

Abstract
Fourier transform infrared (FTIR) spectroscopy has been established as a fast spectroscopic method for biochemical analysis of cells and tissues. In this research we aimed to investigate FTIR's utility for identifying and characterizing different modes of cell death, using leukemic cell lines as a model system. CCRF-CEM and U937 leukemia cells were treated with arabinoside and doxorubicin apoptosis inducers, as well as with potassium cyanide, saponin, freezing-thawing, and H2O2 necrosis inducers. Cell death mode was determined by various gold standard biochemical methods in parallel with FTIR-microscope measurements. Both cell death modes exhibit large spectral changes in lipid absorbance during apoptosis and necrosis; however, these changes are similar and thus cannot be used to distinguish apoptosis from necrosis. In contrast to the above confounding factor, our results reveal that apoptosis and necrosis can still be distinguished by the degree of DNA opaqueness to infrared light. Moreover, these two cell death modes also can be differentiated by their infrared absorbance, which relates to the secondary structure of total cellular protein. In light of these findings, we conclude that, because of its capacity to monitor multiple biomolecular parameters, FTIR spectroscopy enables unambiguous and easy analysis of cell death modes and may be useful for biochemical and medical applications.
Introduction
Apoptosis is the process by which cells self-destruct without evoking an inflammatory response, and is characterized by nuclear and cytoplasmic shrinkage, chromatin condensation, internucleosomal DNA cleavage, and plasma membrane blebbing (1,2). Apoptosis occurs as part of various normal and pathological processes, and can be triggered by chemotherapeutic drugs (3,4). In contrast, necrosis results from severe stress, physicochemical injury, osmotic imbalance, or energy deprivation, and is associated with pathologies such as inflammatory diseases, ischemia, hypoxia, and oxidative stress (5–7). Recent research indicates that necrosis is a programmed and controlled cell death rather than merely an uncontrolled process (5,6). Death via necrosis is characterized by the loss of plasma membrane integrity, cell swelling, and DNA degradation, and is associated with inflammatory responses (5). Human disease progression and response to treatment are often indicated by the degree and type of cell death that occurs (8,9). Therefore, a rapid and simple method of identifying the cell death mode is urgently required to improve clinical diagnoses.
At present, assays such as Annexin V, TdT-mediated deoxyuridine triphosphate nick end labeling, and caspase activity are used as gold standards for differentiating modes of cell death. However, most of these methods are expensive, complicated, and time-consuming, and cannot independently determine the type of cell death with high certainty. The uncertainty stems from the limited capacity of each assay to measure only a single biochemical parameter, which might be common to several death modes, such as plasma membrane permeability common to late apoptosis and necrosis (9). An additional limitation of these current methods is that they require fixation and thus cannot be used for in vivo analysis. Therefore, a simple diagnostic method is needed that does not involve sample processing and can monitor the numerous biochemical changes that take place during cell death.
Fourier transform infrared (FTIR) spectroscopy is emerging as a promising clinical diagnostic method because it enables general biochemical changes in samples to be monitored without the need for reagents or complicated sample handling (10,11). However, the applicability of FTIR spectroscopy to assaying cell death mode is controversial. FTIR methods have been used to study apoptosis, as well as cell cycle, differentiation, and proliferation of different cell lines and tissues (10–17), but many questions remain concerning the interpretation of IR data. Liu et al. (13), Gasparri and Muzio (14), and others (15–17) have reported contradictory results concerning apoptosis in different systems. In the research presented here, we use leukemic cell lines as a model system to resolve the controversy regarding FTIR spectroscopic analysis of apoptosis, and apply this method to study necrotic cell death.
Materials and Methods
Cell culture and treatments
Human leukemic cell lines U937 and CCRF-CEM were cultured as described previously (18). U937 and CCRF-CEM cells were incubated with Ara-C (Hospira Australia Proprietary Ltd., Melbourne, Australia) or doxorubicin (Teva Pharmaceutical, Netanya, Israel) for 24 h and 48 h, respectively, to induce apoptosis. To induce necrosis, U937 cells were subjected to a freeze-thaw procedure (19) or treated for 10 min with 0.2 mg/mL saponin (19) or incubated with KCN (Sigma, St. Louis, MO) in glucose-free RPMI medium for 7 h. CCRF-CEM cells were induced to undergo apoptosis and necrosis by treatment with H2O2 (Sigma) for 7 h.
Cell viability was assessed by diluting an aliquot of cell suspension with the same volume of 0.4% trypan blue solution (Sigma). A lactate dehydrogenase (LDH) release assay (Promega, Madison, WI) was performed according to the manufacturer's instructions.
