

Abstract
Excessive water uptake through aquaporins can be life threatening, and disregulation of water permeability causes many diseases. Therefore, reversible aquaporin inhibitors are highly desired. In this paper, we identified the binding site for tetraethylammonium (TEA) of the membrane water channel aquaporin-1 by a combined molecular docking and molecular dynamics simulation approach. The binding site identified from docking studies was independently confirmed with an unbiased molecular dynamics simulation of an aquaporin tetramer embedded in a lipid membrane, surrounded by a 100-mM tetraethylammonium solution in water. A third independent assessment of the binding site was obtained by umbrella sampling simulations. These simulations, in addition, revealed a binding affinity of more than 17 kJ/mol, corresponding to an IC50 value of << 3 mM. Finally, we observed in our simulations a 50% reduction of the water flux in the presence of TEA, in agreement with water permeability measurements on aquaporin expressed in oocytes. These results confirm TEA as a putative lead for an aquaporin-1 inhibitor.
Keywords: Water, Inhibition, Membrane, Tetraethylammonium, Transport
Introduction
Aquaporins (AQPs) are integral membrane proteins that facilitate passive and highly efficient water transport across biological membranes [19, 27]. They form a large family of integral membrane channels together with the aquaglyceroporins (e.g., the bacterial glycerol facilitator GlpF), which also facilitate the permeation of small molecules such as glycerol and urea. In the human body, 13 aquaporins and aquaglyceroporins have been identified. They are expressed in tissues such as kidney, brain, lung, eye, and red blood cells. Malfunction of these proteins may lead to water balance disorders, which can severely affect the quality of life and can even be life threatening. Aquaporins have been shown to play an important role in diseases that are characterized by excessive water transport such as glaucoma, brain edema, nephrogenic diabetes insipidus (NDI), tumor growth, and cancer [9, 13, 23].
Considering the important role of aquaporins in those diseases, reversible AQP-specific blockers as potential drugs are clinically highly relevant, but not yet available. Mercury, silver, and gold have been shown to inhibit AQPs [17, 20], but the inhibition is not reversible and these metals are highly toxic. Recently, tetraethylammonium (TEA) was suggested as a putative inhibitor for hAQP1, but its binding affinity and potential inhibition of water permeability was controversially discussed in the literature [6, 25, 26]. In oocyte systems, TEA shows potential inhibition for hAQP1 [4, 26], whereas no effect of TEA on AQP1 was observed in erythrocytes and epithelial cell cultures [25]. It is important to note that binding does not necessarily imply inhibition. We present in this paper independent approaches that address the identification of a putative binding site for TEA in hAQP1, the binding affinity for TEA, as well as the effect of bound TEA on hAQP1 water permeability on an atomistic level.
Methods
The simulation system (Fig. 1) setup, the docking of TEA to hAQP1, and molecular dynamics (MD) simulations starting from the putative docking sites were described in detail in a previous publication [6]. We simulated hAQP1 in the deglycosylated form to mimic experimental conditions where none or at most one monomer [20] is glycosylated. For the reader's convenience, the main steps are summarized here.
Fig. 1.

a Simulation system of an hAQP1 tetramer (green, gray, red, and yellow space-filling representation) embedded in a POPE lipid bilayer (gray) and surrounded by water (red, white). b Starting positions for TEA_dockMD as described in the text. The tetramer is represented as cartoons and TEA in space-filling representation. c Starting positions for TEA_20random. Twenty TEA molecules were randomly placed in the bulk water. d TEA binding site defined via the distribution of TEA nitrogen positions obtained from TEA_dockMD (gray spheres), TEA_20random (magenta spheres), and umbrella sampling (orange spheres)
Docking of TEA
The molecular docking package DOCK 4.0 [7] was used to dock TEA into the X-ray structure of bovine AQP1 (Protein Data Bank code 1J4N). The DOCK 4.0 package generated poses in the defined docking region in the upper part of the vestibule. The poses were scored and ranked using the energy scoring function of DOCK 4.0. The 100 best ranked positions were visually inspected. Four representative positions of TEA were used as starting positions for subsequent MD simulations [6].
