

Abstract
Protein-mediated bridging is ubiquitous and essential for shaping cellular structures in all organisms. Here we dissect this mechanism for a model system: the Histone-like Nucleoid-Structuring protein (H-NS). We present data from two complementary single-molecule assays that probe the H-NS-DNA interaction: a dynamic optical-trap-driven unzipping assay and an equilibrium H-NS-mediated DNA looping scanning force microscopy imaging assay. To quantitatively analyze and compare these assays, we employ what we consider a novel theoretical framework that describes the bridging motif. The interplay between the experiments and our theoretical model not only infers the effective interaction free energy, the bridging conformation and the duplex-duplex spacing, but also reveals a second, unresolved, cis-binding mode that challenges our current understanding of the role of bridging proteins in chromatin structure. We expect that this theoretical framework for describing protein-mediated bridging will be applicable to proteins acting in chromatin and cytoskeletal organization.
Introduction
Cells employ an eclectic array of biopolymers to perform a diverse set of tasks: from actin and intermediate filaments, which provide cellular structure, and microtubules, which form the intercellular transport network, to DNA, which is the molecular basis of the genetic code. Each of these biopolymers is regulated and structured by proteins that remodel these filamentous networks. One of the most common structuring mechanisms is the bridging motif, where molecular bridges cross-link the individual biopolymers to form loops, super-filaments, gels, and condensates. In this article, we investigate the role of the molecular bridging mechanism in the organization of cellular structures using the dsDNA -bridging-protein H-NS as a model system. H-NS is an abundant nucleoid associated protein (NAP) that exhibits both a structural and a global regulatory role in Gram-negative bacteria (1,2).
In our analysis, we employ two single-molecule techniques to probe the dsDNA bridging: 1), optical tweezers to unzip the protein bridges, as previously described (3); and 2), scanning force microscopy (SFM) to image protein-induced dsDNA loops (for simplicity, we will refer to dsDNA as DNA in the remainder of the text). To quantitatively compare these assays, we employ a theoretical framework that describes protein-mediated biopolymer bridging. Our model allows us to infer the effective interaction among free energy, the bridging conformation, and the bridged spacing between biopolymers. We previously reported strong evidence that bridging by H-NS arises from trans binding (3); of interest, our current approach shows that the same data also strongly suggest the existence of an additional nonbridging cis-binding mode. This second, previously unresolved cis-binding mode predicts a surprisingly dynamic and flexible adhesion motif whose general equilibrium adhesion strength depends only on the density of bridging states. Cis binding challenges our current understanding of the role of bridging proteins in genome folding and organization and has general implications for bridging as an essential feature in biopolymer organization in all organisms.
Materials and Methods
The materials and methods used in this work are described in the Supporting Material.
Results
The high-resolution imaging of protein-DNA complexes has proved an important tool in determining the function and mechanism of DNA architectural proteins (4,5). SFM imaging has shown that H-NS (6) and functionally-homologous NAPs bridge DNA duplexes (7). Here we use the SFM imaging assay to infer both the H-NS-mediated bridging free energy and the spacing in the DNA–H-NS–DNA complex. At low DNA concentrations, H-NS bridging results in the formation of DNA loops, which are favored kinetically and entropically over interduplex associations. The size of these DNA loops is the result of two competing processes: the adhesive free-energy density that drives H-NS-mediated DNA bridging and the conformational free-energy penalty that results from DNA-loop formation. The distribution of loop sizes can be directly measured by SFM imaging of H-NS–DNA complexes (Fig. 1 A). The number of contour-length L loops observed in equilibrium is related to the looping free energy, Gloop(L), by the Boltzmann Distribution: , where is a normalization constant, k is the Boltzman constant, and T is the temperature.
Figure 1.
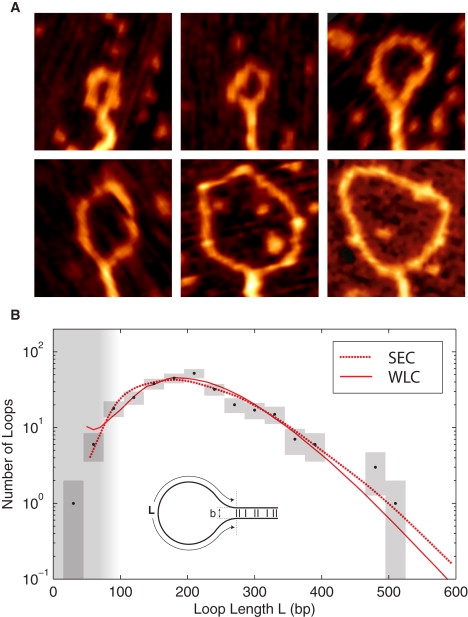
(A) Representative SFM images of H-NS-induced DNA loop complexes. The images are 125 nm by 125 nm. (B) The probability of H-NS-mediated DNA loops length L. A fit of the observed loop-length distribution (black), determined from SFM images of 286 loops, to the predicted loop-length distribution (red) results in excellent agreement with both chain statistics models. These fits predict nearly identical values for the adhesion energy: ρ = 0.13 ± 0.03 kT nm−1 (SEC) and ρ = 0.14 ± 0.03 kT nm−1 (WLC). The SEC model predicts a loop spacing of b = 4 ± 1 nm, whereas the WLC suggests a slightly higher value of b = 6 ± 1 nm . The chain statistical models are discussed in the Supporting Material. (The vertical range of the boxes represents the Poisson error, and the width represents the bin size.)