Subcellular fractionation
Treated and control U937 cells were harvested and washed twice with cold phosphate-buffered saline. Nuclei were purified as described previously (see Gardella et al. (21)). The nuclear pellet was resuspended in 50 μL ice-cold buffer (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF) (20). The mixture was vigorously rocked at 4°C for 15 min on a shaking platform; then 5 μL of 10% NP40 was added and the nuclear extract was centrifuged for 5 min at 14,000 g. The supernatant was frozen in aliquots at −80°C. The protein content of the nuclear and cytoplasmic extracts was determined by a colorimetric Lowry method (Bio-Rad, Munchen, Germany).
Western blot
Immunoblotting was conducted using various primary antibodies specific for HMBG1, caspase 8, actin, and lamin (Santa Cruz Biotechnology, Santa Cruz, CA) as described previously (21).
Fluorescent microscopy
Ethidium bromide (EB; Sigma) and acridine orange (AO; Sigma) staining was performed as described previously (22). Briefly, cultured cells were centrifuged at 650 g for 10 min and stained with AO and EB with a final concentration of 0.05 mg/mL. The death mode for each cell type was evaluated with the use of a fluorescent microscope.
FTIR microspectroscopy
For FTIR microspectroscopy (FTIR-MSP), 1 mL of control or treated cells was washed twice with saline (0.9% NaCl) and resuspended in 5 μL fresh saline. Then 1.5 μL of the washed cells were deposited on a zinc selenide (ZnSe) slide to form approximately a monolayer of cells and then air-dried for 15 min under laminar flow to remove water. Measurements were performed using an FTIR microscope IR scope 2 as previously described (23).
The spectra were baseline-corrected using a polynomial rubberband with 64 points (OPUS software; Bruker Optik GmbH, Ettlingen, Germany) and vector-normalized in the 700–4000 cm−1 region. To obtain precise absorbance values at a given wavenumber with minimal background interference, the second derivative spectra were used to determine concentrations of biomolecules of interest. This method is highly susceptible to changes in the full width at half-maximum (FWHM) of the IR bands. However, in the case of biological samples, all cells from the same type are composed of similar basic components, which give relatively broad bands. Thus, it is possible to neglect the changes in the bands' FWHM (24).
Statistical analysis
Data are presented as the mean ± SE derived from at least three independent experiments. Statistical analysis was performed using Student's t-test; p-values < 0.05 were considered significant. Linear regressions were done by the least-squares method, and a data analysis package (Origin; MicroCal, Northampton, MA) was used to compare series of data.
Results
To better understand the spectral changes that occur during chemotherapy of leukemia patients, we chose cultured leukemia cells treated with cytotoxic drugs as our model system. Cell death mode was analyzed by classical as well as spectroscopic methods to enable characterization of spectral fingerprints for apoptotic versus necrotic cell death.
IR spectral characterization of leukemia cells undergoing apoptosis
Human promonocytic leukemia U937 cells were treated for 48 h with Ara-C, an inducer of apoptosis. The mode of cell death was determined classically by EB and AO staining patterns observed using fluorescence microscopy (Fig. 1). Living cells have normal-shaped nuclei and the chromatin appears green (Fig. 1 a). Early apoptotic cells exhibit shrunken nuclei and condensed chromatin that appears green (Fig. 1 b). In contrast, late apoptotic cells display condensed, fragmented nuclei, and the chromatin therein is brightly stained with EB and thus appears orange (Fig. 1 c). Necrotic cells exhibit a normal shape but the chromatin is stained with EB, which makes the nuclei appear orange (Fig. 1 d). In parallel, control untreated and treated U937 cells were subjected to IR spectral analysis (Fig. 2).
Figure 1.
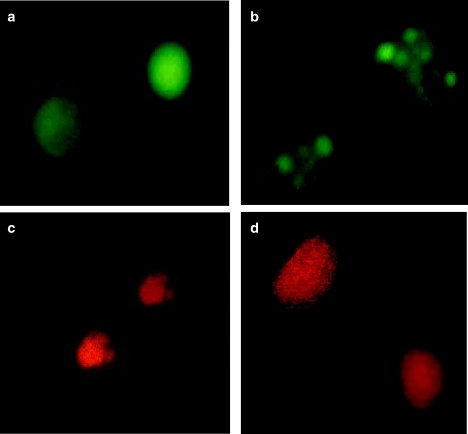
Evaluation of cell death mode by AO and EB staining. U937 cells were cultured for 48 h with different concentrations of Ara-C. The cells were stained with EB and AO and observed using fluorescence microscopy. (a) Living cells have normal-shaped nuclei with green chromatin. (b) Early apoptotic cells have shrunken nuclei with condensed green chromatin. (c) Late apoptotic cells have condensed and fragmented nuclei containing orange chromatin. (d) Necrotic cells have normal-shaped nuclei containing orange chromatin.
