

Abstract
The ahl locus, shown to be a strain-specific Cdh23 dimorphism, contributes to age-related hearing loss in many inbred mouse strains. A/J mice begin to lose hearing by four weeks of age, much earlier than C57BL/6J (B6) mice, although both strains have the same Cdh23ahl variant. Here, we use recombinant inbred strains, chromosome substitution strains, and a linkage backcross to map a locus on distal Chromosome 10, designated ahl4, that contributes to the early-onset hearing loss of A/J mice. Cochleae of 9-week-old A/J mice exhibit inner and outer hair cell loss from the basal turn through the apical turn, with outer hair cell loss at the base being severest. To quantify the progression of hair cell loss, cytocochleograms were evaluated from 0 to 20 weeks of age. AJ mice showed evidence of hair cell loss in the base of the cochlea as early as 14 days of age and the magnitude and extent of loss increased rapidly during the following 2–5 months. Hair cell loss occurred earlier and was much more severe and widespread in A/J mice than in B6 mice during the first 5 months of age. Spiral ganglion neurons, cells of the stria vascularis, and vestibular hair cell densities, however, appeared normal in 20-week-old A/J mice.
Keywords: genetic mapping, mouse, inbred strain, deafness, presbycusis
1. Introduction
Presbycusis or age-related hearing loss (AHL) is one of the most common disorders of elderly individuals and adversely affects the quality of life. Thirty to thirty-five percent of adults between 65 and 75 years of age exhibit hearing loss and by 75 years of age 40–50 percent experience AHL (Gorlin et al., 1995; Morton, 1991). Genetic variation in humans probably plays a role in determining the range of individual susceptibility to AHL (DeStefano et al., 2003). However, no contributing loci have been identified in human populations because of the difficulties in controlling for environmental factors such as noise trauma, toxic agents or disease. Another challenge is the long life span of humans that makes it difficult to carry out prospective longitudinal studies of AHL. Inbred laboratory mice provide a means for researchers to overcome these difficulties.
Genetically well-defined inbred mice, including C57BL/6J, DBA/2J, and BALB/cJ, have been productively used in studies of auditory anatomy, physiology, and pathology, including presbycusis (Erway et al., 1993; Ohlemiller, 2006; Willott et al., 1998). Auditory pathology and function of these strains has been extensively studied and can be used to elucidate mechanisms of human AHL. C57BL/6J (abbreviated B6) is the mouse strain most widely used in studies of AHL. Our auditory brainstem response (ABR) analysis of this strain has shown a progressive hearing loss beginning as early as 9 months of age for high frequencies, but not until 12 months for mid-frequencies (Li and Borg 1991, Keithley et al., 2004, Henry, 1982; and Willott et al., 1998). In contrast, A/J strain mice have a much earlier onset of hearing loss, exhibiting elevated auditory thresholds by 25 days of age (Henry, 1982; Zheng et al., 1999).
The ahl locus is an important contributor to AHL in many inbred mouse strains (Johnson et al., 2000) and was shown to be a strain-specific dimorphism of Cdh23 (Noben-Trauth et al., 2003). Although hearing loss occurs much earlier in A/J mice than in B6 mice, both strains are homozygous for the Cdh23ahl allele. Thus, it is likely that additional genetic factors contribute to the hearing loss phenotype observed in strain A/J. A DNA variant of the mitochondrial tRNA-Arg gene (mt-Tr) also contributes to hearing loss in A/J mice (Johnson et al., 2001); however, its effects are not great enough to fully explain the severe degree of hearing loss in A/J mice compared with B6 mice.
To further investigate the genetic basis of hearing loss in A/J mice, we analyzed recombinant inbred (RI) strains, chromosome substitution (CS) strains, and a linkage backcross. These complementary approaches all showed strong evidence for an additional AHL locus on distal Chromosome (Chr) 10, which we designate ahl4. To test whether the early onset and severe hearing loss of A/J mice has a distinctive pathological basis, we examined cochleae from A/J and B6 mice at multiple ages for cochlear pathologies that underlie human presbycusis (Schuknecht et al., 1993). Hearing loss in both strains correlated with a progressive base-to-apex loss of outer hair cells followed by inner hair cell loss; however, hair cell loss occurred earlier and progressed more rapidly in A/J mice.
2. Materials and Methods
2.1. Mice
A total of 399 mice of either sex were used for this study. Mice of the standard A/J and B6 inbred strains and their derivative recombinant inbred strains and chromosome substitution strains were housed in the Research Animal Facility of The Jackson Laboratory (http://www.jax.org/). The Jackson Laboratory is accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC) and is registered with the United States Department of Agriculture as a research facility. All mice were from The Jackson Laboratory (TJL) research facilities and all procedures were approved by the Institutional Animal Care and Use Committee.
2.2. Evaluation of auditory function in mice
The ABR methodology has been previously described and includes criteria for evaluating hearing loss in response to various stimuli (Zheng et al., 1999). Mice were anesthetized and body temperature was maintained at 37–38 °C by placing mice on an isothermal pad in a sound-attenuating chamber. Sub-dermal needles were used as electrodes, inserted at the vertex and ventrolaterally to each ear. Stimulus presentation, ABR acquisition, equipment control and data management were coordinated using the computerized Intelligent Hearing Systems (IHS; Miami, Florida). Clicks, and 8, 16, and 32 kHz tone-bursts were respectively channeled through plastic tubes into the animals’ ear canals. ABR thresholds were obtained for each stimulus by reducing the sound pressure level (SPL) at 10 dB steps and finally at 5 dB steps up and down to identify the lowest level at which an ABR pattern can be recognized. Samples of CBA/CaJ mice were tested periodically as references for normal hearing, and for monitoring the reliability of the equipment and testing procedures. Based on our previous work (Zheng et al., 1999), any mouse with average ABR threshold values above 55 (for click stimulus), 40 (for 8 kHz), 35 (for 16 kHz), or 60 (for 32 kHz) dB SPL was classified as hearing impaired. Mice with average ABR values below these values for all four test stimuli were considered to have normal hearing. Written and electronic records were made of the ABR recordings and thresholds, the strain background, genotype, and birth and test dates for each mouse tested.
