutional Animal Care and Use Committee and performed in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Cell Lines
Murine pre-B cell ALL was previously isolated from a BCR/ABL transgenic mouse (“8093 cells” 20, 21). A subset of these was transduced on retronectin-coated plates with a retroviral vector containing pMIG_GFP (provided by M. Muschen). Human leukemia cell lines included RCH-ACV (pre-B ALL with an E2A-PBX1 fusion protein22), BV173 (Pre B Ph+ ALL23), SD-1 (pre-B Ph+ ALL, DSMZ, Braunschweig, Germany) and RS4;11 (pre-B t(4;11) ALL, ATCC, Manassas, VA).
Murine fibroblasts and adipocytes were derived from 3T3-L1 cells (ATCC). 3T3-L1 cells were differentiated into adipocytes based on optimization of a method previously described24. Briefly, cells were grown to confluence in DMEM (Invitrogen) with 10% FBS, Glutamax, sodium pyruvate, and antibiotics in poly-D-lysine coated wells (day −2). On day 0, the media were changed to DMEM with supplements plus 15% fetal bovine serum, 20 mM HEPES, 150 nM insulin, 250 nM dexamethasone, and 0.5 mM isobutylmethylxanthine (IBMX, Sigma, St. Louis, MO). On day +2, dexamethasone and IBMX were removed from the media, and on day +4 insulin was removed. Media changes were performed every two days until use. Adipocytes were used for co-culture experiments between days +7 and +14. Undifferentiated 3T3-L1 fibroblasts were irradiated and then plated at confluence. Control experiments were also performed with non-irradiated 3T3-L1 cells at confluence. In different experiments, we used adipocytes differentiated from OP-9 murine bone marrow mesenchymal cells (ATCC) as described above25, and undifferentiated (non-irradiated) OP-9 cells as control.
In vivo Leukemia Model
To test whether obesity would impact proliferation of leukemia in vivo, we injected 5,000 8093 cells into mice via a retro-orbital route. Animals were euthanized upon development of progressive leukemia (weight loss >10%, paralysis, hunched posture, or visible masses > 1cm). Leukemia was verified by necropsy and/or by DNA qPCR for BCR/ABL in peripheral blood, using DNeasy kits (Qiagen, Valencia, CA) and Power SYBR Green master mix on an ABI Prism 7722 Sequence Detector (Applied Biosystems, Foster City, CA).
To examine the effects of DIO on leukemia treatment, we injected 10,000 8093 cells into 12 DIO, 12 control, and 7 vehicle mice as above. Eight days after the injection, mice were treated with vincristine in proportion to body weight (0.5 mg/kg/week via intraperitoneal injection × 4 weeks, Vincasar, Teva Pharmaceuticals, North Wales, PA)26.
Five additional DIO mice were transplanted with GFP+ 8093 cells and treated for 3–4 weeks with vincristine after an 8–10 day engraftment period. Mice were perfused with paraformaldehyde (PFA, n=2) or PBS (n=3) at time of leukemia relapse and sacrifice. Fat pads from the PFA perfused mice were removed, cut into small (~1 mm) pieces, and examined en bloc for the presence of GFP+ leukemia cells. Fat pads from the PBS perfused mice were fixed in PFA, frozen in OCT, and sliced to 10 μm thickness (at −20°C). Additional fat pads were digested with Liberase TM (Roche) at 2U/mL for 30 minutes, spun at 350g × 5 minutes, washed, and pelleted cells were cultured for 3–5 days to test for viability and vincristine sensitivity.
Co-culture Experiments
Leukemia cells were seeded into 24 well plates with fibroblasts, adipocytes, or no feeder layer. In experiments of drug resistance, 5 nM vincristine, 20 nM nilotinib, 35 nM daunorubicin, or 25 nM dexamethasone was added (all at the IC90 in our culture system without feeder layers). After 72 hours, the wells were triturated forcefully to remove cells within and below the feeder layers, and counted by a blinded observer by trypan blue exclusion.
The importance of cell-cell contact was assessed with 8093 leukemia cells in the upper chambers of polycarbonate 0.4 μm pore size TransWells (Corning, Inc., Lowell, MA). TransWell experiments were repeated with BV-173, SD1, RS4;11, and RCH ACV human leukemia cell lines. To assess the importance of adipocyte viability, feeder layers were fixed with PFA prior to being used in TransWells. Layers were exposed to 4% PFA for 1–12 hours, and then rinsed 3 times with PBS followed by RPMI media overnight. All experiments were done in triplicate and at least 3 times unless otherwise noted.
Cell Cycle and RNA Expression Studies
8093 cells cultured in TransWells as above were analyzed for cell cycle and apoptosis by BrdU incorporation (BrdU flow kit; BD Biosciences, San Jose, CA), and cells were analyzed on a FACScan (BD Biosciences, CellQuest software). Lymphocytes were defined based on forward and side scatter, and the percentage of this gated population in each cell cycle phase was determined. To assess gene expression, cells from a single TransWell experiment performed in triplicate were harvested and resuspended in RNAProtect (Qiagen, Valencia, CA), and RNA extracted and purified with RNEasy Mini Kits (Qiagen). RNA was reverse transcribed to cDNA with High Capacity 1st Strand Synthesis Kit (Applied Biosystems). cDNA was combined with a SYBR Green master mix and applied to an Apoptosis PCR Array RT2 Profiler (SABiosciences, Frederick, MD). The cycling program was based on the manufacturer’s instructions.