Figure 2.
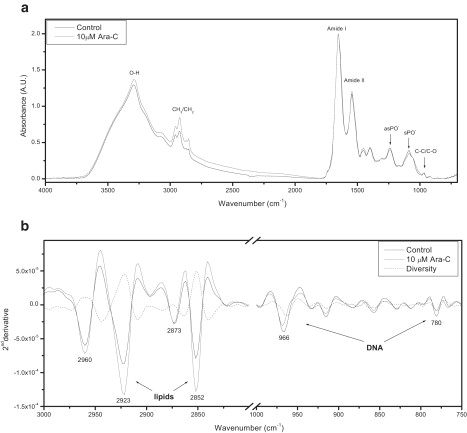
FTIR-MSP spectra of apoptotic U937 cells. U937 cells treated with 10 μM Ara-C for 48 h and control untreated cells were subjected to FTIR spectroscopic analysis. (a) Representative IR absorbance spectra of U937 cells after baseline correction and min-max normalization. Each spectrum represents the average of five measurements at different sites for each sample. The main absorbance bands used in data analysis are marked. (b) Second derivative extended spectra of U937 cells after baseline correction and vector normalization.
Certain absorbance bands relate to specific macromolecules as follows: the region 3000–2830 cm−1 reflects symmetric and antisymmetric stretching of the CH3 and CH2 groups of proteins and lipids; the region 1800–1500 cm−1 relates to amide 1 and amide 2, often an indicator of the secondary structure of proteins; and the region 1150–750 cm−1 reflects a variety of vibrations due to proteins, carbohydrates, lipids, and nucleic acids (Fig. 2 a) (10,12). To optimize accuracy when interpreting spectral data, we analyzed the second derivative of the vector-normalized spectra as presented in Fig. 2 b. When the second derivative spectral patterns of untreated control and apoptotic cells are compared, the following features can be easily discerned as characteristic of apoptosis: an increase in lipid absorbance at 2852 cm−1 and 2923 cm−1 (symmetric and antisymmetric stretching of CH2, respectively) and a decrease in DNA absorbance at 966 cm−1 (due to C-C/C-O stretching of deoxyribose-ribose vibration) and 780 cm−1 (sugar-phosphate vibrations) (13,14,25,26).
Having identified specific spectral features associated with apoptotic cells, we wished to determine how the spectra change during ongoing apoptosis. To look at different stages of apoptosis, we treated U937 cells with various concentrations of Ara-C (0.01–10 μM) for 24 and 48 h. The percentage of apoptotic cells and the stages of apoptosis were evaluated by trypan blue (TB) staining (data not shown) as well as AO and EB fluorescence staining (Fig. 3, a and b), and, in parallel, samples were subjected to IR spectral analysis (Fig. 3, c–h). Treatment with 0.1 μM or 0.5 μM Ara-C induces early stages of apoptosis, which are characterized by condensed chromatin and intact plasma membrane. Higher concentrations induce both early and late apoptosis, the latter indicated by disrupted plasma membrane, with ∼35% of each stage detectable at 24 h (Fig. 3 a). After treatment with 10 μM Ara-C for 48 h, the proportion of apoptotic cells reaches 70% (Fig. 3 b). The percentage of necrotic cells was not significantly increased following Ara-C treatment. Analysis of the FTIR spectra reveals that lipid absorbance (at 2852 cm−1, mainly corresponding to lipids) increases by 35% and 60% after 24 h and 48 h, respectively. In line with our aforementioned data, these increases correlate well with the percentage increases in apoptotic cells (R2 = 0.93 for 24 h and R2 = 0.95 for 48 h treatment, respectively). DNA absorbance at 966 cm−1 decreases up to 15% and 35% following Ara-C treatment (10 μM) for 24 h and 48 h, respectively. A similar pattern is observed for absorbance at 780 cm−1. The correlation of the decrease in DNA absorbance and apoptotic cell percentage is R2 = 0.83 for 24 h and R2 = 0.95 for 48 h. In addition, there are distinctive spectral changes in another region of interest, the amide I at ∼1654 cm−1, which corresponds to the secondary structure of proteins (Fig. 3, g and h). Specifically, the amide I band comprises several major absorption bands, such as 1654 cm−1, 1639 cm−1 and 1627 cm−1, due to α-helix, random coil, and parallel β-strand structures, respectively (27). After 24 h, the amount of β-structure increases at low Ara-C concentrations (0.01 μM), but it increases dramatically at higher concentrations (1–10 μM) by >60% (Fig. 3 g). After 48 h of treatment, the amount of β-structure increases even further: at 10 μM there is ∼200% more than observed in control untreated cells (Fig. 3 h). Of note, these spectral changes related to β-structure indicate a gradual progression of biochemical change rather than a biochemical process with distinct early and late stages.