2.3. Genetic mapping
Three complementary approaches were used to evaluate the genetic basis of the early-onset hearing loss of A/J mice. The first was recombinant inbred (RI) lines, produced by inbreeding individual F2 progeny from an intercross of two inbred strains. RI lines are useful as a first approach to estimate the number and approximate map locations of major genes affecting a complex trait (Williams et al., 2001). Mice from AXB and BXA RI lines were analyzed for associations with ABR thresholds, treated as quantitative traits. There are 14 independent AXB (A/J female progenitor) and 13 BXA (B6 female progenitor) RI strains available from The Jackson Laboratory (Sampson et al., 1998). Collectively, the AXB and BXA RI strains are called AXBXA. A web-based QTL (quantitative trait locus) mapping procedure for RI strains (WebQTL) has been developed by Kenneth Manley and Robert Williams (http://www.genenetwork.org/) based on the MapManager QTX computer program (Manly et al., 2001). The quantitative trait values entered at the WebQTL website are statistically compared with verified genotypes collected at a set of micro-satellite markers in each RI set. The edited strain distribution pattern for the AXBXA strains now includes over 700 loci (Williams et al., 2001).
Chromosome substitution (CS) strains, in which a single chromosome from one inbred strain (donor) has been transferred onto a second strain (host) by repeated backcrossing, may be used to identify chromosomes containing significant QTLs (Nadeau et al., 2000). One of the advantages of mapping with CS strains is that the effects of a QTL are limited to the donor chromosome, eliminating contributions of loci on all other chromosomes. At least eight mice from each of 21 B6.A CS strains (19 autosomes plus X and Y chromosomes) were analyzed for ABR thresholds to determine the individual effects of A/J-derived donor chromosomes on hearing loss in mice with an otherwise B6 host genome (Singer et al., 2004). For clarity, we use a modified nomenclature to designate CS strains, for example the official strain name C57BL/6J-1A/Na is designated B6.A-Chr1.
A linkage backcross was used to confirm the effect of ahl4 on hearing loss and to improve the genetic resolution of the map position of ahl4 on Chr 10. Matings between A/J and CAST/Ei mice produced F1 hybrid progeny that were then backcrossed to A/J mice to generate N2 mice for genotyping and linkage analyses. The same (A/J x CAST/Ei) x A/J backcross used in this study to map ahl4 was used previously to analyze hearing loss associated with ahl and mtDNA variants (Johnson et al., 2000; Johnson et al., 2001).
2.4. Cross-section preparation of inner ears
For the histological sections, anesthetized mice were perfused through the left ventricle of the heart with phosphate-buffered saline (PBS) followed by Bouin’s fixative (Fisher Diagnostics, Middletown, VA, USA). The animals were then decapitated and the bulla was quickly removed. The round window and oval window were opened and the cochlea was immersed in fixative for 24–48 hr at 4 °C. The cochleae were decalcified with Cal-EX Decalcifier (Fisher Scientific, Pittsburg, PA) solution for 6 hr. Specimens were routinely immersed in 2% osmium tetroxide in 0.1 M phosphate buffer for 2 h at 4°C, dehydrated through a graded series of ethanol solutions ending with 100% and embedded in Epon 812 (Electron Microscopy Sciences, Hatfield, PA). After polymerization, samples were cut parallel to the modiolar axis at a thickness of 3 μm, stained with 0.5% toluidine blue and mounted on glass slides.
2.5. Cytocochleograms
Mice were subjected to an overdose of carbon dioxide, decapitated, and the inner ear quickly removed. The tympanic bullae were opened and a small hole made in the round window of the exposed cochleae. Fixative was gently perfused through the round window, followed by immersion of cochleae in fixative. The fixative for routine analyses was 10% buffered formalin as described previously (McFadden et al., 1999a). Details of our experimental methods for preparing mouse cytocochleograms can be found in earlier publications (Ding et al., 2001; McFadden et al., 1999b). Briefly, the organ of Corti was carefully microdissected out and mounted in glycerin on glass slides. The surface preparations were stained with Ehrlich’s hematoxylin solution and examined with light microscope (Zeiss Standard, 400 X) using differential interference contrast optics. Hair cells were counted as present if the cell body and cuticular plate were intact. Inner and outer hair cell counts were made over 0.12–0.24-mm intervals of the organ of Corti, beginning at the apex and continuing towards the base. Individual cochleograms were constructed to show the percentage of hair cells missing as a function of distance from the apex, relative to our laboratory norms for young CBA mice (Ding et al., 2001, Spongr et al., 1997).
2.6. Ganglion cells and auditory nerve fibers
Cochleae were immersed in Decal (VWR Scientific Products) for 2 days for decalcification, embedded in Epon 812 resin, and serially sectioned (3-μm-thickness) parallel to the modiolar axis with a Reichert Super Nova microtome and glass knives as described (McFadden et al., 1999a). Sections were mounted on glass slides, stained with toluidine blue (Fisher Scientific, Pittsburg, PA), and examined under an Axioskop light microscope (Carl Zeiss). For auditory nerve fibers, every fifth section containing the osseous spiral lamina was examined at 1000X in samples from A/J mice and B6 mice. Sections were visually inspected for gross evidence of fiber degeneration.
2.7. Vestibular hair cells
To determine if hair cell lesions extended to the vestibular system, we evaluated hair cell densities in the vestibular sensory epithelium as described previously (Ding et al., 2001). After removal of the inner ear (see cytocochleogram above) and perfusion of the cochlea, the bony walls of the superior and horizontal semicircular canals were opened. Then 10% buffered formalin in 0.1 M cold phosphate buffer was perfused through the oval window. After immersion in cold fixative for 24 hr, the vestibular end organ was placed in PBS and dissected out. The superior, horizontal and posterior portions of the vestibular cavity were opened and the saccule was carefully removed from the underlying bone. The utricle was separated from surrounding tissue and the otoconia of the saccule and utricle were removed to expose the two maculae. The ampullae were separated from their semicircular canals by cutting the attached nerve fibers, blood vessels and connective tissue to expose the cristae. Surface preparations were stained with hematoxylin or toluidine blue. Surface preparations were viewed with an Axioskop light microscope (Carl Zeiss) at magnification of 1000X as described in our earlier publications (Ding et al., 2001).
3. Results
3.1. Hearing assessment of A/J and C57BL/6J mice
ABR thresholds were evaluated for six mice of each strain, A/J and B6. As shown in Fig. 1, A/J mice exhibited hearing loss immediately following weaning, as defined by an increase of three standard deviations or more from the empirically determined mean threshold of hearing (Zheng et al., 1999). At 25 days, the high-frequency 16 and 32 kHz thresholds were elevated indicating hearing-impairment. By three months of age the A/J mice had severe hearing loss as indicated by ABR thresholds >50 dB above normal. In contrast, B6 mice exhibited normal hearing at 6 months of age, and even as late as 12 months gave thresholds only slightly above the mean for normal hearing. Thus, although both strains carry the Cdh23ahl allele and eventually exhibit AHL, the time course for hearing loss in each strain is drastically different and the degree of impairment is more severe for A/J than for B6.