The expression of selected genes was confirmed with rtPCR on five biological replicates, each in triplicate, using 25 ng of cDNA, Power SYBR Green PCR Master Mix (Applied Biosystems) and 200 nM primers generated using NCBI Primer-BLAST. Murine β-actin was amplified using MBACTU (5′-TCATGAAGTGTGACGTTGACATCCGT-3′) and MBACTD (5′-CCTAGAAGCATTTGCGGTGCACGATG-3′), which yielded a product of 285 bp. Murine Pim 2 was amplified using MPIM2U (5′-AGCACCTCCTCCATGTTGAC-3′) and MPIM2D (5′-GATGGCCACCTGACGTCTAT-3′), which yielded a product of 192 bp. Murine Bcl-2 was amplified using MBCL2U (5′-GCAGGTGCCTACAAGAAAGC-3′) and MBCL2D (5′-GCATTTTCCCACCACTGTCT-3′), which yielded a product of 162 bp. Gene expression levels were quantified using the ABI 7900HT Sequence Detection System with the following thermal profile: 10 minutes at 95.0 followed by 40 repeats of 95.0, 15 sec and 60.0, 1 minute, and a final dissociation stage of 95.0 for 15 sec, 60.0 for 15 sec, and 95.0 for 15 sec. Transcript levels were normalized to β-actin.
Western Blots
8093 cells were cultured in TransWells and collected 24 hours after exposure to no drug or 5 nM vincristine. Cells were washed in ice cold PBS and lysed in SSB buffer [62.5 mM Tris-HCl, 2% w/v SDS, 1% v/v Igepal CA-630 (Sigma), 10% glycerol, 0.01 mg/mL aprotinin, 1 mM phenylmethanesulphonylfluoride, and Phosphatase Inhibitor Cocktail Set II (Calbiochem)] by sonication. Protein concentration was measured via the BCA method (Pierce), bromophenol blue and NuPage Reducing Agent (Invitrogen) were added to the lysate, and the resulting mixture was heated. Equal amounts of total protein were run on a 12% SDS polyacrylamide gel and blotted onto a nitrocellulose membrane (Invitrogen). The membranes were blocked in TBST (1x TBS and 0.1% Tween-20) with 5% non-fat dry milk and incubated with the antibodies to Pim-2 (1:200, Santa Cruz Biotechnology, sc-28778), phospho-Bad (1:1000, Ser112, Cell Signaling Technology), Bad (1:1000, Cell Signaling Technology), and β-actin (1:2000, Cell Signaling Technology), and then washed and incubated with HRP-conjugated secondary antibodies. The membranes were developed using enhanced chemiluminescence (Pierce).
Calculations and Statistics
Body weights were compared with unpaired t-tests. Survival curves were generated by Kaplan Meier Life Tables. Due to the low number of animals in the transplantation experiments, p-values for the difference in survival were calculated from a permutation distribution of the log rank test with 10,000 replicates, using SAS Version 9.1 (SAS Institute Inc, Cary, NC). Each co-culture experiment was performed on different days or using different cell thaws, and the averages of three triplicate wells for each condition were calculated. Paired t-tests were used to compare number of viable leukemia cells over the various feeder layers. One-sided p-values were used to test the a priori hypotheses that obese animals would have increased relapse rates compared to lean animals, and adipocytes would protect leukemia cells from chemotherapies.
Results
Modeling leukemia in obese mice
Male C57Bl/6 mice were made obese (DIO) by a high-fat diet27 and used as recipients of syngeneic 8093 BCR/ABL+ leukemia cells, to test for effects of obesity on leukemia development and treatment outcome. At the time of transplantation, the DIO mice were significantly heavier than control mice (39.5±4.7 vs. 30.6±4.9 g, p<0.0001). In our initial experiment, 16 DIO and 16 control mice were transplanted with 5,000 leukemia cells and observed until progressive leukemia developed (weight loss >10%, paralysis, hunched posture, or visible masses > 1cm); we found that obesity did not affect the time to development of progressive leukemia (21.5 vs. 22.0 days, p=0.22, Figure 1A). Next, animals were transplanted with 10,000 leukemia cells and treated with 0.5 mg/kg/week vincristine or vehicle. Independently from obesity, mice treated with vincristine survived longer than vehicle-treated mice (p<0.0001, Fig. 1B). Interestingly, obesity impaired the effect of vincristine: progressive leukemia developed in 7 of 12 DIO mice treated with vincristine, but in only 3/12 control mice (p=0.03). Indeed, one DIO mouse even developed progressive leukemia before receiving the final dose of vincristine.
Figure 1.