Figure 3.
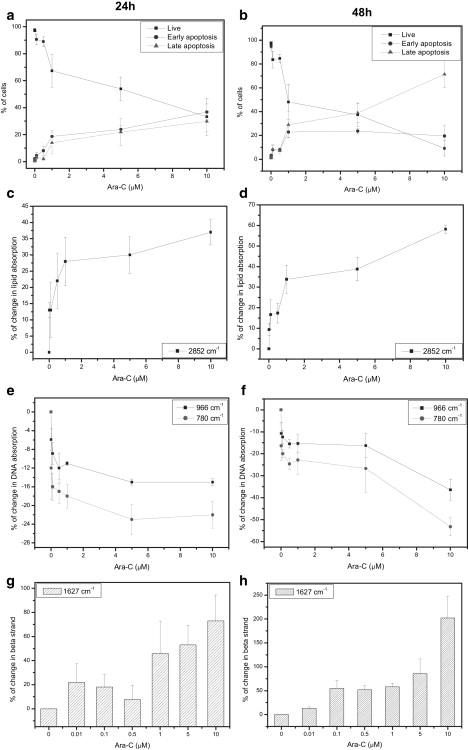
FTIR-MSP spectra of U937 cells undergoing apoptosis. U937 cells were treated with Ara-C (0.01–10 μM) for 24 h (left panels) and 48 h (right panels). (a and b) The percentage of live, apoptotic, and necrotic cells was determined by AO and EB staining. (c–h) Relative absorbance values, derived from FTIR-MSP analysis of untreated versus treated U937 cells, for (c and d) lipids at 2852 cm−1, (e and f) DNA quantified at 970 cm−1 and 780 cm−1, and (g and h) proteins at 1627 cm−1 (β-sheet). Values plotted are the mean ± SE derived from three independent experiments.
We performed similar experiments using human T-lymphoblastic leukemia CCRF-CEM cells and doxorubicin, another cytotoxic drug that induces apoptosis, and generated comparable data (not shown).
IR spectral characterization of necrotic cells
Necrosis of U937 cells was induced by two different methods: freeze-thawing or treatment with 0.2 mg/mL saponin, a reagent known to induce gently membrane permeabilization. TB staining was the classical method employed to evaluate the proportion of necrotic cells, which after either treatment was >95%. Once again, in parallel, samples were subjected to FTIR spectral analysis, and the second derivative spectral patterns from treated and untreated cells were compared (Fig. 4, a and b). After induction of necrosis by saponin or freeze-thawing, there were increases in lipid absorbance at 2852 cm−1 and 2923 cm−1, as well as increases in DNA absorbance at 966 cm−1 and 780 cm−1.
Figure 4.
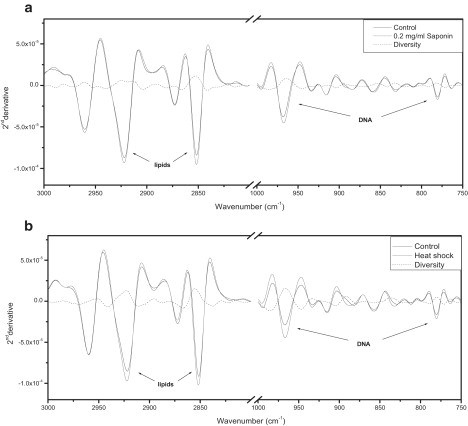
FTIR-MSP spectra of necrotic cells. (a) Untreated U937 cells and treated with 0.2 mg/mL saponin for 10 min and (b) subjected to freeze-thawing. Second derivative IR spectra after baseline correction and vector normalization are shown.
To look at the spectral changes that occur during necrotic cell death, we exposed U937 cells for 7 h to various concentrations of potassium cyanide (KCN), which is a common necrosis inducer in glucose-free conditions (28). Cell viability and the type of death were determined classically by AO and EB staining, as well as by TB staining (Fig. 5, a and b, respectively). In addition, we assayed LDH release and caspase activity to validate the cell death mode (data not shown) as well as the release of high-mobility group B1 (HMGB-1) from the nucleus (Fig. 5 c). The latter is a marker for necrosis, since this protein is known to be involved in chromatin conformation and is released from the nucleus during necrotic cell death. Examination of the second derivative IR spectral patterns reveals that KCN treatment results in increased absorbance due to lipids, with a ∼67% increase at high concentrations relative to control untreated cells (Fig. 5 d). Similarly, DNA absorbance increases by 56% (Fig. 5 e), but notably reaches a plateau after 10 mM. The correlation of lipids and DNA absorbance with the percentage of necrotic cells was R2 = 0.92 and R2 = 0.91, respectively. Additionally, in general, KCN treatment appears to reduce the random coil structure content of proteins, as indicated by reduced absorption at 1639 cm−1, although this trend is not seen at 5 mM (Fig. 5 f).