Fig. 1.

ABR thresholds of A/J and B6 mice. ABR thresholds (dB SPL=sound pressure level in decibels) of A/J and B6 mice were determined for the four stimuli shown on the X-axis. Each data point represents an average threshold value (calculated as the arithmetic mean) for each age group (n=6 mice) described in the adjacent data point legend. Error bars indicate the standard deviation from the mean, shown only in the positive direction for simplicity. Rectangles ranging from 0 to a positive value delineate the range of threshold values empirically determined to fall within a normal hearing range (Zheng et al., 1999). All points lying outside the rectangles are considered to be hearing impaired.
3.2. Genetic analysis of AXB and BXA recombinant inbred strains
As a first approach to map loci contributing to the early onset hearing loss of A/J mice, we analyzed AXB and BXA recombinant inbred strains derived from A/J and B6 parental strains. Because both of these strains have the same Cdh23ahl allele, the effects of this locus on hearing loss are eliminated in the RI strain analysis. None of these RI strains had been previously assessed for hearing. We tested mice at 6–9 months of age, an age when inbred mice of one parental strain (A/J) exhibit hearing loss but mice of the other strain (B6) do not. From a comparison of the total variance of 16-kHz ABR thresholds (=716 among all 170 mice tested) with the between-strain variance (=591 among the 27 strains tested), we estimated the broad-sense heritability of this trait to be quite high (about 0.8) in the AXBXA RI line mice, which improves the likelihood of detecting linkage. Figure 2 shows the variation in mean and standard error of ABR thresholds among these RI strains, and Supplementary Table 1 gives the means and standard deviations of click, 8 kHz, 16 kHz, and 32 kHz ABR threshold estimates for each of the 27 RI strains tested.
Fig. 2.
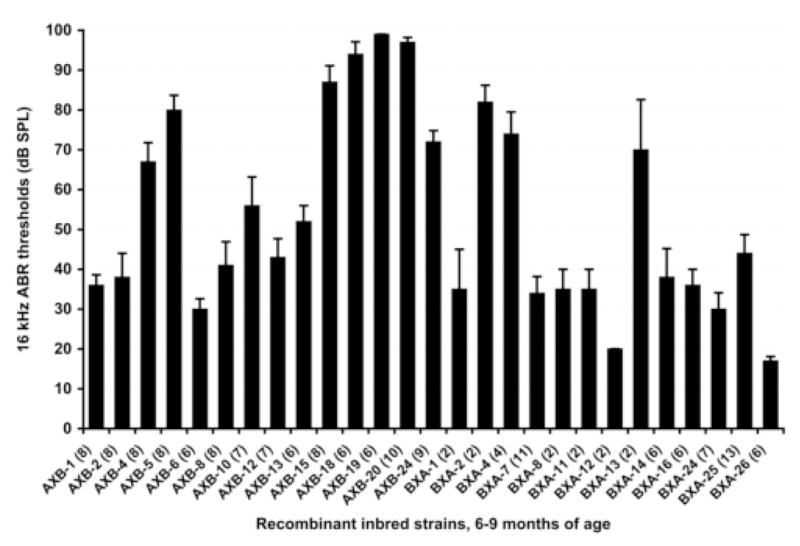
ABR threshold variation among 14 AXB and 13 BXA recombinant inbred (RI) strains tested between 6 and 9 months of age. Means and standard error bars for 16 kHz thresholds and the numbers of mice tested (in parentheses) are shown for each strain. At this age, average thresholds of parental strain A/J mice are greater than 90 dB, whereas those of B6 mice are less than 40 dB. A statistically significant association of ABR thresholds was found with loci on distal Chr 10 (max LOD 4.3).
We used the web-based QTL mapping program WebQTL to analyze ABR threshold measurements as quantitative traits for the 27 AXBXA RI strains, which have been genotyped for hundreds of markers on all chromosomes (Williams et al., 2001). The only statistically significant associations detected were those of 16-kHz ABR thresholds with markers on distal Chr 10 (LOD scores 3.8 and 4.3 for markers D10Mit35 and D10Mit205). All other locus associations were below the 95th percentile significance level determined empirically from 1000 permutations (Manly et al., 2001). We assigned the symbol ahl4 to designate this quantitative trait locus (QTL) on distal Chr 10 that affects AHL.
3.3. Genetic analysis of B6.A chromosome substitution strains
To evaluate the isolated effects of individual A/J-derived chromosomes on hearing loss, we tested a panel of B6.A CS strains in which each strain has a single chromosome from the A/J donor strain substituting for the corresponding chromosome in the B6 host strain (Singer et al., 2004). We measured ABR thresholds of at least 8 mice from each of the 21 B6.A CS strains, all tested at 5–7 months of age (Fig. 3A, Supplementary Table 1). None of the B6.A CS strain mice, with the exception of B6.A-Chr10 and B6.A-Chr11 mice, exhibited thresholds significantly above normal according to previously established criteria (Zheng et al., 1999). As predicted from the RI strain results, mice of the B6.A-Chr10 strain, which contains an A/J-derived ahl4 allele on the transferred Chr 10, exhibited elevated thresholds. The 16 kHz ABR thresholds of these mice were on average about 30 dB higher than normal. The thresholds of B6.A-Chr11 mice were about 10 dB above normal and suggest that another AHL locus might be located on Chr 11, although with smaller effects than ahl4. The onset of hearing loss in B6.A-Chr10 mice occurs earlier and progresses more rapidly than that of control B6 mice (Fig 3B). While 16 kHz thresholds of B6.A-Chr10 mice are only slightly higher than those of B6 mice at 3 months of age, they are about 30 dB higher at 6 months and about 50 dB higher at 9 months.
Fig. 3. ABR thresholds of B6.A chromosome substitution strains.