Diet-induced obesity impairs vincristine treatment. A: Obese (solid line) and non-obese (dashed line) mice transplanted with 5,000 syngeneic 8093 leukemia cells and left untreated (p=0.22). B: Mice transplanted with 10,000 8093 cells and treated with 0.5 mg/kg/week vincristine (gray bar). Solid line = obese mice (n=12), dashed black line = control mice (n=12), gray dotted line = vehicle-treated controls (n=7). C: GFP+ 8093 ALL cells in the perirenal fat pad of a transplanted obese mouse which developed progressive leukemia during vincristine treatment. Lipid is stained with Nile Red, and the image is counterstained with Dapi. Image was taken on a Leica DM RXA2 with a 20x objective with 1.6x Optovar magnification (32x final). Picture is representative of fat pads obtained from the other 4 mice after vincristine treatment. D: GFP+ 8093 leukemia cells (green) and adipocyte feeder layer (lipid stained with Nile Red) after 48 hours in co-culture. Images obtained with a Zeiss LSM-510 laser scanning confocal/multiphoton microscope with a 40X objective lens (Carl Zeiss Inc. Thornwood NY). Image represents Z-stack transformation (“side view”) of the adipocyte monolayer and leukemia cells (Volocity 4.3, Improvisation Inc., Waltham, MA).
To address whether obesity impaired leukemia treatment via an interaction between adipocytes and leukemia cells, we next investigated whether adipose tissue could act as a “sanctuary” for leukemia cells during or after chemotherapy. Five obese mice were injected with GFP+ ALL cells and then treated with vincristine. Animals were sacrificed when signs of leukemia developed, which occurred during the vincristine treatment period in 3 of the 5 mice. Numerous GFP+ leukemia cells were visible in fat pads from all mice (Fig. 1C). These cells remained viable and proliferated in culture, and after 3–5 days, <10% were apoptotic, as measured by Annexin V and 7-AAD (n=3, Figure S1). These cells also retained their sensitivity to 5 nM vincristine (n=3, not shown). These findings support the concept that adipose tissue can be a “sanctuary” site for leukemia during vincristine treatment.
Blood and tissue vincristine concentration
To exclude the possibility that altered vincristine pharmacokinetics in obese mice confounded our survival results, we measured blood and tissue concentrations of vincristine after a 0.5 mg/kg vincristine injection in groups of DIO and control mice (see Supplementary Methods). As discussed, vincristine was given at doses proportional to body weight, so obese mice on average received ~28% more vincristine in total than control mice (19.7±2.2 vs. 14.7±0.8 μg/mouse). However, vincristine profiles in blood and tissues were very similar between groups (Fig. S2). Thus, poorer survival in obese mice was not likely due to altered exposure to the drug after dosage adjustment using body weight.
Adipocytes protect leukemia cells against drug treatment
To further characterize the possible effects of adipocytes on vincristine-induced cytotoxicity of leukemia cells, we developed an in vitro co-culture system. Murine embryonic fibroblasts (MEFs) have been shown to provide significant protection to leukemia cells against some drugs28, 29. 3T3-L1 fibroblasts were differentiated into adipocytes by exposure to a mixture of insulin, dexamethasone and isobutylmethylxanthine (see Methods), and cultured together with 8093 leukemia cells. Irradiated, undifferentiated 3T3-L1 cells, which have a fibroblast phenotype, were used as controls.
The differentiated 3T3-L1 cells accumulated large lipid droplets as has been previously described30. Leukemia cells rapidly (within 72 hours) migrated into and beneath the adipocyte layer (Fig. 1D), similar to what we had previously observed with MEFs. The proliferation of 8093 cells in co-culture tended to be less with either 3T3-L1 fibroblasts (p=0.27) or adipocytes (p=0.09) after 3 days than without feeder layer, perhaps due to depletion of nutrients from the media (not shown). However, co-culture with adipocytes significantly decreased vincristine cytotoxicity toward leukemia. After 72 hours of vincristine exposure, the mean number of viable ALL cells was higher in cultures with adipocytes (52.8±16.0 × 103) than with fibroblasts (21.4±7.0 × 103, p=0.048) or no feeder (8.5±1.5 × 103, p=0.054, Fig. 2A, top panel). Similar co-culture experiments were performed with 3 other anti-leukemia agents, each with different modes of antitumor activity (dexamethasone, daunorubicin, or nilotinib, Fig. 2A, top and bottom panel). For each drug there were more surviving leukemia cells in co-culture with adipocytes than with fibroblasts or no feeder.
Figure 2.

Co-culture with 3T3-L1 adipocytes provides significant protection against drug treatment to 8093 leukemia cells. A: Number of viable leukemia cells after 72 hours of exposure to vincristine (5 nM), nilotinib (20 nM), daunorubicin (35 nM), or dexamethasone (25 nM) while in co-culture with 3T3-L1 fibroblasts (hatched bars) or adipocytes (solid bars), compared to culture alone (gray bars). B: Proliferation of 8093 cells in TransWells over no feeder (dotted gray line, triangles), fibroblasts (dashed line, open circles), and adipocytes (solid line, closed circles, p=n.s. for all comparisons; n=5 experiments performed in triplicate). C: Viable 8093 cells during exposure to 5 nM vincristine while in TransWells over adipocytes (solid line), fibroblasts (black dashed line), or no feeder layer (gray dotted line; n=5 expts done in triplicate) *p<0.05, ***p<0.005 adipocyte vs. fibroblast feeder layer, paired t-test.