Stability test for TEA
All MD simulations were based on the X-ray structure of bovine AQP1. The structure was modified to adopt the structure of human AQP1 using the WHATIF package [24]. The sequence identity between human and bovine AQP1 is 90.63%. None of the 21 mutations and two insertions constituting the difference between human and bovine AQP1 is located in the pore region. AQP1 was simulated as a tetramer embedded in a solvated palmitoyloleoyl phosphatidylethanolamine (POPE) lipid bilayer. The simulation system contained 8,340 protein atoms, 14,093 lipid atoms, 19, 769 SPC water molecules [2], and four chloride ions to neutralize the simulation system (see Fig. 1a). MD simulations were carried out using the Gromacs simulation suite version 3.1.4 [15]. Lincs and Settle [8, 16] were applied to constrain covalent bond lengths, allowing an integration time step of 2 fs. Electrostatic interactions were calculated every step using the Particle Mesh Ewald method [8]. The temperature was kept constant by separately coupling the protein, lipids, ions, and solvent to an external temperature bath (τ = 0.1 ps) [1]. The pressure was kept constant by weak coupling (τ = 1.0 ps) in the z-direction (normal to the bilayer plane) to a pressure bath [1]. The Gromacs force field, which is the Gromos87 force field [22] with slight modifications [21] and explicit hydrogens on aromatic residues, was applied. The simulation setup and conditions were identical to those described before [5]. As the starting configuration for the subsequent simulations, a snapshot from this simulation was taken.
To equilibrate the system, 500 ps of MD were performed with harmonic position restraints on all non-hydrogen protein atoms (k = 1,000 kJ/(mol nm2)). All subsequent simulations, except for the umbrella simulations and water permeability simulations (see below), started from the resulting structure.
For the identification of the TEA binding site and a comparison of the water mobility without TEA and with bound TEA, three simulations were performed, each of 15 ns length. The first simulation was carried out without TEA to obtain a reference water mobility along the pore axis of hAQP1. For the second simulation, TEA was placed at the four different docking positions identified above, within the monomeric channels of the tetramer. Water molecules that showed a significant overlap with TEA were removed after TEA insertion. The system was reequilibrated for 500 ps with position restraints (k = 1,000 kJ/(mol nm2)) on the central nitrogen atom of the TEA, before the production run started. Only in one of the four positions (see [6]) did TEA remain stably bound to hAQP1. Therefore, a second simulation was set up (TEA_dockMD), with TEA molecules placed at this position in all four monomers of the aquaporin (see Fig. 1b).
First principles docking
To independently predict the TEA binding site, we developed a MD-based first principles approach. The aim of this approach is to find spontaneous binding events of TEA on hAQP1 in an unbiased MD simulation. To this end, 20 TEA molecules were placed at randomly chosen positions in bulk water (see Fig. 1c), generating a TEA solution of 100 mM. The large TEA concentration was chosen to raise the statistical significance of the observed (partial) binding events. All water molecules that overlapped with TEA were removed. From this starting system, a 100 ns free MD simulation (TEA_20random) was carried out.
Definition of the binding site
To distinguish bound from unbound TEA in the TEA_20random simulation, a definition of the TEA binding site in hAQP1 is required. Because of the large structural and orientational heterogeneity of the binding site (see “Results and discussion”), the nitrogen TEA positions from TEA_dockMD (see Fig. 1d, gray spheres) were chosen as a reference. The TEA nitrogen distribution was obtained with respect to a reference monomer chosen from the starting structure of TEA_dockMD by the following protocol: (1) backbone fit of the helical part of each monomer to the reference monomer, including the TEA; (2) store the resulting TEA nitrogen positions; (3) perform a nearest neighbor search with a cutoff of 1 Å for each TEA nitrogen position; (4) exclude rarely visited areas or nearly unbound TEA molecules by selecting only those TEA nitrogen positions with more than three TEA neighbors in a 1-Å distance.
To probe binding for TEA molecules in TEA_20random, the nitrogen positions of TEA from this simulation were obtained following steps (1) and (2) above. As a first estimate, we define each TEA molecule from TEA_20random bound if the TEA nitrogen is closer than 5 Å to the reference TEA nitrogen distribution from TEA_dockMD. To exclude positions that do not significantly contribute to binding, only TEA nitrogen positions were selected with more than ten TEA neighbors in a 1 - Å distance (Fig. 1d, magenta spheres).
Umbrella sampling
To quantify the TEA binding affinity, umbrella sampling simulations were set up as follows. The centers of the umbrella potentials were chosen in steps of 0.25 Å perpendicular to the membrane plane, ranging from the AQP pore entrance into the bulk water. For each umbrella center, a random snapshot was chosen from a 20-ns equilibrium simulation of hAQP1 and used as starting configuration. For each of the four monomeric channels, a TEA molecule was placed at the umbrella center. Water molecules that overlapped with TEA were removed.