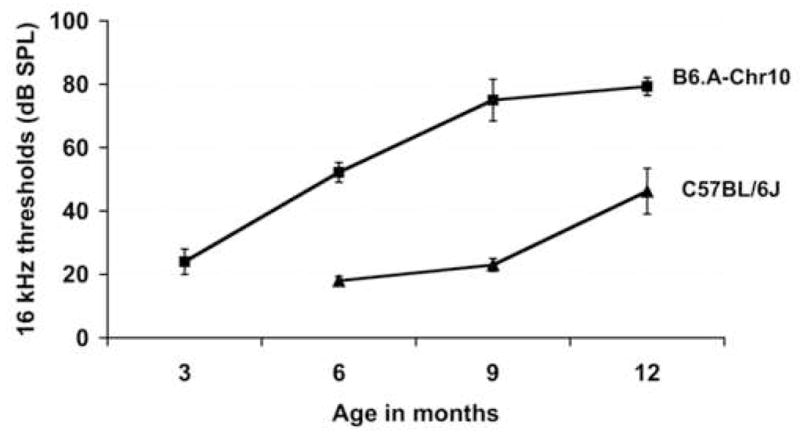
A) ABR threshold variation among 21 B6.A chromosome substitution strains tested between 5 and 7 months of age. Means and standard error bars for 16 kHz thresholds are shown for each strain. A minimum of 8 mice were tested per strain. Note that Chr 10 appears to be the largest contributor to the increased hearing loss of A/J mice relative to B6 mice.
B) The effect of an A/J-derived Chr 10 (containing ahl4) on ABR thresholds of mice with an otherwise B6 genome. Means and standard error bars for 16 kHz ABR thresholds of B6 mice are compared with those of age-matched mice from the B6.A-Chr10 strain. Four or five mice from each strain were tested at each timepoint.
3.4. Genetic analysis of the (A/J x CAST) x A/J backcross
To confirm the effect of the ahl4 locus on ABR thresholds and refine its map position, we analyzed a previously described (A/J x CAST) x A/J backcross (Johnson et al., 2000; Johnson et al., 2001) for markers along the length of Chr 10. We previously reported that the Cdh23ahl locus (ahl) has a major effect on hearing thresholds of (A/J x CAST) x A/J backcross mice (Johnson et al., 2000). Here we refine this analysis using D10Mit138 to mark the ahl locus, and evaluate its effect on hearing loss in 180 N2 mice at 3 and 6 months of age (Fig. 4 panel A). Most N2 mice that were homozygous for the A/J allele at this locus (AA) exhibited elevated hearing thresholds, but thresholds of heterozygous mice (AC) were normal. The ahl locus has a very large effect and can explain about 50% of the total 16 kHz ABR threshold variation exhibited by the N2 mice. The average 16 kHz threshold of N2 mice with AA genotypes at ahl was 49 dB SPL at 3 months of age and 62 dB SPL at 6 months. The average 16 kHz threshold of N2 mice with AC genotypes at ahl was 16 dB SPL at 3 and 6 months of age.
Fig. 4.
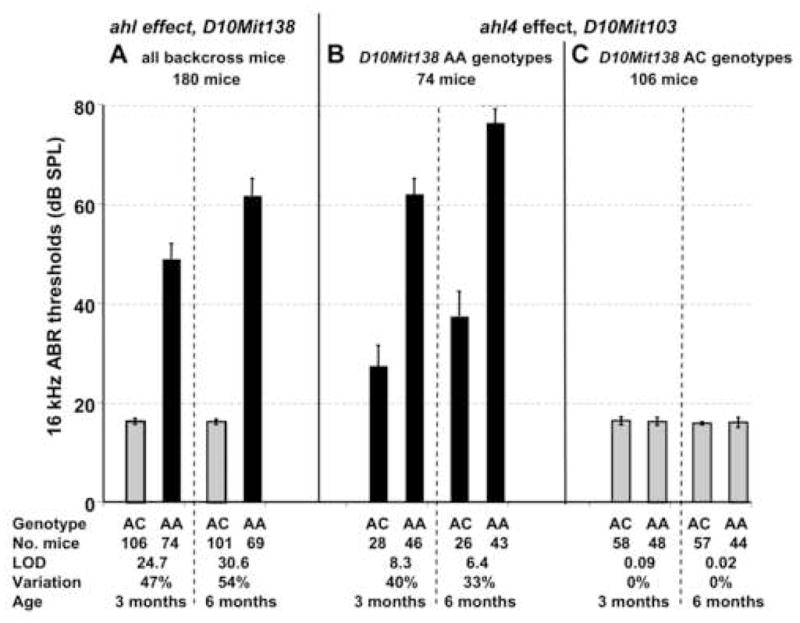
The effects of the ahl and ahl4 loci on ABR thresholds of (A/J x CAST/Ei) x A/J backcross mice. Each bar represents the mean 16 kHz ABR threshold (with its associated standard error) for a given genotype class tested at 3 and 6 months of age. Black bars represent mice with D10Mit138 AA genotypes, and gray bars represent mice with D10Mit138 AC genotypes, where A is the A/J-derived allele and C is the CAST/Ei-derived allele.
A) The ahl locus (marked by D10Mit138) has a highly significant effect on ABR thresholds of (A/J x CAST) x A/J backcross mice. All 180 backcross mice were analyzed for D10Mit138 genotypes (AC, heterozygous for A/J and CAST alleles; AA, homozygous for A/J allele). Note that only mice with an AA genotype at D10Mit138 exhibit elevated thresholds at the ages tested.
B) The ahl4 locus (marked by D10Mit103) has a marked effect on ABR thresholds, but only in backcross mice that are homozygous for the A/J-derived allele at the ahl locus (D10Mit138 AA genotypes). Means and standard error bars for 16 kHz ABR thresholds of N2 backcross mice tested at 3 and 6 months of age. Only the 74 mice that were homozygous for the A/J-derived allele at the D10Mit138 locus were analyzed for threshold associations with D10Mit103 genotypes (AC, heterozygous for A/J and CAST alleles; AA, homozygous for A/J allele). C) The ahl4 locus (marked by D10Mit103) has no effect on ABR thresholds in backcross mice that are heterozygous at the ahl locus (D10Mit138 AC genotypes).
Because the N2 mice that are heterozygous at ahl (AC genotype) exhibit very little threshold variation (Fig. 4A, C), we limited further genetic analysis to the 74 N2 mice that are homozygous for the A/J allele at this locus (AA genotype), which show significant differences in mean ABR thresholds when partitioned by D10Mit103 genotypes (Fig. 4B, Supplementary Table 1). In accordance with the AXBXA RI strain and CS strain results, the (A/J x CAST) x A/J backcross analysis of these mice confirmed a strong association of hearing loss with markers on distal Chr 10 (Fig. 5, Supplementary Table 2). The peak association (LOD = 8.3 at 3 months of age, 6.4 at 6 months of age) was with D10Mit103 at position 125.1 Mb (NCBI Build 36), with the most likely interval containing the ahl4 locus extending from 120 to 130 Mb. For the subset of N2 mice with AA genotypes at ahl, the ahl4 locus can explain about 40% of the 16 kHz threshold variation at 3 months of age and 33% at 6 months (Fig. 4B). The average 16 kHz threshold of N2 mice with AA genotypes at both ahl and ahl4 was 62 dB SPL at 3 months of age and 76 dB SPL at 6 months. The average 16 kHz threshold of N2 mice with AA genotypes at ahl and AC genotypes at ahl4 was 27 dB SPL at 3 months of age and 37 dB SPL at 6 months.