To relate the observed H-NS–mediated looping free energy to the molecular-level interactions of H-NS, we need to model DNA loop formation. The free energy associated with an H-NS–mediated DNA loop is: Gloop(L;b,ρ) = 1/2 ρ L + Gconf (L,b), where ρ is the H-NS–mediated adhesion energy per unit length of DNA and Gconf is the conformational free energy of the DNA loop, which depends on the loop spacing b and the contour length of the unbridged loop L (the factor of one-half that appears in the looping free energy is a consequence of the loop geometry: a dL reduction in the adhesion length results in a 2dL increase in the loop length). We applied the following coarse-grained binding configuration: 1), the polymer tangents at contour lengths 0 and L are antiparallel; and 2), the duplexes are displaced in the normal direction by the loop spacing b (see inset of Fig. 1 B). The conformational free energy of DNA bound to mica has been the subject of extensive investigation (8–10). We systematically calculated the conformational free energy of all 2D configurations for a wide range of contour lengths, employing two 2D DNA statistical models: SEC (10) and WLC (11).
Fig. 1 B shows that the loop-size distribution predicted by both models closely match the observed distribution for an adhesion energy of ρ = 0.13 ± 0.03 kT nm−1 and a loop spacing of b between 4 and 6 nm (13). Although the small-loop limit depends sensitively on the loop spacing, the large loop limit is adhesion dominated. Therefore, errors associated with the failure of the short-contour-length chain statistics or errors in determining the traced loop length do not significantly affect the measured adhesion energy, although they limit the accuracy of the inferred loop spacing. Note that the loop spacing b is too small to be resolved directly via SFM imaging, nevertheless, a rough estimate, based on the apparent width of the bridged duplexes, confirms that the loop spacing is between 3 and 6 nm (see the Supporting Material). This spacing suggests that the H-NS tail domains associate in a compact parallel conformation, excluding one of the two previously proposed dimerization models for H-NS (3,12,13) according to which the dimer is an extended structure.
The SFM assay measures the equilibrium interaction between H-NS and DNA. We have previously probed the nonequilibrium interaction between H-NS and DNA in a dynamic unzipping assay (this experiment is described in detail in Ref (3)). In these studies, H-NS–DNA complexes were assembled by allowing the H-NS proteins to bind between two aligned DNA molecules at saturating concentrations of H-NS. These complexes were unzipped employing optical tweezers at constant pulling rates. The force-response was found to be pulling-rate dependent: ∼1 pN at 6.5 nm s−1, ∼7 pN at 22 nm s−1, and ∼25 pN at 88 nm s−1 (3). In the high-force limit, the individual H-NS unbinding events, or steps, are resolved and both the step-size distribution and the force-dependent off rate can be directly measured. The zero force off rate was estimated from an Arrhenius fit to off rates measured at different forces. The estimated value is 1.5 ± 0.2 s−1 (3). The step-size distribution, captured at a constant pulling rate of 88 nm s−1, is of particular relevance to our current analysis and is shown in Fig. 2. The Fourier transform of this step-size distribution reveals a peak corresponding to the helical repeat of DNA which implies that H-NS can bridge DNA duplexes every helical repeat (3).
Figure 2.
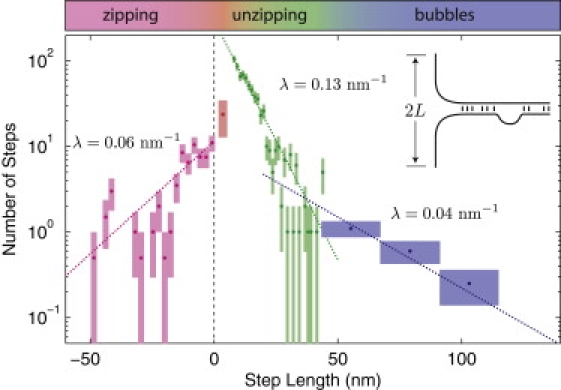
Step-size distribution for saturated, bridged duplexes upon optical-tweezers driven unzipping. In this assay, the step lengths are determined by the bridge spacing in the DNA–H-NS–DNA complex. The number of observed contour length steps is plotted as a function of the step length. The Poisson error is represented by the vertical box range. The bin size is represented by the box width. (Where the bin size has been expanded, the observed event number has been renormalized, resulting in fractional events.) The first step bridging probability (red) appears suppressed relative to the amplitude of subsequent steps in the unzipping regime which are well described by an exponential decay (green). The decay length fit from n > 1 steps is λ = 0.13 ± 0.02 nm−1.