Figure 5.
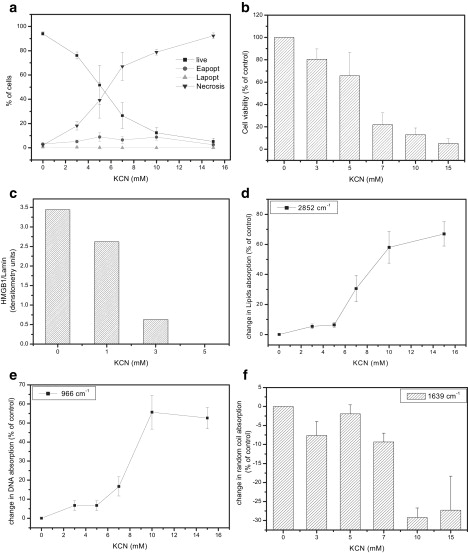
FTIR-MSP spectra of U937 cells undergoing necrosis. Cells were treated with KCN (0–15 mM) for 7 h. The percentage of live, apoptotic, and necrotic cells was determined by (a) AO and EB staining, (b) TB staining, and (c) HMGB1 release. (d–f) Relative absorbance values, derived from FTIR-MSP analysis of untreated versus treated U937 cells, for (d) lipids at 2852 cm−1, (e) DNA at 966 cm−1, and (f) proteins at 1639 cm−1 (random coil). Values plotted are the mean ± SE derived from three independent experiments.
Apoptotic/necrotic cell death inducer
Next, we took advantage of a treatment known to induce both modes of cell death in a concentration-dependent manner (18) to investigate whether FTIR spectra do indeed reflect differentially apoptosis versus necrosis. Human T-lymphoblastic leukemia CCRF-CEM cells were incubated with various concentrations of H2O2 for 7 h. Cell viability and death mode were determined classically by TB, AO, and EB staining. At low H2O2 concentrations, early-stage apoptotic cells and a few necrotic cells are evident, whereas at higher concentrations necrotic cells predominate (Fig. 6 a). Of importance, the FTIR spectral patterns mirror the induction of cell death and, in particular, the H2O2 concentration-dependent switch from apoptosis to necrosis (Fig. 6, b–d). Increased lipid absorption at 2852 cm−1, a marker common to both cell death modes, is detectable at all concentrations tested, as expected. Notably, there is a plateau around 1 mM, but at higher concentrations there are further increases in lipid absorption relative to control untreated cells. DNA absorbance at 966 cm−1 decreases slightly at low H2O2 concentrations (∼0.1 mM), which is in line with our earlier finding that decreased DNA absorbance is associated with apoptosis. But then at higher H2O2 concentrations, in accord with the switch from apoptosis to necrosis, DNA absorbance increases (Fig. 6 c). Finally, parallel to the increasing proportions of necrotic cells at higher H2O2 concentrations, the random coil structure content of proteins becomes lower, as indicated by the reduced absorbance at 1639 cm−1 in the Amide I region (Fig. 6 d).
Figure 6.
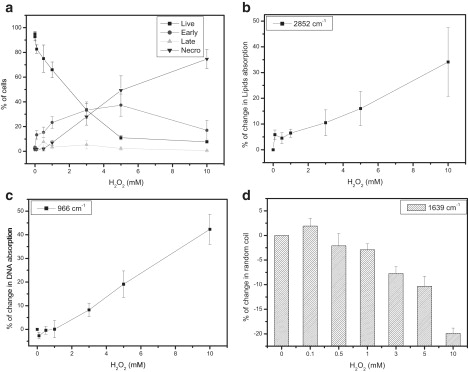
FTIR-MSP spectra of CCRF-CEM cells undergoing cell death induced by an apoptosis/necrosis-inducing agent. Cells were treated with various concentrations of H2O2 for 7 h. (a) The percentage of live, apoptotic, and necrotic cells was determined by AO and EB staining. (b–d) Relative absorbance values, derived from FTIR-MSP analysis of untreated versus treated CCRF-CEM cells, for (b) lipids at 2852 cm−1, (c) DNA at 966 cm−1, and (d) proteins at 1639 cm−1 (random coil). Values plotted are the mean ± SE derived from at least three independent experiments.