The simulation box contained the hAQP1 tetramer and four TEA molecules solvated in a membrane of 271 POPE lipids and approximately 19,300 TIP4P water molecules [10]. The OPLS all-atom force field [11, 12] was applied to describe the protein, and the lipid parameters were taken from Berger et al. [3]. The simulation parameters were chosen as described in the section on the stability test (see above). Umbrella simulations were carried out by restraining the nitrogen atom of TEA with a harmonic umbrella potential. A force constant of 1,000 kJ/(mol nm2) was applied.
In addition, the TEA nitrogens were confined into a cylinder (radius rc = 11 Å) of which the axis was centered along the corresponding monomeric pore. The cylindrical confinement was implemented via an additional force F (r) = −kc(r − rc) H(r − rc) pointing towards the cylinder axis. Here, r denotes the distance of the TEA nitrogen from the cylinder axis, kc = 400 kJ/(mol nm2) the force constant, and H the Heaviside step function. Umbrella simulations with the TEA close to the binding region (30 umbrella sections) were run for 6 ns each, simulations with the TEA in bulk water or in loose contact with AQP loops were run for 600 ps each. Umbrella histograms collected from the simulations were corrected in case of fluctuations of the corresponding monomer within the AQP tetramer.
As reference, the z-coordinate (perpendicular to the membrane) of the center of mass of the backbone atoms of the Asn-Pro-Ala (NPA) motifs was applied and defined as z = 0 in Fig. 2. For each of the pores, a free-energy profile was computed separately using the weighted histogram analysis method [14]. Subsequently, the probability for a TEA to be located at some position z along the reaction coordinate was averaged over the four monomers. To this aim, the four profiles Gi(z) were combined into an effective profile Geff (z) via exp(−Geff (z)/kBT) = (1/4)∑ i4 = 1 exp(−Gi(z)/kBT). Here, T = 300 K denotes the temperature and kB the Boltzmann constant.
Fig. 2.

Potential of mean force for TEA binding along the channel axes in the upper vestibule of the water pore. The inset shows IC50 values computed from PMFs for different equilibration times. For a detailed description, see text
In addition, the location of the TEA binding site was determined from the umbrella simulations. The location of the TEA nitrogen (see Fig. 1d, orange spheres) was extracted from three umbrella simulations, with the TEA located around the minimum in Geff (z). The last 2 ns of these three 6-ns simulations were analyzed. The TEA positions at the four monomers were combined into an effective binding site. The number of TEA positions at a monomer i that contribute to the effective binding site was chosen proportional to the probability exp(−Gi(z)/kBT) for the TEA to be located at the position z in monomer i.
IC50 estimates
From the effective profile Geff (z) the IC50 concentration was calculated, i.e., the TEA concentration where the TEA binding probability is 50%. To this end, the TEA was considered as bound if the distance z between the NPA site and the TEA nitrogen was smaller than zb = 25 Å (compare Fig. 2). As the TEA nitrogen was confined within a cylinder of radius rc during the umbrella simulations, the IC50 estimation reduces to the determination of the length L (or volume πrc2L) of the cylinder that leads a 50% binding probability.
The probability for the TEA for being bound is given by 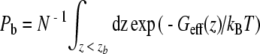 . The probability for being unbound is
. The probability for being unbound is 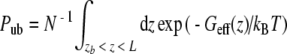 . Here, N is a normalization constant. Applying Pb = Pub allows to solve for L and to determine the corresponding TEA concentration IC50 = 1/(πrc2L).
. Here, N is a normalization constant. Applying Pb = Pub allows to solve for L and to determine the corresponding TEA concentration IC50 = 1/(πrc2L).
Permeability estimates
To quantify the inhibitory effect of TEA, an equilibrium simulation of hAQP1 with TEA bound into the binding site was compared to a reference simulation without any TEA present. The same simulation system, force field, and simulation parameters as for the umbrella simulations were chosen, except that no umbrella potential and cylindrical confinement was applied. The simulations with and without TEA were run for 10 and 20 ns, respectively. As a measure for water permeability, the number of water molecules were counted that permeated completely across a single-file section of the pore of 9.5 Å length including the NPA site and the aromatic/arginine constriction region. This pore section displays the lowest water diffusion constant (data not shown), thus limiting the water flux. Hence, the number of complete permeation events across this section is directly expected to be proportional to the osmotic permeability coefficient pf and the diffusive permeability coefficient pd.