The step sizes fall into three regimes: the zipping regime (back steps), the unzipping regime (steps shorter that 50 nm), and the bubble regime (steps longer than 50 nm). The zipping regime corresponds to back steps, or re-zipping events where bridges, that have been disrupted, reform. All back steps are short since the formation of bridges by rezipping is suppressed by the force applied by the optical trap. The unzipping and bubble regimes both correspond to unzipping events. At a pulling rate of 88 nm s−1, the H-NS-DNA complex cannot re-equilibrate and step-size distribution is a snapshot of the step length distribution between H-NS bridges in the unloaded complex. In the unzipping regime, the step size decays exponentially with decay constant λ = 0.13 ± 0.02 nm−1 for step lengths longer than a helical repeat. For shorter steps, the bridging probability is significantly smaller than predicted by the exponential decay. The mean step size in the unzipping regime is B = 10.8 nm (the mean step size is not equivalent to the decay length of the step-size distribution, since short steps are suppressed). For steps longer than 50 nm, the decay rate is significantly less than λ, suggesting that a different mechanism may control this bubble regime. Interestingly, the transition between the unzipping and bubble regime occurs at roughly the persistence length of DNA, the length scale on which DNA becomes flexible. We therefore believe the bubble regime correspond to regions where binding is frustrated during the assembly of the bridged duplex due to the stiffness of DNA.
Discussion
Do the results from these two complementary assays, probing H-NS function, lead to a consistent picture of H-NS-mediated bridging? The most direct method for comparing the looping and unzipping assays is to compare the estimated equilibrium force required to unzip the bridged duplexes to the measured unzipping force. The equilibrium force is defined as the work-per-unit-length required to overcome the adhesion energy: Feq ≡ ρ/2. (When length dL of duplex is unzipped, the additional contour length between beads increases by 2dL.) The looping assay predicts that this equilibrium force is just 0.25 pN (see the Supporting Material), only a quarter of the average force measured at the slowest measured pulling rate (6.5 nm s−1). We expect the pulling force to approach the equilibrium force for rates slow compared to the equilibration time. If we assume the equilibration time is the inverse of zero-force off rate, we expect to measure equilibrium forces for unzipping speeds less than koff B = 16.5 nm s−1, more than twice as fast as the slowest pulling rate we tested. Thus, the looping assay predicts a pulling force much smaller than measured.
The results from the unzipping experiment itself also appear to contradict our expectation that saturating H-NS concentrations imply saturated bridging. The step-size distribution is nontrivial: there are unbridged binding sites, which remain empty as the concentration of H-NS is increased. Could these unbridged sites correspond to transiently unbound H-NS protein? We analyzed the step-size distribution with the use of a statistical mechanics model (for a description of the model, see Section SD of the Supporting Material.)
These calculations reveal that the energy difference between unbridged and bridged states is ∼1 kT with a penalty for bridges at adjacent sites. The small energy penalty associated with bridging adjacent sites is compatible with a free energy penalty arising from excluded volume (see Subsection SC4). Thus it appears that two H-NS dimers do not bind cooperatively to adjacent sites by direct protein-protein interactions (3). If we interpret the bridged and unbridged states as a trans-bound H-NS dimer and a singly bound dimer, respectively, the free-energy loss on protein binding is just 1 kT, which predicts a significantly higher off-rate and a lower unzipping force than observed (see Subsection SD9 of the Supporting Material).
On the other hand, there is a subtle connection between the looping assay and the step-size distribution: In an adhesion model, the probability of a region length L being open is proportional to exp(−ρL/kT), where ρ is the adhesion energy density. In the fit to the step-size distribution, we find that the step-size distribution decays like exp(−λ L). Therefore, the adhesion energy, measured in the looping experiment, predicts the value of the decay length of step-size distribution: λ = ρ/kT. This prediction is satisfied by the experimental data, since the measured decay length from the step-size distribution and the decay length predicted from the looping assay are equal (to experimental precision). The step-size distribution and loop formation are subject to the same H-NS-induced adhesive free energy.