Discussion
In this work, we demonstrate that the FTIR-MSP spectral pattern of dying leukemia cells reflects their cell death mode (necrosis versus apoptosis). Changes in three IR biomarkers characterize apoptosis: 1), an increase in lipid absorbance (Fig. 3, c and d); 2), a decrease in DNA absorbance (Fig. 3, e and f); and 3), an increase in the β-secondary structure of total cellular protein (Fig. 3, g and h). Similarly, during necrosis there are characteristic changes in three IR biomarkers. Similarly to apoptosis, an increase in lipid absorbance is seen (Figs. 4, a and b, 5 d, and 6 b), but in contrast to apoptosis, a dramatic increase in DNA absorbance (Figs. 4, a and b, 5 e, and 6 c) and a decrease in the random coil structure of proteins (Figs. 5 f and 6 d) are also observed.
Many, though not all (14,16), earlier studies reported increased lipid-related methylene absorbance after treatment with various apoptosis inducers, as observed in our experiments (13,15,17). At this time, the specific biochemical process responsible for these lipid-related IR spectral changes is unknown. Membrane changes associated with apoptosis, such as phosphatidylserine exposure, membrane blebbing, and vesicle formation (2), could explain the increased methylene absorbance. However, we find that increased lipid-related methylene absorbance is also characteristic of the IR spectral patterns of necrotic cells. Membrane changes typical of necrotic cells include swelling of cellular organelles and loss of plasma membrane integrity. Since apoptosis and necrosis do not involve similar macro membrane changes, one possibility is that the increased lipid absorbance observed during each process is caused by unrelated membrane events. Alternatively, it is possible there is a more subtle process occurring at the membrane shared by both cell death modes that underlies the common lipid absorbance changes, perhaps signaling via ceramide, FAS, or other death domain receptors (28–31).
Lipid absorbance does not distinguish apoptosis from necrosis; however, other spectral features do characterize the cell death mode. DNA absorbance appears to be a uniquely useful parameter for discerning the death mode since it decreases during apoptosis, as reported here and previously (13,14), and, in contrast, increases during necrosis as reported here for the first time, to our knowledge. Various explanations have been proposed as to why apoptotic DNA absorbs less IR, i.e., becomes opaque, despite DNA degradation during apoptosis. Primarily, this opacity is attributed to the condensation of DNA that occurs during apoptosis (32,33). More specifically, the opaque nature of apoptotic chromatin is likely due to its tight association with HMGB-1, which masks the chromatin from neighboring cells and prevents activation of the inflammatory response (34). In contrast, during necrosis the DNA is degraded but not compacted, which accords with our observation that necrotic DNA absorbs more IR, i.e., becomes less opaque (35).
Previous investigators encountered the phenomenon of a decline in DNA absorbance during the cell cycle and hypothesized that it resulted from DNA opaqueness or “non-Beer-Lambert absorption” due to the high density of DNA in the nucleus (32,33,36). However, this hypothesis remains to be validated (37). Furthermore, it is not yet clear what the DNA's opaqueness degree is.
Our results regarding apoptosis and necrosis identification may partly solve this enigma. In necrotic cell death, the DNA is completely unwound, and thus we may assume that 100% of the DNA is visible to IR at this stage. We observed an increase of ∼65% in DNA absorbance in necrosis relativel to the control. Therefore, the degree of opaqueness of DNA in its native state is only ∼61%. This can be further validated by the ∼16% decrease in absorption after DNA condensation that is observed in apoptotic cell death. Thus, by these experiments we can confirm the “non-Beer-Lambert absorption” theory. This confirmation may raise further important questions for future research regarding the possibility of opaqueness of other dense biomolecules such as lipids (or lipid rafts) and proteins.
Our study reveals that the capacity of FTIR spectroscopy to monitor global changes in protein secondary structure is also useful for identifying cell death mode, as apoptosis and necrosis appear to affect protein structures differentially. The significant increase of up to 75% in β-sheet structures associated with apoptosis reported here corroborates one earlier study (35) but contradicts another (13). The conflicting data could be due to the use of different model systems or cell death inducers, or may reflect the existence of alternative apoptosis pathways. In contrast, during necrosis we observe a decrease in the random coil structure of total protein. Reductions in random coil structure are seen after both KCN and H2O2 treatments, but to different extents (Figs. 5 f and 6 d) despite similar levels of necrosis (Figs. 5 a and 6 a). This quantitative difference may be related to the dissimilar target and action of each agent (28,29).
In summary, our data show that FTIR spectroscopy can distinguish cell death mode based on changes in DNA conformation and protein secondary structure. Furthermore, given these results, we propose a solution for a fundamental issue regarding IR opaqueness of DNA, which has further implications for other biomolecules. Since FTIR spectroscopy provides distinctive biochemical information and requires minimal sample handling and no reagents, it may provide a promising new tool to monitor cell death in the clinic.
Acknowledgments
This study was supported in part by grant 3-3680 from the Chief Scientist Office of the Ministry of Health, Israel.