Results and discussion
Identification of the binding site for TEA
From a previously published set of simulations of TEA binding to hAQP1 [6], we first characterized in detail the TEA binding site. From the combined docking/stability test MD approach, it was found that TEA exhibits an unusually large structural heterogeneity, both for the protein and for the TEA positions and orientations. Rather than representing the bound state by one structure, as usually done, we therefore represented the binding site as a distribution of all TEA nitrogen atom positions (see Fig. 1d, gray spheres) from the trajectory of the simulation in which TEA was bound to each of the four channels for the duration of the simulation (TEA_dockMD). The TEA distribution for TEA_dockMD was obtained as described in the “Methods” section. A 3-Å radius around the TEA distribution from TEA_dockMD includes parts of the C-loop (especially ASP 128 and ASP 131), parts of the E-loop (especially ASP 185), and the A-loop of the neighboring monomer, which were described as a putative binding site for TEA in hAQP1 before [6]. It was observed that the flexible A-loop of the neighboring monomer seems to function as a lid, which stabilizes TEA binding. This binding site, identified from TEA_dockMD simulation, was compared to the TEA distribution obtained from a 100-ns MD simulation with 20 TEA placed at random in bulk water (TEA_20random), which corresponds to a TEA concentration of about 100 mM. During the simulation time, three binding and two unbinding events of TEA to the previously determined binding site occurred.
In the TEA_20random simulation (Fig. 1d, magenta spheres) the binding sites in three different monomers were occupied by TEA for ∼80%, ∼30%, and ∼15% of the simulation time, respectively.
The fact that the two complementary and independent approaches identified similar binding sites provides strong support for this binding site. The overlap is not complete, however. Each of the two methods also suggests putative binding sites, which are not identified by the other method. These differences indicate insufficient sampling in at least one but likely both of the approaches, rendering a straightforward affinity estimate for TEA binding to hAQP1 from probability densities problematic. Therefore, we employed umbrella sampling simulations as a third, independent approach to assess the binding affinity.
Binding affinity
Figure 2 displays the potential of mean force (PMF) for TEA moving into the pore of hAQP1, as derived from umbrella sampling simulations. The solid curve shows the PMF that was derived by sampling each umbrella window for 1 ns after an equilibration of 5 ns. The PMF displays a clear minimum at z ∼ 18 Å. The TEA nitrogen distribution obtained from the umbrella simulations (Fig. 1d, orange spheres) overlaps significantly with the TEA nitrogen distribution from TEA_dockMD (Fig. 1d, gray spheres). This is the third independent identification of the TEA binding site in hAQP1.
Conformational motions of loops occur typically on timescales of tens of nanoseconds and may therefore be insufficiently sampled in nanoseconds umbrella simulations. In particular, the A-loop conformation of the bound state might not be converged, and therefore the actual binding affinity might be larger. To investigate this effect, we calculated the PMF from the 6 ns of simulation with an increasing time for equilibration before analysis. Between 1 and 5 ns in steps of 0.5 ns were removed at the beginning of the umbrella trajectories, while the remaining parts of the trajectories were used for the computation of the ‘non-equilibrium’ PMFs (dashed lines in Fig. 2). The position of the minimum does not change with equilibration time and therefore reflects the (converged) location of the binding site. The binding affinity is observed to increase with simulation length, with an increasingly pronounced minimum in the PMF around z ∼ 18 Å. These findings demonstrate that the TEA binding site rearranges with time in the presence of TEA, displaying a clear example of induced-fit binding at the atomic level.
The inset of Fig. 2 quantifies the induced fit. The graph displays IC50 concentrations, i.e., TEA concentrations at which 50% of the hAQP1 monomers bind a TEA molecule. The IC50 values are shown as a function of the inverse equilibration time 1/tequil that was used for the calculation of the respective PMF. These IC50 concentrations are rather large, in the range of several millimolars. In the experiments from Detmers et al. [6], the IC50 is measured as 1.4 μM (diamond). As expected from the PMFs, the IC50 values continuously decrease with increasing equilibration time. Moreover, the plot suggests that the computed IC50 value is likely to decrease further with longer equilibration, as indicated by the dashed line. In this sense, the simulations reveal an upper limit of the IC50 value of ∼3 mM, which is compatible to, but would not yet enable one to predict, the experimentally derived value.