Fig. 5.

The most likely position of ahl4 on mouse Chr 10 as determined from the (A/J x CAST) x A/J backcross. LOD scores for the associations of DNA markers with 16 kHz ABR thresholds of three-month-old N2 mice. To control for the epistatic effect of the ahl locus, only the subset of backcross mice that were homozygous for the A/J-derived allele at the D10Mit138 locus were evaluated (74 mice). Backcross mice heterozygous for A/J and CAST-derived alleles of D10Mit138 exhibited normal thresholds and so were not included in this analysis.
3.5. Pathology of inner ears of A/J mice
Cochlear sections were examined microscopically for an initial gross assessment of cochlear morphology (data not shown). Compared to the normal morphology observed in the postnatal day 30 (P30) B6 cochlea, the P30 A/J cochlea displayed a hair cell lesion in the basal turn of the cochlea. However, the spiral ganglion appeared normal in all turns including the basal turn. In addition, the stria vascularis, Reissner’s membrane and tectorial membrane appeared essentially normal by gross inspection of mid-modiolar cross sections.
To further characterize the hair cell pathology identified in A/J mice at P30, we examined cochlear surface preparation in the mid-basal and mid-apical turns of 9-week-old A/J and B6 mice (Fig. 6), a time point at which the mice have reached sexual maturity and the cochlea is fully developed. In the mid-basal turn, B6 mice (Fig. 6A) showed a full complement of inner hair cells (IHC) and nearly normal population of outer hair cells (OHC). In contrast, A/J mice (Fig. 6C) showed a massive loss of both IHC and OHC in the mid-basal turn and a moderate loss of IHC and OHC in the mid-apical turn (Fig. 6D). These observations indicate that hair cell loss begins near the base of the cochleae and spreads toward the apical turn, a pattern typical of AHL.
Fig. 6.
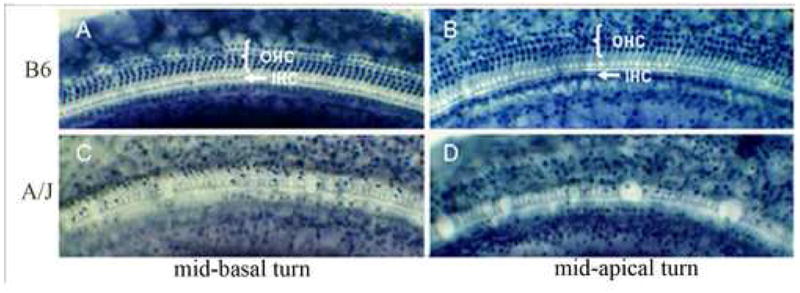
Organ of Corti surface preparations from A/J and B6 mice at two months of age. Photomicrographs of the organ of Corti from the mid-basal and mid-apical turns of 9 week old A/J (C and D) and B6 mice (A and B). Magnification is 1000X.
The cytocochleograms in Figure 7 illustrate the pattern of hair cell loss seen in B6 and A/J mice at 9 weeks and 20 weeks of age. To aid in comparing the hearing loss and hair cell loss data, the frequency-place locations of the 8, 16 and 32 kHz test stimuli are shown by the vertical bars (Ehret, 1975). Nine-week-old A/J mice showed 90–100% OHC loss and ~70% IHC loss over the basal two-thirds of the cochlea (Fig. 7B); these regions are associated with the 8, 16 and 32 kHz stimuli for ABR testing. In contrast, age-matched B6 mice showed only a minor loss of OHC in the extreme base of the cochlea (Fig. 7A), a region that corresponds to none of the test frequencies. At 20 weeks of age, A/J mice showed massive IHC and OHC loss throughout the cochlea (Fig. 7D) except for the apical 10%. In contrast, B6 mice displayed significant OHC loss in the basal 30% of the cochlea and moderate to severe IHC loss in the basal 20% of the cochlea (Fig. 7C); the 32 kHz test frequency lies just within the region of OHC loss.
Fig. 7.

Cytocochleograms showing the percentage of OHC and IHC loss in typical A/J mice at 9 weeks (B) and 20 weeks (D) of age and B6 mice at 9 weeks (A) and 20 weeks (C) of age. Vertical bars show the frequency-to-place map for the 8, 16 and 32 kHz test tones (Ehret, 1975).
To determine the developmental time course of the hair cell loss, we obtained cytocochleograms from 23 A/J mice between P0 to P150. Similar data were obtained from seven B6 cohorts between P0 and P165. For each cytocochleogram, we calculated the percentage of missing OHC and IHC at 0–25%, 25–50%, 50–75% and 75–100% distance from the apex of the cochlea. Scatter plots were constructed showing the percentage of IHC and OHC loss as function of age for A/J and B6 mice (Figure 8A) and the data were fit with a Boltzmann function (GraphPad Prism 5.01). A/J mice showed no evidence of OHC loss between P0–P8. However, OHC loss in the 75–100% region increased from around 20% at P16 to 100% at P42 (Figure 8A, left column). In the 50–100% region, OHC loss increased from roughly 10% at P16 to approximately 90% at P42. OHC loss in the 25–50% region reached 90% around P150 whereas OHC loss in the 0–25% regions developed more slowly. OHC data from the seven B6 mice are shown in Figure 8A (right) along with mean (n=10) data from B6 mice from an earlier study from our lab (Spongr et al., 1997). Between P0 and P165, OHC lesions were negligible (<20%) except for the 75–100% regions where the OHC lesion slowly increased from around 20% at P65 to 90% at P165.
Fig. 8.


(A) Scatter plots showing the percentage of OHC missing as a function of age at locations 0–25%, 25–50%, 50–75% and 75–100% distance from the apex of the cochlea. Boltzmann sigmoidal function fit to the data (GraphPad Prism 5). Left column and right columns display data for A/J and B6 mice respectively. Filled circles show data from individual animals in the current study. Open squares in right column plots show mean (n=10) data from B6 mice from Spongr et al. (1998). (B) Scatterplots for the percentage of IHC missing as in Figure 8A.