Protection of leukemia cells does not depend on cell-cell contact, but requires living adipocytes
Because the leukemia cells established a close physical interaction with the feeder layers in co-culture, we next investigated whether direct contact between adipocytes and leukemia cells is necessary for protection. Leukemia cells were co-cultured in TransWells over the feeder layers, so that the two cell types were separated by a porous membrane which prevents physical contact. The feeder layers did not influence the leukemia proliferation rate in TransWells (Fig. 2B, p=n.s. for all comparisons). However, adipocytes in the lower chamber provided significant protection against vincristine (Fig. 2C). In fact, 5 nM vincristine suppressed viable leukemia cells over adipocytes by only 11±9% from the initial plated value, while those over fibroblasts or no feeder were much more significantly suppressed (by 55±3%, and 85±2%, p=0.009 and p=0.001 vs. adipocytes, respectively).
To confirm that these co-culture results are relevant to human leukemia, and not particular to the culture models used, we performed several additional experiments. To ensure that irradiation of the fibroblast feeder layers was not responsible for the lack of protection compared to adipocytes, we performed TransWell experiments with non-irradiated, senescent 3T3-L1 fibroblasts. These fibroblasts offered similar protection to leukemia cells against vincristine as the irradiated layers (not shown). We also found that 3T3-L1 adipocytes caused resistance to vincristine in 3 out of 4 human leukemia cell lines tested (Fig. 3A). Finally, OP-9 cells, which are murine bone marrow derived mesenchymal cells that can be differentiated into adipocytes, also protected 8093 cells against vincristine, though to a lesser degree than 3T3-L1 adipocytes (Fig. 3B).
Figure 3.

Adipocytes protect other ALL cell lines against vincristine. A: Viable SD-1, RS(4;11), BV173, and RCH ACV cells after 72 hours of exposure to 5 nM vincristine in TransWells over adipocytes (black bars), fibroblasts (hatched bars), or no feeder (gray bars). B: Number of viable 8093 cells after 72 hour exposure to 5 nM vincristine in TransWells over OP9 bone marrow derived adipocytes (striped bar), 3T3-L1 derived adipocytes (black bar), and no feeder (gray bar). One of two representative experiments shown, each done in triplicate. C, D: Number of viable 8093 cells after 72 hours of co-culture in TransWells over PFA fixed or unfixed fibroblasts or adipocytes, without (C) or with (D) 5 nM vincristine (n=3 experiments, each done in triplicate; *p<0.05, **p<0.01, ***p<0.005)
To verify that living adipocytes are needed to provide protection to the leukemia cells, we also tested vincristine-induced cytotoxicity in TransWell cultures, in which the feeder layers had been fixed with PFA. The presence of fixed adipocytes or fibroblasts in TransWell co-culture did not alter leukemia proliferation rates (Fig. 3C). However, neither fixed fibroblasts nor fixed adipocytes protected 8093 leukemia cells against vincristine treatment (Fig. 3D).
Mechanisms of adipocyte-induced vincristine resistance
We considered the possibility that adipocytes might protect leukemia by sequestering the lipophilic vincristine, decreasing its availability. To examine this, vincristine levels were measured in fibroblasts and adipocytes after 48 hours of drug exposure (see Supplementary methods). We found that adipocytes in TransWells did accumulate significantly more vincristine than fibroblasts (1.28±0.28 vs. 0.49±0.12 nM vincristine, p=0.002, Fig. S3A). The accumulation of drug into these monolayers of cells, however, was not reflected by a detectible decrease in the concentration of vincristine in the media (Fig. S3B) or leukemia cells (not shown).
To further explore how adipocytes protect leukemia cells, we assessed the leukemia cell cycle and apoptotic status in our co-culture system. 8093 cells were exposed to BrdU while in TransWells over the various feeder layers, and their cell cycle state assessed using FACS analysis. Neither feeder layer altered the cell cycle kinetics under baseline conditions (Table). Addition of vincristine decreased the proportion of cells in S-phase and increased the proportion of cells undergoing apoptosis. Compared to leukemia cells over no feeder (Fig. 4A) or fibroblasts (Fig. 4B), adipocytes partially reversed the effects of vincristine, by decreasing apoptosis and increasing the proportion of cells in G0/G1 and S-phase during vincristine exposure (Fig. 4C, Table).
Figure 4.

Adipocytes prevent 8093 apoptosis in response to 5 nM vincristine. A–C: Leukemia cells were grown in TransWells for 48 hours over no feeder (A), fibroblasts (B), or adipocytes, (C) with 5 nM vincristine. Top panels show the composite plots from all 3 replicates of BrdU vs. 7-AAD with gating used to define cell cycle phases: A = apoptosis, S = synthesis, G0/G1 = G0/G1 phase, G2 = G2 + M phase.