Inhibitory effect of TEA on hAQP1
Now that the TEA binding site was reliably identified, we will subsequently address the inhibitory effect on permeation by TEA binding to this site. As described in the “Introduction”, the binding of TEA at the pore entrance of hAQP1 does not necessarily imply inhibition of water permeation through the channel. To investigate inhibition, we analyzed the influence of TEA binding on the water permeability of hAQP1. As a robust measure for permeability, the number of water molecules permeating between the NPA site and the Arg195 was counted (see “Methods” section), i.e., the permeation events across the narrowest single-file part of the pore. This number is directly proportional to the osmotic and diffusive permeation coefficients pf and pd [18].
In a simulation with TEA present in the binding site of hAQP1 we observed only nine permeation events within 9.5 ns of simulation corresponding to 0.9 events per nanosecond. In a 19-ns reference simulation of hAQP1 without TEA, we observed a total of 36 permeation events corresponding to 1.9 permeation events per nanosecond. Hence, TEA reduced the water flux by ∼50%, which, given the number of events, is significant. This result confirms the observation from oocyte experiments [6] that TEA is an inhibitor for human AQP1 and seems to contradict experiments with erythrocytes where no inhibitory effect of TEA could be observed [25]. The presented computational approach does not provide an explanation for the contradicting experimental observations and is hence proposed to be addressed by future experimental studies.
The binding site for tetraethylammonium (TEA) in human aquaporin-1 (hAQP1) was identified by the combination of three independent approaches: molecular docking with subsequent molecular dynamics simulations, first principles molecular dynamics docking, and umbrella sampling. We stress that combined computational approaches as presented in this paper are in general an effective tool to predict and to characterize binding sites for ligand molecules, in particular if co-crystallization is challenging for the protein of interest.
There are regions (Fig. 1d) that are identified by all three approaches (overlap of gray, magenta, and orange spheres), which we therefore consider as the most likely binding site. However, there are also regions explored by first principles docking, but not identified by TEA_dockMD simulations or umbrella sampling (Fig. 2d, magenta spheres). We suggest that these states are intermediate states that need to be visited before binding. Alternatively, but in our view less likely, one of these states might be the actual binding site, which in this case would not have been identified by the two other methods because of incomplete sampling. There are also regions explored by TEA_dockMD simulations, which were not identified by the two other methods (Fig. 2d, gray spheres). Again, incomplete sampling by the other two methods provides a possible explanation. Another possibility is a scenario in which these starting configurations generated by the docking procedure are metastable, but the 15 ns simulation time was too short for the TEA to overcome local barriers. Overall, the differences between the TEA binding regions identified by the three methods indicate that all these methods suffer from incomplete sampling. From this point of view, the mere fact that likely an overlap was found puts already substantial weight to the binding site identified in our combined approach. It further highlights the strength of the combined approach, which is not shown by any of the individual approaches alone.
Conclusion
We presented a combination of three different theoretical approaches to identify the binding site for TEA in human aquaporin-1. Differences and intermediates observed in molecular docking with subsequent molecular dynamics simulations, first principles molecular dynamics docking, and umbrella sampling underscore the complexity of TEA binding to hAQP1. The observed binding site is not simply either occupied or not; rather, binding is suggested to proceed via a network of intermediate states, and the binding kinetics is complicated by slow induced fit motions of the protein. The A-loop, in particular, seems to play a prominent role here. Our combined approach allowed a tentative characterization of this complexity.
Finally, it was demonstrated that bound TEA reduces water permeability through hAQP1, in agreement with previous MD simulations and experiments [6]. In summary, this combined approach strongly suggests that TEA is indeed a putative lead for hAQP1 inhibitors.
Acknowledgments
We thank Gerrit Groenhof for help with calculating partial charges for TEA and Daniel Seeliger for support with the Pymacs software tool.
Footnotes
Müller and Hub contributed equally to this article.