To resolve the mystery of how the adhesion energy could be just 1 kT per bridge in equilibrium, we consider two models: the Flexible-Linker model and the Rigid-Linker model. If H-NS bridges the ∼5 nm space between duplexes in trans, it may also permit cis binding between adjacent binding sites, spaced by a helical repeat, 3.6 nm (Fig. 3). Although structural and mutational studies suggest that each H-NS monomer has a flexible linker of 25 residues (15), this does not necessarily imply that cis binding is accessible. The Flexible-Linker model posits that both a cis- and a trans-binding site are accessible and that the two states have roughly the same binding energy since the distance to each state is comparable. After the first head binds, the effective concentration of the second H-NS binding domain at the respective binding sites is approximately equal (see Subsection SD2 of the Supporting Material for an extended discussion of the effective concentration). In the Rigid-Linker model, the cis-binding mode is not accessible.
Figure 3.
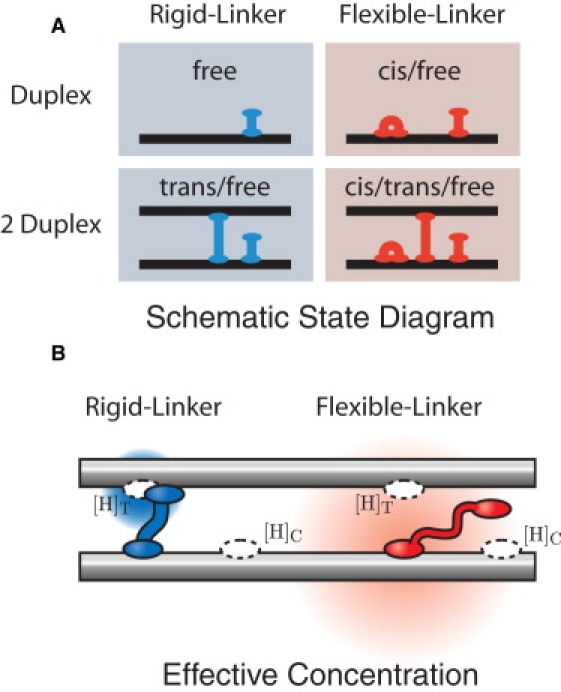
(A) Schematic state diagram of H-NS–DNA interactions. In our schematic drawings, we represent the H-NS dimer as a single unit. We consider two simple models for H-NS–DNA interaction: the Rigid and Flexible-Linker models. In the Flexible-Linker model, the H-NS dimer can bind to DNA in three states: free-head, trans, and cis. When H-NS interacts with two DNA duplexes, there is a competition between the three modes of binding. DNA adhesion is driven only by entropic effects (since H-NS can assume the cis conformation at the same energetic cost). In the Rigid-Linker model, the linker is too stiff to efficiently permit cis binding and the H-NS–DNA interaction is dominated by the free-head and trans binding modes. (B) The effective concentration illustrated for the Flexible and Rigid-Linker models. When H-NS binds a head to a DNA duplex, the diffusion of the second head is constrained. The physical concentration ([H]) of the second head in the proximal volume is dramatically increased. (The concentration is schematically illustrated by the red and blue gradients where the deeper hue corresponds to a higher local concentration.) This effective concentration is predicted by a polymer model for the linker domain. Structural and mutational studies of the H-NS protein suggest that the dimerization and DNA-binding domains are connected by a flexible linker (15). The Flexible-Linker model makes use of the fact that the linker domain can be modeled as a Gaussian chain with the generic amino-acid Kuhn length. In contrast, the linker domain of the Rigid-Linker model is assumed to have a structure which localizes the head domain in proximity to the trans-binding site. We therefore expect the trans head concentration to be much greater than that computed in the Flexible-Linker model.
The Flexible-Linker model naturally explains a number of otherwise remarkable aspects of H-NS–DNA interactions:
-
1.
Cis binding can naturally account for the failure of all binding sites to be bridged at saturating H-NS concentrations. In the Flexible-Linker model, unbridged sites are cis bound, not unbound, under saturating concentrations of H-NS and therefore the number of bridges does not increase with H-NS concentration. In the Rigid-Linker model, we would expect an equilibrated duplex of duplexes to be bridged at every site. Thus the Flexible-Linker model predicts that the step-size distribution is the result of the competition between cis and trans binding rather than trans binding and singly-bound H-NS dimers (Rigid-Linker model).
-
2.
In equilibrium, the Flexible-Linker model predicts that the adhesion between duplexes is entropic in nature because the free energy difference between cis and trans binding states is small. (Two states of equal free energy are available in a duplex of duplexes whereas only one of those states is available when H-NS binds a single duplex.) As a result, the adhesion energy is ∼1 kT per bridge, as observed (see Subsection SD8 of the Supporting Material for an extended discussion of this prediction). In contrast, we expect the adhesion energy in the Rigid-Linker model to be greater on account of the appreciable difference between the free energies of one-head and trans binding (see Subsection SD7 of the Supporting Material).