We next investigated the leukemia cell expression of apoptosis-related genes that might be altered by the presence of adipocytes. Adipocytes upregulated Bcl-2 and Pim-2 expression, both with and without vincristine, and these results were verified by rtPCR (Fig. 5A). Overall Pim-2 protein level was upregulated by adipocytes, particularly in the presence of vincristine (Figs. 5B and S4). Since Pim-2 prevents apoptosis via inactivation of Bad, we assessed Bad phosphorylation, and found that adipocytes increased the level of phosphorylated Bad in 8093 leukemia cells. Thus, adipocyte protection of leukemia cells from vincristine was associated with upregulation of Bcl-2 and Pim-2, and an increased phosphorylation of Bad.
Figure 5.
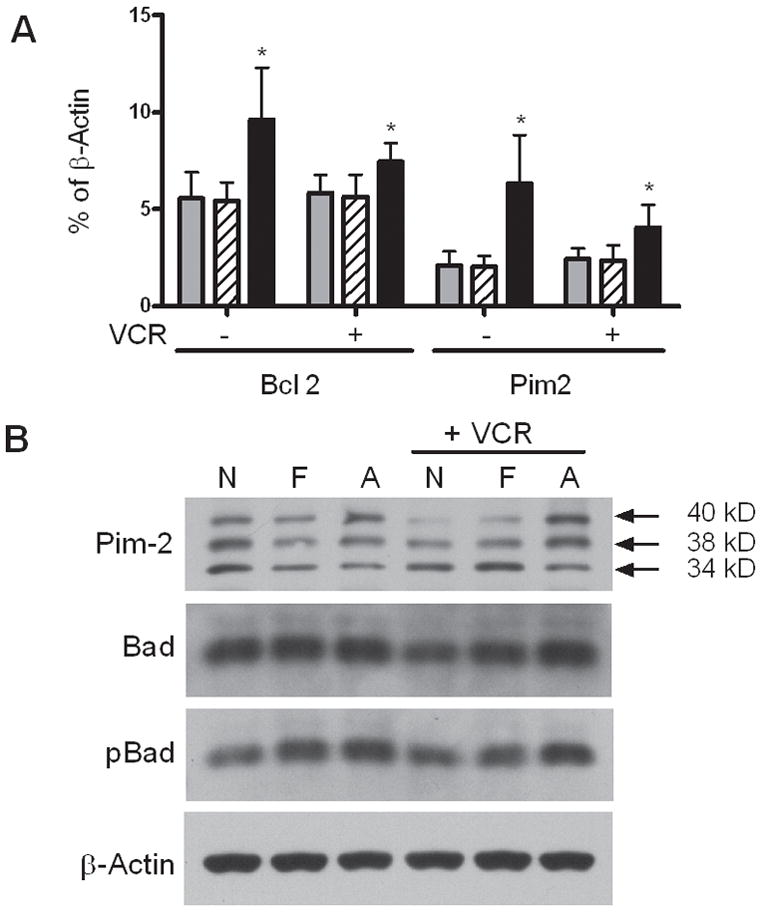
Effect of adipocytes on leukemia cell expression of the survival genes Pim-2 and bcl-2. A: Gene expression of leukemia cells was quantified by rtPCR after 24 hour exposure to no stroma (gray bars), fibroblasts (hatched bars) or adipocytes (black bars), with or without vincristine. Results are expressed as % of β-actin gene expression. The averages and SD of 5 biological replicates are shown. *p<0.05 vs. both no feeder and fibroblast layers. B: Cropped western blots of 8093 protein lysates showing the 3 murine isoforms of Pim-2, Bad, and phospho-S112 Bad, after exposure to no feeder layer (N), fibroblasts (F), or adipocytes (A), with or without 5 nM vincristine. β-actin is shown as a loading control. 1 of 2 representative experiments is shown. Full length blots are in Supplemental Figure S4.
Discussion
Elucidating the mechanism(s) linking obesity and leukemia relapse is complicated by the large number of effects of obesity, and the long list of potential factors which could lead to relapse. Therefore, we have developed cell culture and animal models of obesity and leukemia to explore this relationship. In our mouse model, we have recapitulated the increased ALL relapse observed in obese patients—obese mice injected with highly malignant pre-B lymphoblastic leukemia cells had a worse treatment outcome. Interestingly, we found leukemia cells associated with fat pads in vincristine-treated mice, suggesting that adipose tissue may be partly responsible for the effect of obesity to increase leukemia relapse. Although the fat depots were examined only once the mice developed progressive leukemia, 3 of the 5 mice developed progressive disease during the vincristine treatments period, showing that fat depots can harbor leukemia cells during chemotherapy. Interestingly, subcutaneous lymphoma cells have been noted to form a “rim” around adipocytes, implying a cell-cell interaction also between these two cell types31.