Figure 8B shows the time course of IHC loss for A/J and B6 mice. In A/J mice, IHC lesions in the 75–100% region increased from around 18% at P42 to roughly 78% at P63. IHC lesions in A/J mice developed in a pattern similar to the most basal region in the 50–75% and 25–50% regions, but the onset of the loss was delayed by 15–20 days. Little IHC loss occurred in the 0–25% region even as late as P150. IHC lesions were negligible in B6 mice except in the 75–100% region where the loss increased to 35% at P165. These results show that hair cell loss in A/J mice begins postnatally around P16 and that OHC are more vulnerable than IHC, Furthermore the lesion spreads from base to apex and that the losses are much more severe in A/J mice than B6 mice over the age range examined.
To investigate whether the early onset of hearing loss might be the result of gross stereocilia disarray, cochleae from P0 and P8 A/J mice were labeled with FITC-phalloidin that heavily labels F-actin in the stereocilia and circumferential ring of the cuticular plate (Data not shown). Propidium iodide was used to label the nuclei. The IHC and OHC were arranged in orderly rows in P0 and P8 cochleae. The stereocilia bundles on the IHC and OHC appeared qualitatively normal and no gross abnormalities were observed at these ages. Moreover, the hair cell nuclei were large, round and homogenously labeled with propidium iodide, features characteristic of healthy cells (Ding et al., 2007).
To determine if hair cell lesions were also present in the vestibular sensory epithelium of A/J mice, we dissected out the sensory epithelium from the crista ampullaris and macula of the utricle as a flat surface preparation and stained the specimens with hematoxylin. At 5 months of age, hair cell density in the crista ampullaris and macula of the utricle of A/J mice appeared normal and similar to that seen in the crista and macula of B6 mice (data not shown).
To determine if the hair cell lesions seen in A/J mice were accompanied by significant loss of spiral ganglion neurons, we examined mid-modiolar cross-sections of cochleae of A/J and B6 mice at 9 weeks and 20 weeks of age (5 months). At 9 weeks, 70–80% of the IHC were missing in the basal half of the A/J cochleae, whereas less than 5% were missing in B6 cochleae (Fig. 8B). Cross sections through the basal turn showed hair cell loss in a 9-week-old A/J mouse (Fig 9A) and intact hairs cells in a B6 mouse (Fig. 9B); Despite the loss of hair cells in the A/J cochleae, the density of spiral ganglion neurons in A/J (Fig 9A) and B6 cochleae (Fig. 9B) appeared similar. The stria vascularis in 9-week-old A/J mice also appeared qualitatively similar to that of B6 mice. At 5 months of age, nearly all of the IHC were missing in the basal half of the A/J cochlea (Fig. 8B) whereas IHC losses in the basal 25% of the B6 cochleae ranged from 15–60%. Despite the massive loss of hair cells in the basal half of the A/J cochlea, the density and appearance of the spiral ganglion neurons in this region were similar to those in B6 mice (Fig. 9C, D).
Fig. 9.

Cross sections (3 μm) through the modiolus of cochleae from a 9-week-old A/J (A) and B6 (B)mouse. Hair cells were missing in the A/J mouse, but present in the B6 mouse. The density of SGN in the A/J mouse was similar to that in the B6 mouse (400X). Cross sections (3 μm) through Rosenthal’s canal at mid-basal turn (represents 2–4 kHz) shown in 5-month-old A/J (C) and B6 (D) mice; 1000X magnification. The density and gross appearance of the SGN in the A/J cochlea were similar to the B6 cochlea.
Nerve fiber density in the habenular perforata was also examined in 9-week- and 5-month-old A/J and B6 mice. Nerve fiber densities in the basal half of the cochlea of 9-week- (Fig. 10A) and 5-month-old (Fig. 10B) A/J mice appeared normal and qualitatively similar to those from B6 mice of the same age (Fig. 10C–D).
Fig. 10.

Cross sections (3 μm) taken through the habenula perforata at mid-basal turn (represents 2–4 kHz) showing the nerve fiber density in a 9-week- and a 5-month-old A/J (A, B) and B6 mouse (C, D) respectively (1000X magnification). Nerve fiber density in habenular openings of A/J mice was similar to the density in B6 mice of the same age.
4. Discussion
4.1. ahl4, a new locus affecting age-related hearing loss in A/J mice
The combined results from the genetic analyses of the AXBXA RI strains, the B6.A CS strains, and the (A/J x CAST) x A/J backcross provide compelling evidence for a QTL on distal Chr 10, designated ahl4, that contributes significantly to the early onset of AHL in A/J strain mice. The map position of this new locus is distinct from the chromosomal locations of all previously reported AHL loci in inbred mouse strains, including ahl (Johnson et al., 1997), ahl2 (Johnson et al., 2002), ahl3 (Nemoto et al., 2004), ahl5 and ahl6 (Drayton et al., 2006), and Phl1 and Phl2 (Mashimo et al., 2006). Although Phl2 was mapped to Chromosome 10, its map position between D10Mit170 (47.8 Mb, NCBI build 36) and D10Mit115 (69.7 Mb) is clearly distinct from that of ahl4, which is distal to 110 Mb (Fig. 5). The ahl4 locus on distal Chr 10 showed no association with ABR thresholds in previously described linkage backcrosses involving CAST/Ei and the following strains: BUB/BnJ, DBA/2J, NOD/LtJ, 129P1/ReJ, SKH2/J, BALBcByJ, and B6 (Johnson et al., 2000). In contrast to the Cdh23ahl allele, which is common among multiple inbred strains (Johnson et al., 2000; Noben-Trauth et al., 2003), the ahl4 allele conferring increased hearing-loss susceptibility appears to be unique to the A/J inbred strain.
Analysis of the (A/J x CAST) x A/J backcross showed that heterozygosity at the ahl locus (AC genotype, Fig. 4) precludes ahl4 manifestation. N2 mice with AC genotypes retain normal hearing even at 6 months of age (16 kHz ABR threshold mean = 16.3 dB SPL, standard deviation = 4.5); therefore, nearly all of the ABR threshold variation among N2 mice is limited to those with homozygous AA genotypes (mean = 61.8 dB SPL; standard deviation = 28.2). Within this subgroup, the ahl4 locus has a large effect and can explain up to 40% of the threshold variation (LOD 8.3) at three months of age and up to 33% (LOD 6.4) at 6 months (Fig. 4B). The ahl locus has been shown to have an epistatic effect on other AHL-related loci, including ahl2 (Johnson et al., 2002) and mt-Tr (Johnson et al., 2001), and a modifying effect on the hearing impairment caused by some mutations (Johnson et al., 2006), including Atp2b2dfw (Zheng et al., 2001) and Gpr98frings (Johnson et al., 2005).