References
- 1.Berendsen HJ, Postma JP, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690 [DOI]
- 2.Berendsen HJ, Postma JP, van Gunsteren WF, Hermans J (2004) Intermolecular forces. D. Reidel Publishing Company, Dordrecht
- 3.Berger O, Edholm O, Jähnig F (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J 72:2002–2013 [DOI] [PMC free article] [PubMed]
- 4.Brooks HL, Regan JW, Yool AJ (2000) Inhibition of aquaporin-1 water permeability by tetraethylammonium: involvement of the loop e pore region. Mol Pharmacol 57:1021–1026 [PubMed]
- 5.de Groot BL, Grubmüller H (2001) Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science 294:2353–2357 [DOI] [PubMed]
- 6.Detmers FJM, de Groot BL, Müller EM, Hinton A, Konings IBM, Sze M, Flitsch SL, Grubmüller H, Deen PMT (2006) Quaternary ammonium compounds as water channel blockers: specificity, potency, and site of action. J Biol Chem 281:14207–14214 [DOI] [PubMed]
- 7.Ewing TJ, Kuntz ID (1997) Critical evaluation of search algorithms for automated molecular docking and database screening. J Comp Chem 18:1175–1189 [DOI]
- 8.Hess B, Bekker H, Berendsen HJ, Fraaije JG (1997) LINCS: a linear constraint solver for molecular simulations. J Comp Chem 18:1463–1472 [DOI]
- 9.Hoque MO, Soria JC, Woo J, Lee T, Lee J, Jang SJ, Upadhyay S, Trink B, Monitto C, Desmaze C, Mao L, Sidransky D, Moon C (2006) Aquaporin 1 is overexpressed in lung cancer and stimulates NIH-3T3 cell proliferation and anchorage-independent growth. Am J Pathol 168:1345 [DOI] [PMC free article] [PubMed]
- 10.Jørgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935 [DOI]
- 11.Jørgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–11236 [DOI]
- 12.Kaminski GA, Friesner RA, Tirado-Rives J, Jørgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105:6474–6487 [DOI]
- 13.King LS, Kozono D, Agre P (2004) From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol 5:687 [DOI] [PubMed]
- 14.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comp Chem 13:1011–1021 [DOI]
- 15.Lindahl E, Hess B, van der Spoel D (2004) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7:306–317
- 16.Miyamoto S, Kollman PA (1992) Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comp Chem 13(8):952–962 [DOI]
- 17.Niemietz CM, Tyerman SD (2002) New potent inhibitors of aquaporins: silver and gold compounds inhibit aquaporins of plant and human origin. FEBS Lett 531:443–447 [DOI] [PubMed]
- 18.Pohl P, de Groot BL, Tieleman DP, Grubmüller H (2002) Water permeation through gramicidin A: desformylation and the double helix; a molecular dynamics study. Biophys J 82:2934–2942 [DOI] [PMC free article] [PubMed]
- 19.Preston GM, Carroll TP, Guggino WB, Agre P (1992) Appearance of water channels in Xenopus oocytes expressing red–cell CHIP28 protein. Science 256:385–387 [DOI] [PubMed]
- 20.Preston GM, Jung JS, Guggino WB, Agre P (1993) The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J Biol Chem 268:17–20 [PubMed]
- 21.Van Buuren AR, Marrink SJ, Berendsen HJ (1993) A molecular dynamics study of the decane/water interface. J Phys Chem 97:9206–9212 [DOI]
- 22.van Gunsteren WF, Berendsen HJC. GROMOS Manual. BIOMOS b.v., Biomolecular Software, Groningen, The Netherlands
- 23.Verkman AS (2005) More than just water channels: unexpected cellular roles of aquaporins. J Cell Sci 118:3225–3232 [DOI] [PubMed]
- 24.Vriend G (1990) WHATIF: a molecular modeling and drug design program. J Mol Graph 8:52–56 [DOI] [PubMed]
- 25.Yang B, Kim JK, Verkman AS (2006) Comparative efficacy of HgCl2 with candidate aquaporin-1 inhibitors DMSO, gold, TEA+ and acetazolamide. FEBS Lett 580:28–29 [DOI] [PMC free article] [PubMed]
- 26.Yool AJ, Brokl OH, Pannabecker TL, Dantzler WH, Stammer WD (2002) Tetraethylammonium block of water flux in aquaporin-1 channels expressed in kidney thin limbs of Henle’s loop and kidney-derived cell line. BMC physiol 2:4 [DOI] [PMC free article] [PubMed]
- 27.Zeidel ML, Ambudkar SV, Smith BL, Agre P (1992) Reconstitution of functional water channels in liposomes containing purified red-cell CHIP28 protein. Biochemistry 31:7436–7440 [DOI] [PubMed]