Thus, we developed in vitro models which demonstrated that the adipocyte microenvironment may be a protective niche for leukemia cells during chemotherapy. Firstly, we show that adipocytes are as competent as fibroblasts in supporting leukemia cell steady-state growth. This complements recent studies which seem to suggest that adipocytes impair the proliferation of normal hematopoietic cells32, while they support the growth of malignant cells such as multiple myeloma cells33. Secondly, we show that adipocytes confer a significant protection to leukemia cells against vincristine, daunorubicin, dexamethasone, and nilotinib. Thus, adipocytes may actively contribute to leukemia cell survival in the face of multi-agent chemotherapy. Adipocytes also protected some, though not all established human pre-B ALL cell lines from vincristine. Interestingly, the one cell line tested which was not protected by adipocytes, RCH-ACV, harbors the (1;19) translocation, which is known to be associated with increased sensitivity to many chemotherapies including vincristine34. Thus, the inability of these cells to resist vincristine in the presence of adipocytes may reflect an overall inability of cells with this translocation to resist chemotherapy.
We next found that the presence of adipocytes decreases leukemia apoptosis and increases cell cycling in the face of vincristine. Adipocytes increased expression of pro-survival signals, which may shift the apoptotic balance of leukemia cells toward survival; this effect was maintained in the presence of vincristine. The main targets of this adipocyte-mediated effect are possibly the survival genes Bcl-2 and Pim-2. Over-expression of Bcl-2 causes leukemia cell resistance to several drugs, including vincristine35, and high expression of Bcl-2 is strongly predictive of survival in adults with AML36. Pim-2 is an oncogene that promotes growth of hematopoietic cells37, as well as leukemia cell survival via multiple mechanisms38, including phosphorylation of Bad39. Indeed, Bad phosphorylation was increased by adipocytes both with and without vincristine, and this may be one of the mechanisms by which adipocytes protect leukemia cells from vincristine. Overall, our finding that adipocytes alter the balance of apoptotic signals toward survival is consistent with our finding that they protect leukemia cells from a variety of chemotherapeutics with different mechanisms of action.
It is unknown which specific adipose depots are relevant to leukemia escape from chemotherapy. Studies on the bone marrow tumor microenvironment generally ignore the role of adipocytes, the most abundant stromal cell in adult bone marrow40. We demonstrated in vitro that bone marrow derived adipocytes (OP9 cells) can protect leukemia cells against vincristine. Since bone marrow adiposity does not increase substantially with obesity41, marrow adipocytes may contribute to leukemia treatment resistance in both lean and obese patients, but are not likely responsible for the increased relapse in obese patients.
We also considered that vincristine accumulation in adipose tissue could be partly responsible for the association between obesity and leukemia relapse. Our measurements confirmed that vincristine accumulates in adipocytes more than in other cells such as fibroblasts, though this did not affect drug levels in the medium or leukemia cells. This was not unexpected, as in this uncomplicated system a single monolayer of adipocytes was exposed to a comparably large volume of media. In vivo, vincristine concentrations in blood and tissue were well-matched between obese and non-obese mice after a single intravenous injection. However, this injection was dosed proportional to body weight. In clinical practice, chemotherapies such as vincristine are dosed in proportion to body surface area, which leads to lower doses per kg body weight in older and more overweight children—patients at higher risk of relapse. This underdosing may be compounded by the fact that chemotherapy doses are often “capped” to prevent dose-dependent toxicities. If obesity does lead to lower vincristine exposure in children, then this could be a significant factor in clinical outcome, particularly given the increasing prevalence of obesity worldwide. Pharmacokinetic experiments in obese patients will be needed to rigorously address this issue42–44.
In summary, our findings demonstrate that obesity can directly impair the anti-leukemia efficacy of the first-line chemotherapy, vincristine. This effect is likely due in part to adipocytes interacting with leukemia cells, as their presence in co-culture leads to impaired leukemia killing by this and other drugs. The protective effects of adipocytes may also contribute to poorer prognosis of obese patients with other malignancies, as adipose tissue has been suggested to play a protective role in the microenvironment of breast45 and colon46 cancer. However, further studies are necessary to elucidate the effects of adipocytes to alter chemotherapy pharmacokinetics, secrete cancer survival factors, or both. Given that 32% of children are overweight and 16% are obese47, understanding the associations between adiposity and increased morbidity from leukemia and other cancers (breast, colon, prostate) will be crucial in preventing a significant number of patient deaths.
Supplementary Material
Table.
Cell cycle and apoptosis in 8093 cells over various feeder layers, with and without vincristine. Numbers indicate percentage of cells in different states.
| Condition | Feeder Layer | G0/G1 | S | G2+M | Apoptotic |
|---|---|---|---|---|---|
| Baseline (24 hr, n=2) | None | 39±2 | 58±2 | 3±0 | 0±0 |
| Fibroblasts | 48±1 | 48±1 | 3±0 | 0±0 | |
| Adipocytes | 39±11 | 53±3 | 3±0 | 2±3 | |
| Vincristine (72 hr, n=3) | None | 19±4 | 2±1 | 2±1 | 76±6 |
| Fibroblasts | 38±4 | 4±1 | 4±1 | 53±4 | |
| Adipocytes | 58±4*** | 23±10 | 10±5 | 9±1*** |
p<0.005 vs Fibroblasts
Acknowledgments
The authors thank Markus Muschen for the generous gift of the pMIG retrovirus, use of reagents and equipment, and helpful comments on the manuscript, and Daniel Trageser for assistance with the transfection. We also thank Donna Foster for expert animal care, Xingchao Wang for histology assistance, Nick Mordwinkin for vincristine measurements, and Lingyun Ji for assistance with statistics.