The candidate interval surrounding ahl4 (Fig. 5) extends from 120 to 130 Mb on Chr 10 (NCBI Build 36). This genomic region contains more than 100 genes. One of these genes, Myo1a (127.1 Mb), is a particularly attractive candidate for ahl4 because mutations of the orthologous human gene were shown to underlie the progressive nonsyndromic deafness disorder DFNA48 (Donaudy et al., 2003). To examine inbred mouse strains for DNA alterations of Myo1a, we searched for previously identified single nucleotide polymorphisms (SNPs) using the Mouse Phenome SNP Database (http://www.jax.org/phenome/snp.html). This database is derived from many sources, including SNPs identified by the Perlegen/NIEHS project (http://mouse.perlegen.com/mouse/resequencing) to resequence 16 mouse strains, including A/J and B6. The search retrieved only seven SNPs within the Myo1a gene that differ between A/J and B6, and none of them alters the protein coding sequence or splice sites. Although Myo1a was shown to be expressed in the inner ear (Donaudy et al., 2003), knockout mice appear to have normal hearing and vestibular function (Tyska et al., 2005). Thus, Myo1a is unlikely to be the gene responsible for ahl4.
To aid in the identification of the ahl4 gene, efforts are now being made to narrow the candidate region by generating multiple B6 congenic strains harboring different A/J-derived subregions of Chr 10 and testing their effects on AHL. The time and effort required to produce congenic strains is greatly reduced by starting with a specific CS strain as donor (Nadeau et al., 2000). ABR thresholds of B6.A-Chr10 mice are 30 dB above those of B6 mice by 6 months of age (Fig. 3B), indicating that the ahl4 effect on hearing is not dependent on interactions with loci on other chromosomes and supporting the feasibility of the congenic strain approach to isolate the ahl4 locus.
4.2. Rapid progression of cochlear hair cell loss in A/J mice
Cochlear hair cell loss occurred earlier and progressed more rapidly in A/J mice than in B6 mice (Fig. 8A–B). Hair cell densities were lower in 9-week-old A/J mice than in 20-week-old B6 mice. A/J mice show minimal loss of OHC and IHC at postnatal day 8, but by 9 weeks of age, massive OHC and IHC loss was seen in the basal half of the cochlea. These observations are in agreement with an earlier report of extensive hair cell loss in aging A/J mice (Chole et al., 1983). Hearing loss of A/J mice selectively starts with high frequencies, which corresponds to the progression of cochlear hair cell loss from base to apex. Although cochlear hair cell loss was extensive, hair cell density in the vestibular system appeared normal in 5-month-old A/J mice. Cross sections through A/J cochleae failed to reveal any gross morphological damage to the stria vascularis or other non-sensory structures. Except for hair cell loss, the gross morphological features of the A/J mouse cochlea appeared qualitatively similar to that of the B6 mouse of the same age (Fig. 9).
IHC loss progressed more slowly than OHC loss. The delayed IHC loss could be the result of defective gene(s) that are expressed at higher levels in OHCs than IHCs. By 40–50 days of age, most of the IHC were missing in the basal half of the cochlea (Fig. 8B) whereas most of the spiral ganglion neurons and nerve fibers in this region were still present and appeared normal (Fig 9–10). The most parsimonious explanation for the survival of spiral ganglion neurons in the basal half of the cochlea is that they simply have not had sufficient time to undergo secondary degeneration due to the loss of trophic support from missing hair cells in the basal half of the cochlea (Fritzsch et al., 2004). Alternatively, the residual IHC and OHC present in the apical half of the A/J cochlea could promote the survival of spiral ganglion neurons in the base by release of neurotrophic substance such as NT-3 and BDNF. However, this would require the diffusion of these neurotrophic factors from the apical toward the basal half of the cochlea (Fritzsch et al., 2004). The rapid progression of hearing loss but retention of spiral ganglion cells suggest that A/J mice could potentially serve as an animal model for cochlear implantation (Kretzmer et al., 2004), because a minimal density of spiral ganglion cells is required for effective cochlear implants (Wake et al., 1998).
In summary, we have genetically mapped a locus (ahl4) on distal Chr 10 that is a significant contributor to the early onset AHL of A/J mice. The major pathological effect of ahl4 appears to be an increase in the progression of cochlear hair cell loss. Identification of the ahl4 gene and determination of its function will help elucidate molecular mechanisms involved in the cochlear degeneration associated with presbycusis.
Supplementary Material
Acknowledgments
This research was supported by NIH grants DC005827 (KRJ), DC007392 (QYZ). We thank Dr. Cindy Benedict-Alderfer for assistance in preparation of this manuscript.
Footnotes
Disclosure Statements
a) There are no actual or potential conflicts of interest regarding the authors of this manuscript and the work described herein.