-
3.
Of particular importance is the cis lifetime in the Flexible-Linker model binding, which is expected to be on the order of many minutes due to the cooperative binding of the two heads (see Subsection SD5 of the Supporting Material) (14).
This long Flexible-Linker model cis lifetime implies that unzipping experiments, even at lowest pulling rate of 6.5 nm s−1, are faster than the relaxation time. The force difference between the equilibrium force and the forces measured at the slowest pulling rate are a result of a failure to reach equilibrium. The high force in the Rigid-Linker model (and unequilibrated Flexible-Linker model) results from the necessity of leaving H-NS heads unbound. In the equilibrated Flexible-Linker model, nearly all heads are bound, regardless of whether H-NS can bind in trans. Therefore the failure of the unzipping force to approach half the equilibrium force density makes it difficult to reconcile the Rigid-Linker model with the pulling and looping results. 4) Precoated H-NS–DNA complexes do not adhere (3). Even if the complexes were initially saturated, the Rigid-Linker model predicts that binding sites would become free after 1 s, resulting in adhesion. In contrast, the Flexible-Linker model predicts that these binding sites appear only after the lifetime of the cis state. Adhesion of precoated DNA molecules was not observed over the timescale of minutes (see Subsection SD5 of the Supporting Material). These four independent lines of experimental evidence strongly support the existence of a hidden cis-binding state as predicted by Flexible-Linker model binding by H-NS. Notably, a flexible linker domain has also been suggested by structural studies of H-NS (15). This finding thus reconciles the apparently contradictory results from the two single-molecule bridging assays.
Since a key prediction of the cis-binding model is the long lifetime of H-NS bound to an extended DNA molecule, we devised a force-extension experiment to probe the lifetime of these complexes. (If H-NS can only bind in trans, H-NS is predicted to diffuse away at the off-rate (2/s), which predicts that, after a few minutes, virtually no H-NS will remain bound to an extended DNA molecule if H-NS cannot rebind. In contrast, FLM predicts that H-NS can bind in cis to an extended DNA molecule with a lifetime of minutes.) This experiment was carried out in a sophisticated flow system, in which a DNA molecule can be rapidly moved between independent channels containing different solutions (3,16,17). A single DNA molecule is loaded with H-NS and subsequently transferred to a buffer solution without H-NS. The DNA molecule is then stretched to test if H-NS induced bridges are present. Next, this extended conformation is maintained for >3 min before relaxing the molecule. After multiple of these stretch-relaxation cycles with a total time up to 20 min, DNA-H-NS interactions are still detected (see Fig. 4). This clearly demonstrates that H-NS remains bound to DNA in a buffer containing no H-NS with a lifetime at least an order of magnitude greater than the lifetime of a single H-NS-DNA binding domain (∼1 s) (3), as predicted by the Flexible Linker model. Analogous observations have been reported by Skoko et al. (14), who argued that the low off rate of the architectural proteins in their studies is due to cooperative binding, induced either by changes in DNA conformation or direct protein-protein interactions. In the case of H-NS, the protein is not thought to significantly deform DNA. While cooperative binding has been reported, the mechanism of this cooperativity is not well understood. Cooperativity may arise from bridging, which facilitates the formation of neighboring bridges (3,6), direct interactions between adjacent dimers through residues located in the linker region and the N-terminal domain (13) or a combination thereof. Direct protein interactions implies a reduction in free energy between adjacent H-NS bridges, whereas the analysis of the step-size distribution suggests that in fact these configurations are energetically penalized. Therefore cis binding offers the most attractive model for the observed low off rates in the lifetime experiment. Although cis binding extends the lifetime of protein-DNA complexes, it still permits rapid diffusion. Flexible-Linker model bridging proteins are predicted to undergo two-tier diffusion: cis-mediated stepwise one-dimensional diffusion along DNA duplexes, and trans-mediated intersegmental transfer (18).
Figure 4.