Reference List
- 1.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 2.McTiernan A. Obesity and cancer: the risks, science, and potential management strategies. Oncology (Williston Park) 2005;19:871–81. [PubMed] [Google Scholar]
- 3.El Serag HB. Obesity and disease of the esophagus and colon. Gastroenterol Clin North Am. 2005;34:63–82. doi: 10.1016/j.gtc.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Ross JA, Parker E, Blair CK, Cerhan JR, Folsom AR. Body mass index and risk of leukemia in older women. Cancer Epidemiol Biomarkers Prev. 2004;13:1810–3. [PubMed] [Google Scholar]
- 5.Hursting SD, Margolin BH, Switzer BR. Diet and human leukemia: an analysis of international data. Prev Med. 1993;22:409–22. doi: 10.1006/pmed.1993.1034. [DOI] [PubMed] [Google Scholar]
- 6.Larsson SC, Wolk A. Overweight and obesity and incidence of leukemia: a meta-analysis of cohort studies. Int J Cancer. 2008;122:1418–21. doi: 10.1002/ijc.23176. [DOI] [PubMed] [Google Scholar]
- 7.Whiteman MK, Hillis SD, Curtis KM, et al. Body mass and mortality after breast cancer diagnosis. Cancer Epidemiol Biomarkers Prev. 2005;14:2009–14. doi: 10.1158/1055-9965.EPI-05-0106. [DOI] [PubMed] [Google Scholar]
- 8.Whiteman MK, Hillis SD, Curtis KM, et al. Body mass and mortality after breast cancer diagnosis. Cancer Epidemiol Biomarkers Prev. 2005;14:2009–14. doi: 10.1158/1055-9965.EPI-05-0106. [DOI] [PubMed] [Google Scholar]
- 9.Freedland SJ, Grubb KA, Yiu SK, et al. Obesity and risk of biochemical progression following radical prostatectomy at a tertiary care referral center. J Urol. 2005;174:919–22. doi: 10.1097/01.ju.0000169459.78982.d7. [DOI] [PubMed] [Google Scholar]
- 10.Lange BJ, Gerbing RB, Feusner J, et al. Mortality in overweight and underweight children with acute myeloid leukemia. JAMA. 2005;293:203–11. doi: 10.1001/jama.293.2.203. [DOI] [PubMed] [Google Scholar]
- 11.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 12.Pui CH. Childhood leukemias. N Engl J Med. 1995;332:1618–30. doi: 10.1056/NEJM199506153322407. [DOI] [PubMed] [Google Scholar]
- 13.Butturini A, Vignetti M, Gubbiotti S, et al. Obesity Independently Predicts Event Free Survival (EFS) in Adults with BCR-ABL-Negative Acute Lymphoblastic Leukemia (ALL). A Retrospective Analysis of Two GIMEMA Studies ASH Annual Meeting Abstracts. 2005;106:1828. [Google Scholar]
- 14.Baillargeon J, Langevin AM, Lewis M, et al. Obesity and survival in a cohort of predominantly Hispanic children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2006;28:575–8. doi: 10.1097/01.mph.0000212985.33941.d8. [DOI] [PubMed] [Google Scholar]
- 15.Hijiya N, Panetta JC, Zhou Y, et al. Body mass index does not influence pharmacokinetics or outcome of treatment in children with acute lymphoblastic leukemia. Blood. 2006;108:3997–4002. doi: 10.1182/blood-2006-05-024414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butturini AM, Dorey FJ, Lange BJ, et al. Obesity and outcome in pediatric acute lymphoblastic leukemia. J Clin Oncol. 2007;25:2063–9. doi: 10.1200/JCO.2006.07.7792. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Daquinag A, Traktuev DO, et al. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer Res. 2009;69:5259–66. doi: 10.1158/0008-5472.CAN-08-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaspers GJ, Veerman AJ, Pieters R, et al. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood. 1997;90:2723–9. [PubMed] [Google Scholar]
- 19.Lock RB, Liem N, Farnsworth ML, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood. 2002;99:4100–8. doi: 10.1182/blood.v99.11.4100. [DOI] [PubMed] [Google Scholar]
- 20.Heisterkamp N, Jenster G, ten Hoeve J, et al. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–3. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- 21.Mishra S, Zhang B, Groffen J, Heisterkamp N. A farnesyltransferase inhibitor increases survival of mice with very advanced stage acute lymphoblastic leukemia/lymphoma caused by P190 Bcr/Abl. Leukemia. 2004;18:23–8. doi: 10.1038/sj.leu.2403203. [DOI] [PubMed] [Google Scholar]
- 22.Jack I, Seshadri R, Garson M, et al. RCH-ACV: a lymphoblastic leukemia cell line with chromosome translocation 1;19 and trisomy 8. Cancer Genet Cytogenet. 1986;19:261–9. doi: 10.1016/0165-4608(86)90055-5. [DOI] [PubMed] [Google Scholar]
- 23.Pegoraro L, Matera L, Ritz J, et al. Establishment of a Ph1-positive human cell line (BV173) J Natl Cancer Inst. 1983;70:447–53. [PubMed] [Google Scholar]