References
- 1.Chowdhury I., Tharakan B., Bhat G.K. Current concepts in apoptosis: the physiological suicide program revisited. Cell. Mol. Biol. Lett. 2006;11:506–525. doi: 10.2478/s11658-006-0041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hengartner M.O. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 3.Rudin C.M., Thompson C.B. Apoptosis and disease: regulation and clinical relevance of programmed cell death. Annu. Rev. Med. 1997;48:267–281. doi: 10.1146/annurev.med.48.1.267. [DOI] [PubMed] [Google Scholar]
- 4.Fischer U., Schulze-Osthoff K. New approaches and therapeutics targeting apoptosis in disease. Pharmacol. Rev. 2005;57:187–215. doi: 10.1124/pr.57.2.6. [DOI] [PubMed] [Google Scholar]
- 5.Proskuryakov S.Y., Konoplyannikov A.G., Gabai V.L. Necrosis: a specific form of programmed cell death? Exp. Cell Res. 2003;283:1–16. doi: 10.1016/s0014-4827(02)00027-7. [DOI] [PubMed] [Google Scholar]
- 6.Vanlangenakker N., Berghe T.V., Krysko D.V., Festjens N., Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008;8:207–220. doi: 10.2174/156652408784221306. [DOI] [PubMed] [Google Scholar]
- 7.Hou S.T., MacManus J.P. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int. Rev. Cytol. 2002;221:93–148. doi: 10.1016/s0074-7696(02)21011-6. [DOI] [PubMed] [Google Scholar]
- 8.Debatin K.M., Stahnke K., Fulda S. Apoptosis in hematological disorders. Semin. Cancer Biol. 2003;13:149–158. doi: 10.1016/s1044-579x(02)00132-3. [DOI] [PubMed] [Google Scholar]
- 9.Fink S.L., Cookson B.T. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diem M., Griffiths P., Chalmers J. John Wiley & Sons; New York: 2008. Vibrational Spectroscopy for Medical Diagnosis. [Google Scholar]
- 11.Liu K.Z., Xu M., Scott D.A. Biomolecular characterisation of leucocytes by infrared spectroscopy. Br. J. Haematol. 2007;136:713–722. doi: 10.1111/j.1365-2141.2006.06474.x. [DOI] [PubMed] [Google Scholar]
- 12.Mourant J.R., Gibson R.R., Johnson T.M., Carpenter S., Short K.W. Methods for measuring the infrared spectra of biological cells. Phys. Med. Biol. 2003;48:243–257. doi: 10.1088/0031-9155/48/2/307. [DOI] [PubMed] [Google Scholar]
- 13.Liu K.Z., Jia L., Kelsey S.M., Newland A.C., Mantsch H.H. Quantitative determination of apoptosis on leukemia cells by infrared spectroscopy. Apoptosis. 2001;6:269–278. doi: 10.1023/a:1011383408381. [DOI] [PubMed] [Google Scholar]
- 14.Gasparri F., Muzio M. Monitoring of apoptosis of HL60 cells by Fourier-transform infrared spectroscopy. Biochem. J. 2003;369:239–248. doi: 10.1042/BJ20021021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jamin N., Miller L., Moncuit J., Fridman W.H., Dumas P. Chemical heterogeneity in cell death: combined synchrotron IR and fluorescence microscopy studies of single apoptotic and necrotic cells. Biopolymers. 2003;72:366–373. doi: 10.1002/bip.10435. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J., Wang Z., Sun S., Liu M., Zhang H. A rapid method for detecting conformational changes during differentiation and apoptosis of HL60 cells by Fourier-transform infrared spectroscopy. Biotechnol. Appl. Biochem. 2001;33:127–132. doi: 10.1042/ba20000074. [DOI] [PubMed] [Google Scholar]
- 17.Gault N., Rigaud O., Poncy J.L., Lefaix J.L. Infrared microspectroscopy study of gamma-irradiated and H2O2-treated human cells. Int. J. Radiat. Biol. 2005;81:767–779. doi: 10.1080/09553000500515368. [DOI] [PubMed] [Google Scholar]
- 18.Grusch M., Polgar D., Gfatter S., Leuhuber K., Huettenbrenner S. Maintenance of ATP favours apoptosis over necrosis triggered by benzamide riboside. Cell Death Differ. 2002;9:169–178. doi: 10.1038/sj.cdd.4400937. [DOI] [PubMed] [Google Scholar]
- 19.Didenko V.V., Ngo H. Baskin DS. Early necrotic DNA degradation: presence of blunt-ended DNA breaks, 3′ and 5′ overhangs in apoptosis, but only 5′ overhangs in early necrosis. Am. J. Pathol. 2003;162:1571–1578. doi: 10.1016/S0002-9440(10)64291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang D., Shi Y., Kang R., Li T., Xiao W. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 2007;81:741–747. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardella S., Andrei C., Ferrera D., Lotti L.V., Torrisi M.R. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGahon A.J., Martin S.J., Bissonnette R.P., Mahboubi A., Shi Y. The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol. 1995;46:153–185. doi: 10.1016/s0091-679x(08)61929-9. [DOI] [PubMed] [Google Scholar]