b) All mice were from The Jackson Laboratory (TJL) research facilities and all procedures were approved by the Institutional Animal Care and Use Committee.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chole RA, Henry KR. Disparity in the cytocochleogram and the electrocochleogram in aging LP/J and A/J inbred mice. Audiology. 1983;22:384–392. doi: 10.3109/00206098309072798. [DOI] [PubMed] [Google Scholar]
- DeStefano AL, Gates GA, Heard-Costa N, Myers RH, Baldwin CT. Genome-wide linkage analysis to presbycusis in the Framingham Heart Study. Arch Otolaryngol Head Neck Surg. 2003;129:285–289. doi: 10.1001/archotol.129.3.285. [DOI] [PubMed] [Google Scholar]
- Ding D, McFadden SL, Salvi R. Cochlear hair cell densities and inner-ear staining techniques. In: Willott JF, editor. Handbook of Mouse Auditory Research from Behavior to Molecular Biology. CRC Press; Boca Raton: 2001. pp. 189–204. [Google Scholar]
- Donaudy F, Ferrara A, Esposito L, Hertzano R, Ben-David O, Bell RE, Melchionda S, Zelante L, Avraham KB, Gasparini P. Multiple mutations of MYO1A, a cochlear-expressed gene, in sensorineural hearing loss. Am J Hum Genet. 2003;72:1571–1577. doi: 10.1086/375654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayton M, Noben-Trauth K. Mapping quantitative trait loci for hearing loss in Black Swiss mice. Hear Res. 2006;212:128–139. doi: 10.1016/j.heares.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Ehret G. Masked auditory thresholds, critical ratios, and scales of the basilar membrane of the house mouse (mus musculus) J Comp Physiol. 1975;103:329–341. [Google Scholar]
- Erway LC, Willott JF, Archer JR, Harrison DE. Genetics of age-related hearing loss in mice: I. Inbred and F1 hybrid strains. Hear Res. 1993;65:125–132. doi: 10.1016/0378-5955(93)90207-h. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Beisel KW. Keeping sensory cells and evolving neurons to connect them to the brain: molecular conservation and novelties in vertebrate ear development. Brain Behav Evol. 2004;64:182–197. doi: 10.1159/000079746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Toriello HV, Cohen MM. Hereditary Hearing Loss and Its Syndromes. Oxford University Press; New York, Oxford: 1995. [Google Scholar]
- Greenwood DD. A cochlear frequency-position function for several species--29 years later. J Acoust Soc Am. 1990;87:2592–2605. doi: 10.1121/1.399052. [DOI] [PubMed] [Google Scholar]
- Henry KR. Age-related auditory loss and genetics: an electrocochleographic comparison of six inbred strains of mice. J Gerontol. 1982;37:275–282. doi: 10.1093/geronj/37.3.275. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY. Ahl2, a second locus affecting age-related hearing loss in mice. Genomics. 2002;80:461–464. [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70:171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Erway LC, Cook SA, Willott JF, Zheng QY. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res. 1997;114:83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fischel-Ghodsian N. A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet. 2001;27:191–194. doi: 10.1038/84831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Weston MD, Ptacek LJ, Noben-Trauth K. The Mass1(frings) mutation underlies early onset hearing impairment in BUB/BnJ mice, a model for the auditory pathology of Usher syndrome IIC. Genomics. 2005;85:582–590. doi: 10.1016/j.ygeno.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Noben-Trauth K. Strain background effects and genetic modifiers of hearing in mice. Brain Res. 2006;1091:79–88. doi: 10.1016/j.brainres.2006.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keithley EM, Canto C, Zheng QY, Fischel-Ghodsian N, Johnson KR. Age-related hearing loss and the ahl locus in mice. Hear Res. 2004;188:21–28. doi: 10.1016/S0378-5955(03)00365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzmer EA, Meltzer NE, Haenggeli CA, Ryugo DK. An animal model for cochlear implants. Arch Otolaryngol Head Neck Surg. 2004;130:499–508. doi: 10.1001/archotol.130.5.499. [DOI] [PubMed] [Google Scholar]
- Li HS, Borg E. Age-related loss of auditory sensitivity in two mouse genotypes. Acta Otolaryngol 1991. 1991;111(5):827–34. doi: 10.3109/00016489109138418. [DOI] [PubMed] [Google Scholar]
- Manly KF, Cudmore RH, Jr, Meer JM. Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome. 2001;12:930–932. doi: 10.1007/s00335-001-1016-3. [DOI] [PubMed] [Google Scholar]
- Mashimo T, Erven AE, Spiden SL, Guenet JL, Steel KP. Two quantitative trait loci affecting progressive hearing loss in 101/H mice. Mamm Genome. 2006;17:841–850. doi: 10.1007/s00335-004-2438-5. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Reaume AG, Flood DG, Salvi RJ. Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol Aging. 1999a;20:1–8. doi: 10.1016/s0197-4580(99)00018-4. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Burkard RF, Jiang H, Reaume AG, Flood DG, Salvi RJ. Cu/Zn SOD deficiency potentiates hearing loss and cochlear pathology in aged 129, CD-1 mice. J Comp Neurol. 1999b;413:101–112. [PubMed] [Google Scholar]
- Morton NE. Genetic epidemiology of hearing loss. Ann NY Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Singer JB, Matin A, Lander ES. Analysing complex genetic traits with chromosome substitution strains. Nat Genet. 2000;24:221–225. doi: 10.1038/73427. [DOI] [PubMed] [Google Scholar]
- Nemoto M, Morita Y, Mishima Y, Takahashi S, Nomura T, Ushiki T, Shiroishi T, Kikkawa Y, Yonekawa H, Kominami R. Ahl3, a third locus on mouse chromosome 17 affecting age-related hearing loss. Biochem Biophys Res Commun. 2004;324:1283–1288. doi: 10.1016/j.bbrc.2004.09.186. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK. Contributions of mouse models to understanding of age- and noise-related hearing loss. Brain Res. 2006;1091:89–102. doi: 10.1016/j.brainres.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Sampson SB, Higgins DC, Elliot RW, Taylor BA, Lueders KK, Koza RA, Paigen B. An edited linkage map for the AXB and BXA recombinant inbred mouse strains. Mamm Genome. 1998;9:688–694. doi: 10.1007/s003359900849. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol. 1993;102:1–16. doi: 10.1177/00034894931020S101. [DOI] [PubMed] [Google Scholar]
- Singer JB, Hill AE, Burrage LC, Olszens KR, Song J, Justice M, O’Brien WE, Conti DV, Witte JS, Lander ES, Nadeau JH. Genetic dissection of complex traits with chromosome substitution strains of mice. Science. 2004;304:445–448. doi: 10.1126/science.1093139. [DOI] [PubMed] [Google Scholar]
- Spongr VP, Flood DG, Frisina RD, Salvi RJ. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J Acoust Soc Am. 1997;101:3546–3553. doi: 10.1121/1.418315. [DOI] [PubMed] [Google Scholar]
- Tyska MJ, Mackey AT, Huang JD, Copeland NG, Jenkins NA, Mooseker MS. Myosin-1a is critical for normal brush border structure and composition. Mol Biol Cell. 2005;16:2443–2457. doi: 10.1091/mbc.E04-12-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake M, Takeno S. Spiral ganglion degeneration following induction of inner hair cell lesions. Clin Otolaryngol. 1998;23:272–273. [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-11-research0046. RESEARCH0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott JF, Turner JG, Carlson S, Ding D, Seegers Bross L, Falls WA. The BALB/c mouse as an animal model for progressive sensorineural hearing loss. Hear Res. 1998;115:162–174. doi: 10.1016/s0378-5955(97)00189-5. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR. Hearing loss associated with the modifier of deaf waddler (mdfw) locus corresponds with age-related hearing loss in 12 inbred strains of mice. Hear Res. 2001;154:45–53. doi: 10.1016/s0378-5955(01)00215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.