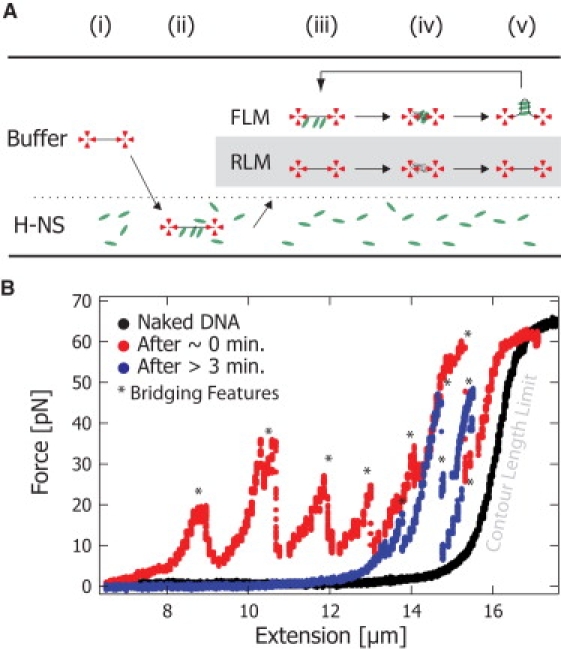
H-NS-DNA complex lifetime experiment. (A) A single DNA molecule is caught with optical tweezers. A force extension curve is taken in buffer solution (i). Subsequently, the DNA is “loaded” with H-NS in a buffer containing H-NS (2 μM) (ii). The DNA molecule is then placed back into the buffer solution without H-NS (iii). To probe the occurrence of bridge formation (and thus detect the presence of H-NS), the beads were brought together, releasing the DNA tension and allowing trans binding (iv). The force-extension curves were taken at time intervals of 1–5 min (iv→v→iii). The Rigid-Linker model (gray box) predicts that when the DNA is extended (iii), the lifetime of the H-NS-DNA complex is the lifetime of a single head (∼1 s). The Flexible-Linker model predicts that when the DNA molecule is extended (iii), the lifetime of the H-NS-DNA complex will be on order minutes due to the cooperative binding of the two heads. (B) The black curve shows the initial force-extension response of naked DNA. The red curve shows the force-extension response for the DNA immediately after incubation with H-NS. Bridging features are force spikes (∗) at extensions shorter than the contour length that result from large regions of trans-bound H-NS. After holding an extended state for >3 min, the blue curve still exhibits bridging features (∗) that demonstrate that H-NS remains bound to DNA for a lifetime an order of magnitude greater than predicted by the single-head off rate. These data strongly support the existence of cis-bound H-NS as predicted by the Flexible-Linker model.
In vivo, the Flexible-Linker model predicts that the structure of the nucleoid as imposed by H-NS bridging is surprisingly dynamic. In addition to condensing the nucleoid, DNA-bridging proteins act as topological insulators to the propagation of twist. The existence of topological domains in prokaryotic nucleoids is widely accepted although both the physical and biological nature of these domains is still uncertain (19). Although Flexible-Linker model bridging proteins would act as a barrier to twist propagation, the domain walls would be diffuse in nature, in contrast to the canonical assumptions (19). Recently, we proposed that H-NS is one of the key proteins in topological domain formation (20). This prediction sheds new light on the observations of Postow and co-workers (19), who attributed the apparently diffuse nature of the topological insulators of twist propagation, in the Escherichia coli genome, to an artifact: the population average of stochastically-positioned sharp domains. In contrast, our results predict that the observed diffuse barriers are an intrinsic property of H-NS domains (although high-affinity sites (21) within regions of preferential binding could result in sharper domain walls (22,20)). H-NS-mediated repression has been attributed to high-affinity sites, which nucleate H-NS binding and the formation of DNA loops. Cis binding suggests a possible mechanism for the rapid relief and recovery of H-NS-mediated silencing (23). H-NS dimers could switch from trans-bound to cis-bound states at an appreciable rate, allowing access to DNA by H-NS antagonists and/or RNA polymerase. H-NS, maintained in the cis-binding state, could then rapidly transition back to the trans-bound silenced state.
The single-molecule in vitro experiments, described above, strongly favor the Flexible-Linker model model to describe H-NS-mediated bridging. The models that we employed are applicable to the description of molecular bridging generically. We will therefore describe the general properties of the Flexible and Rigid Linker models. A generic feature of the Flexible-Linker model is that it limits the equilibrium adhesion energy of flexible bridges to 1 kT per bridge, regardless of the equilibrium constant of head binding. Such small adhesion energies imply that the bridging molecules must be in abundance (like H-NS) to lead to appreciable adhesion energies. As we have argued above, the Rigid-Linker model is not limited by cis binding and can result is much higher adhesion energies per bridge.