- 23.Bogomolny E., Huleihel M., Suproun Y., Sahu R.K., Mordechai S. Early spectral changes of cellular malignant transformation using Fourier transform infrared microspectroscopy. J Biomed. Opt. 2007;12:024003. doi: 10.1117/1.2717186. [DOI] [PubMed] [Google Scholar]
- 24.Toyran N., Turan B., Severcan F. Selenium alters the lipid content and protein profile of rat heart: an FTIR microspectroscopic study. Arch. Biochem. Biophys. 2007;458:184–193. doi: 10.1016/j.abb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 25.Diem M., Romeo M., Boydston-White S., Miljkovic M., Matthaus C. A decade of vibrational micro-spectroscopy of human cells and tissue (1994–2004) Analyst (Lond.) 2004;129:880–885. doi: 10.1039/b408952a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arakawa H., Neault J.F., Tajmir-Riahi H.A. Silver(I) complexes with DNA and RNA studied by Fourier transform infrared spectroscopy and capillary electrophoresis. Biophys. J. 2001;81:1580–1587. doi: 10.1016/S0006-3495(01)75812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petibois C., Déléris G. Chemical mapping of tumor progression by FT-IR imaging: towards molecular histopathology. Trends Biotechnol. 2006;24:455–462. doi: 10.1016/j.tibtech.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Thon L., Möhlig H., Mathieu S., Lange A., Bulanova E. Ceramide mediates caspase-independent programmed cell death. FASEB J. 2005;19:1945–1956. doi: 10.1096/fj.05-3726com. [DOI] [PubMed] [Google Scholar]
- 29.Troyano A., Sancho P., Fernandez C., de Blas E., Bernardi P. The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide (H2O2)-treated and glutathione-depleted human promonocytic cells. Cell Death Differ. 2003;10:889–898. doi: 10.1038/sj.cdd.4401249. [DOI] [PubMed] [Google Scholar]
- 30.Vanden Berghe T., van Loo G., Saelens X., Van Gurp M., Brouckaert G. Differential signaling to apoptotic and necrotic cell death by Fas-associated death domain protein FADD. J. Biol. Chem. 2004;279:7925–7933. doi: 10.1074/jbc.M307807200. [DOI] [PubMed] [Google Scholar]
- 31.Denecker G., Vercammen D., Declercq W., Vandenabeele P. Apoptotic and necrotic cell death induced by death domain receptors. Cell. Mol. Life Sci. 2001;58:356–370. doi: 10.1007/PL00000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boydston-White S., Gopen T., Houser S., Bargonetti J., Diem M. Infrared spectroscopy of Human tissue. V. Infrared spectroscopic studies of myeloid leukemia (ML-1) cells at different phases of the cell cycle. Biospectroscopy. 1999;5:219–227. doi: 10.1002/(SICI)1520-6343(1999)5:4<219::AID-BSPY2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 33.Holman H.Y., Martin M.C., Blakely E.A., Bjornstad K., McKinney W.R. IR spectroscopic characteristics of cell cycle and cell death probed by synchrotron radiation based Fourier transform IR spectromicroscopy. Biopolymers. 2000;57:329–335. doi: 10.1002/1097-0282(2000)57:6<329::AID-BIP20>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Raucci A., Palumbo R., Bianchi M.E. HMGB1: a signal of necrosis. Autoimmunity. 2007;40:285–289. doi: 10.1080/08916930701356978. [DOI] [PubMed] [Google Scholar]
- 35.Gault N., Lefaix J.L. Infrared microspectroscopic characteristics of radiation-induced apoptosis in human lymphocytes. Radiat. Res. 2003;160:238–250. doi: 10.1667/rr3020.1. [DOI] [PubMed] [Google Scholar]
- 36.Mohlenhoff B., Romeo M., Diem M., Wood B.R. Mie-type scattering and non-Beer-Lambert absorption behavior of human cells in infrared microspectroscopy. Biophys. J. 2005;88:3635–3640. doi: 10.1529/biophysj.104.057950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mourant J.R., Yamada Y.R., Carpenter S., Dominique L.R., Freyer J.P. FTIR spectroscopy demonstrates biochemical differences in mammalian cell cultures at different growth stages. Biophys. J. 2003;85:1938–1947. doi: 10.1016/S0006-3495(03)74621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]