- 24.Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–33. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- 25.Wolins NE, Quaynor BK, Skinner JR, et al. OP9 mouse stromal cells rapidly differentiate into adipocytes: characterization of a useful new model of adipogenesis. J Lipid Res. 2006;47:450–60. doi: 10.1194/jlr.D500037-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Liem NL, Papa RA, Milross CG, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103:3905–14. doi: 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 27.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–7. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 28.Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood. 2000;96:1926–32. [PubMed] [Google Scholar]
- 29.Weisberg E, Wright RD, McMillin DW, et al. Stromal-mediated protection of tyrosine kinase inhibitor-treated BCR-ABL-expressing leukemia cells. Mol Cancer Ther. 2008;7:1121–9. doi: 10.1158/1535-7163.MCT-07-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–33. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- 31.Lozzi GP, Massone C, Citarella L, Kerl H, Cerroni L. Rimming of adipocytes by neoplastic lymphocytes: a histopathologic feature not restricted to subcutaneous T-cell lymphoma. Am J Dermatopathol. 2006;28:9–12. doi: 10.1097/01.dad.0000187933.87103.03. [DOI] [PubMed] [Google Scholar]
- 32.Naveiras O, Nardi V, Wenzel PL, et al. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009 doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caers J, Deleu S, Belaid Z, et al. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia. 2007;21:1580–4. doi: 10.1038/sj.leu.2404658. [DOI] [PubMed] [Google Scholar]
- 34.Frost BM, Forestier E, Gustafsson G, et al. Translocation t(1;19) is related to low cellular drug resistance in childhood acute lymphoblastic leukaemia. Leukemia. 2004;19:165–9. doi: 10.1038/sj.leu.2403540. [DOI] [PubMed] [Google Scholar]
- 35.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81:151–7. [PubMed] [Google Scholar]
- 36.Campos L, Rouault JP, Sabido O, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–6. [PubMed] [Google Scholar]
- 37.Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood. 2005;105:4477–83. doi: 10.1182/blood-2004-09-3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;27:4809–19. doi: 10.1038/onc.2008.123. [DOI] [PubMed] [Google Scholar]
- 39.Fox CJ, Hammerman PS, Cinalli RM, et al. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003;17:1841–54. doi: 10.1101/gad.1105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gimble JM, Robinson CE, Wu X, Kelly KA. The function of adipocytes in the bone marrow stroma: an update. Bone. 1996;19:421–8. doi: 10.1016/s8756-3282(96)00258-x. [DOI] [PubMed] [Google Scholar]
- 41.Di IN, Mittelman SD, Gilsanz V. Differential effect of marrow adiposity and visceral and subcutaneous fat on cardiovascular risk in young, healthy adults. Int J Obes (Lond) 2008;32:1854–60. doi: 10.1038/ijo.2008.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox J, Penn N, Masood M, Hancock AK, Parker D. Drug overdose--a hidden hazard of obesity. J R Soc Med. 1987;80:708–9. doi: 10.1177/014107688708001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleming RA, Eldridge RM, Johnson CE, Stewart CF. Disposition of high-dose methotrexate in an obese cancer patient. Cancer. 1991;68:1247–50. doi: 10.1002/1097-0142(19910915)68:6<1247::aid-cncr2820680611>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 44.Herrington JD, Tran HT, Riggs MW. Prospective evaluation of carboplatin AUC dosing in patients with a BMI>or=27 or cachexia. Cancer Chemother Pharmacol. 2006;57:241–7. doi: 10.1007/s00280-005-0012-9. [DOI] [PubMed] [Google Scholar]
- 45.Iyengar P, Combs TP, Shah SJ, et al. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene. 2003;22:6408–23. doi: 10.1038/sj.onc.1206737. [DOI] [PubMed] [Google Scholar]
- 46.Amemori S, Ootani A, Aoki S, et al. Adipocytes and preadipocytes promote the proliferation of colon cancer cells in vitro. Am J Physiol Gastrointest Liver Physiol. 2007;292:G923–G929. doi: 10.1152/ajpgi.00145.2006. [DOI] [PubMed] [Google Scholar]
- 47.Ogden CL, Carroll MD, Flegal KM. High Body Mass Index for Age Among US Children and Adolescents, 2003–2006. JAMA: The Journal of the American Medical Association. 2008;299:2401–5. doi: 10.1001/jama.299.20.2401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.