A generic feature we can predict for the Rigid-Linker model is highly-cooperative bridging (24). This binding cooperativity is the result of effective substrate concentration, not direct interaction between adjacent bridges. The Rigid-Linker model predicts the nucleation of “super” bridges consisting of many adjacent molecular bridges. These super bridges have a long lifetime in the absence of active remodeling. The formation of super bridges has regulatory consequences. For instance, the segregation protein ParB can bind DNA only in trans and is known to bind DNA cooperatively (25). ParB polymerizes along the DNA helix and has been shown to silence genes up to 10 kb away from a single parS nucleation site (26,27). Like ParB, proteins that exhibit Rigid-Linker model DNA-binding are predicted to result in long range silencing around high-affinity binding sites, whereas cis-binding proteins, which bind less cooperatively, are predicted to be dependent on multiple high-affinity binding sites distributed throughout silenced regions (28). (As a result of trans binding, the ParB protein would exhibit cooperative binding even in the absence of protein-protein interactions.) The Rigid-Linker model also predicts a preferred filament orientation for cross-linked filaments. This binding configuration tends to align filaments in a stiff bundle-like structure reminiscent of actin fiber bundles rather than a uniform condensate. In contrast to the Rigid-Linker model, there is a much smaller entropic advantage for binding Flexible-Linker model proteins in series. Although many of the bridging molecules are bound in cis (not bridging), the lack of cooperative binding implies there are many more bridging contacts in the filament networks, resulting in a more uniform condensation with a dynamic structure. Flexible-Linker model cross-linking predicts the binding of all filament orientations with nearly equal probability, resulting in a gel-like network.
The Flexible-Linker and Rigid-Linker models illustrate possible design principles that govern bridging motifs in general, from chromatin structure to the cytoskeleton. There are numerous examples of DNA bridging proteins in all three domains of life (29), many of which, such as SMC family proteins (30) are involved in genome organization and compaction. Molecular bridging is also important in structuring the cytoskeleton. Although many actin-binding proteins are believed to be quite stiff, filamin has a long flexible linker. As predicted by the Flexible-Linker model, it forms actin-filament gels at a significantly lower concentration than other actin bridging proteins with similar head binding affinity (31). Analogously, we anticipate that the physical properties (e.g., flexibility and length) of the neck linker of different classes of multimeric kinesins (32) will be shown to be key determinants in how they structure, move, and bundle microtubules. Given the ubiquity of the bridging proteins in cellular structure, the Flexible-Linker and Rigid-Linker models will be an essential tool for understanding the role of bridging in structuring biopolymers in the cell.
Supporting Material
Methods, results, models, six figures, a table, and references are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01276-4.
Supporting Material
Acknowledgments
We thank Niels Laurens for assistance with the experiments.
This research was supported by the Whitehead Institute for Biomedical Research (P.A.W.), the Netherlands Organization for Scientific Research (NWO) through a NWO VIDI grant (R.T.D.), and a NWO VICI grant (G.J.L.W.).
References
- 1.Dorman C.J. H-NS: a universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2004;2:391–400. doi: 10.1038/nrmicro883. [DOI] [PubMed] [Google Scholar]
- 2.Dorman C.J. H-NS, the genome sentinel. Nat. Rev. Microbiol. 2007;5:157–161. doi: 10.1038/nrmicro1598. [DOI] [PubMed] [Google Scholar]
- 3.Dame R.T., Noom M.C., Wuite G.J.L. Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature. 2006;444:387–390. doi: 10.1038/nature05283. [DOI] [PubMed] [Google Scholar]
- 4.Dame R.T. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol. Microbiol. 2005;56:858–870. doi: 10.1111/j.1365-2958.2005.04598.x. [DOI] [PubMed] [Google Scholar]
- 5.Luijsterburg M.S., Noom M.C., Wuite G.J.L., Dame R.T. The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: a molecular perspective. J. Struct. Biol. 2006;156:262–272. doi: 10.1016/j.jsb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Dame R.T., Wyman C., Goosen N. H-NS mediated compaction of DNA visualised by atomic force microscopy. Nucleic Acids Res. 2000;28:3504–3510. doi: 10.1093/nar/28.18.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dame R.T., Luijsterburg M.S., Krin E., Bertin P.N., Wagner R. DNA bridging: a property shared among H-NS-like proteins. J. Bacteriol. 2005;187:1845–1848. doi: 10.1128/JB.187.5.1845-1848.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivetti C., Guthold M., Bustamante C. Scanning force microscopy of DNA deposited onto mica: equilibration versus kinetic trapping studied by statistical polymer chain analysis. J. Mol. Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 9.Rivetti C., Walker C., Bustamante C. Polymer chain statistics and conformational analysis of DNA molecules with bends or sections of different flexibility. J. Mol. Biol. 1998;280:41–59. doi: 10.1006/jmbi.1998.1830. [DOI] [PubMed] [Google Scholar]
- 10.Wiggins P.A., Heijden T. van der, Moreno-Herrero F., Spakowitz A., Phillips R. High flexibility of DNA on short length scales probed by atomic force microscopy. Nat. Nanotechnol. 2006;1:137–141. doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- 11.Kratky O., Porod G. Rotgenuntersuchung geloster Fadenmolekule. Rec. Trav. Chim. 1949;68:1106–1122. [Google Scholar]
- 12.Bloch V., Yang Y., Margeat E., Chavanieu A., Auge M.T. The H-NS dimerization domain defines a new fold contributing to DNA recognition. Nat. Struct. Biol. 2003;10:212–218. doi: 10.1038/nsb904. [DOI] [PubMed] [Google Scholar]
- 13.Esposito D., Petrovic A., Harris R., Ono S., Eccleston J.F. H-NS oligomerization domain structure reveals the mechanism for high order self-association of the intact protein. J. Mol. Biol. 2002;324:841–850. doi: 10.1016/s0022-2836(02)01141-5. [DOI] [PubMed] [Google Scholar]
- 14.Skoko D., Wong B., Johnson R.C., Marko J.F. Micromechanical analysis of the binding of DNA-bending proteins HMGB1, NHP6A, and HU reveals their ability to form highly stable DNA-protein complexes. Biochemistry. 2004;43:13867–13874. doi: 10.1021/bi048428o. [DOI] [PubMed] [Google Scholar]
- 15.Dorman C.J., Hinton J.C., Free A. Domain organization and oligomerization among H-NS-like nucleoid-associated proteins in bacteria. Trends Microbiol. 1999;7:124–128. doi: 10.1016/s0966-842x(99)01455-9. [DOI] [PubMed] [Google Scholar]
- 16.Noom M.C., van den Broek B., van Mameren J., Wuite G.J. Visualizing single DNA-bound proteins using DNA as a scanning probe. Nat. Methods. 2007;4:1031–1036. doi: 10.1038/nmeth1126. [DOI] [PubMed] [Google Scholar]
- 17.van Mameren J., Modesti M., Kanaar R., Wyman C., Peterman E.J. Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature. 2009;457:745–748. doi: 10.1038/nature07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Broek B., Lomholt M.A., Kalisch S.M., Metzler R., Wuite G.J.L. How DNA coiling enhances target localization by proteins. Proc. Natl. Acad. Sci. USA. 2008;105:15738–15742. doi: 10.1073/pnas.0804248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postow L., Hardy C.D., Arsuaga J., Cozzarelli N.R. Topological domain structure of the Escherichia coli chromosome. Genes Dev. 2004;18:1766–1779. doi: 10.1101/gad.1207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noom M.C., Navarre W.W., Oshima T., Wuite G.J., Dame R.T. H-NS promotes looped domain formation in the bacterial chromosome. Curr. Biol. 2007;17:R913–R914. doi: 10.1016/j.cub.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Bouffartigues E., Buckle M., Badaut C., Travers A., Rimsky S. H-NS cooperative binding to high-affinity sites in a regulatory element results in transcriptional silencing. Nat. Struct. Mol. Biol. 2007;14:441–448. doi: 10.1038/nsmb1233. [DOI] [PubMed] [Google Scholar]
- 22.Navarre W.W., Porwollik S., Wang Y., McClelland M., Rosen H. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science. 2006;313:236–238. doi: 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- 23.Stoebel D.M., Free A., Dorman C.J. Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology. 2008;154:2533–2545. doi: 10.1099/mic.0.2008/020693-0. [DOI] [PubMed] [Google Scholar]
- 24.McGhee J.D., von Hippel P.H. Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 25.Schumacher M.A., Funnell B.E. Structures of ParB bound to DNA reveal mechanism of partition complex formation. Nature. 2005;438:516–519. doi: 10.1038/nature04149. [DOI] [PubMed] [Google Scholar]
- 26.Lynch A.S., Wang J.C. SopB protein-mediated silencing of genes linked to the sopC locus of Escherichia coli F plasmid. Proc. Natl. Acad. Sci. USA. 1995;92:1896–1900. doi: 10.1073/pnas.92.6.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodionov O., Lobocka M., Yarmolinsky M. Silencing of genes flanking the P1 plasmid centromere. Science. 1999;283:546–549. doi: 10.1126/science.283.5401.546. [DOI] [PubMed] [Google Scholar]
- 28.Fang F.C., Rimsky S. New insights into transcriptional regulation by H-NS. Curr. Opin. Microbiol. 2008;11:113–120. doi: 10.1016/j.mib.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luijsterburg, M. S., M. F. White, R. van Driel, and R. T. Dame. 2008. The major architects of chromatin: architectural proteins in bacteria, archaea and eukaryotes. In Critical Reviews in Biochemistry and Molecular Biology. [DOI] [PubMed]
- 30.Nasmyth K., Haering C.H. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 2005;74:595–648. doi: 10.1146/annurev.biochem.74.082803.133219. [DOI] [PubMed] [Google Scholar]
- 31.Stossel T.P., Condeelis J., Cooley L., Hartwig J.H., Noegel A. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 32.Kapitein L.C., Peterman E.J.G., Kwok B.H., Kim J.H., Kapoor T.M. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature. 2005;435:114–118. doi: 10.1038/nature03503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.