

Abstract
Purpose
To screen for mutations of connexin50 (Cx50)/GJA8 in a panel of patients with inherited cataract and to determine the cellular and functional consequences of the identified mutation.
Methods
All patients in the study underwent a full clinical examination and leucocyte DNA was extracted from venous blood. The GJA8 gene was sequenced directly. Connexin function and cellular trafficking were examined by expression in Xenopus oocytes and HeLa cells.
Results
Screening of the GJA8 gene identified a 139 G to A transition that resulted in the replacement of aspartic acid by asparagine (D47N) in the coding region of Cx50. This change co-segregated with cataract among affected members of a family with autosomal dominant nuclear pulverulent cataracts. While pairs of Xenopus oocytes injected with wild type Cx50 RNA formed functional gap junction channels, pairs of oocytes injected with Cx50D47N showed no detectable intercellular conductance. Co-expression of Cx50D47N did not inhibit gap junctional conductance of wild type Cx50. In transiently transfected HeLa cells, wild type Cx50 localised to appositional membranes and within the perinuclear region, but Cx50D47N showed no immunostaining at appositional membranes with immunoreactivity confined to the cytoplasm. Incubation of HeLa cells transfected with Cx50D47N at 27°C resulted in formation of gap junctional plaques.
Conclusions
The pulverulent cataracts present in members of this family are associated with a novel GJA8 mutation, Cx50D47N, that acts as a loss-of-function mutation. The consequent decrease in lens intercellular communication and changes associated with intracellular retention of the mutant connexin may contribute to cataract formation.
Congenital cataracts account for 10% of childhood blindness and are the most common treatable cause of childhood visual impairment. Approximately half are genetically determined. Inherited cataracts are genetically and phenotypically heterogeneous; inheritance is most commonly autosomal dominant although autosomal recessive and X-linked forms have also been reported.1 Thirteen genes have been implicated in hereditary isolated congenital cataracts including the genes encoding the gap junction proteins connexin46 (Cx46), GJA3, and connexin50 (Cx50), GJA8.2-18
Gap junctions are membrane specialisations that contain clusters of intercellular channels. Each channel is formed by docking of two hexamers made of subunit proteins called connexins. These channels allow transfer of ions and molecules up to 1000 Da between adjacent cells.19 In the lens, epithelial cells express mainly Cx43,20 21 whereas fibre cells express Cx4622 and Cx50.23 Here, we report a novel GJA8 mutation, Cx50D47N, in a family with nuclear pulverulent cataract, and characterise its functional and biological properties in order to understand the pathological consequences of the mutation.
METHODS
Patient ascertainment and collection of genetic material
Patients with autosomal dominant inherited cataract seen at Moorfields Eye Hospital, London and the Hospital for Children, Great Ormond Street, London and other family members were invited to take part in a study of the molecular genetics of inherited cataract. Ethical approval for this study was obtained from the local research ethics committee. Written informed consent, which followed the tenets of the Declaration of Helsinki, was obtained from all adult individuals and from the parents of children under 16 years of age. One hundred and fifty families agreed to participate, a full family history was taken, and all individuals underwent a full clinical examination including slit lamp examination after pupillary dilatation. Genomic DNA was extracted from venous blood leucocytes using the Nucleon II DNA extraction kit (Tepnel Life Sciences).
Sequencing
The entire coding region of the GJA8 gene (exon 2) was amplified from genomic DNA by polymerase chain reaction (PCR) using primers:
Cx501F: GCTCAGCTCTTGCCTTCTCC,
Cx501R: GCTGCAGCGGTACAGAGG,
Cx502F: GGCAGCAAAGGCACTAAGAA,
Cx502R: GAACTGATTGAAAGGCTTG,
Cx503F: CCCACTATTTCCCCTTGACC,
Cx503R: TCCTTTCATCTTGCCCTACG.
PCR products were purified from agarose gels using the QIA-quick gel extraction kit (Qiagen) and then subcloned into pGEM-TEasy (Promega). Plasmid DNA was purified by the GenElute plasmid miniprep kit (Sigma). The DNA insert was cycle-sequenced with Big Dye Terminator Ready Reaction Mix (Applied Biosystems) and analysed on an ABI PRISM 3100 Genetic Analyser (Applied Biosystems).
Subcloning of human and mouse Cx50 DNA
Subcloning of human wild type Cx50 into pcDNA3.1/Hygro(+) (Invitrogen Life Technologies) and pSP64TII has been reported.3 The mutant (Cx50D47N) allele was generated by site directed mutagenesis using the Quick Change Site-Directed Mutagenesis Kit (Stratagene, UK) using primers:
5′CAGAGTTCGTGTGGGGGAATGAGCAATCCGACTTC and
3′GAAGTCGGATTGCTCATTCCCCCACACGAACTCTG.
Products were sequenced to check the fidelity of the amplification reaction. The PCR products were subcloned into pcDNA3.1/Hygro(+) or the RNA expression vector, pSP64TII.24 Wild type and mutant (Cx50D47A) mouse Cx5025 26 were subcloned into pcDNA3.1/Hygro(+).
Cell culture and transient transfection
HeLa cells were grown in MEM supplemented with non-essential amino acids, 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin G and 100 μg/ml streptomycin sulfate. Cell transfections with the pcDNA3.1/Hygro(+) constructs were performed using lipofectin and Plus reagent (Invitrogen Life Technologies). For the temperature-shift experiments, cells were transfected and grown at 37°C for 24 h. Then they were either transferred to 27°C or maintained at 37°C for an additional 24 h.
Immunofluorescence
HeLa cells were plated on four-well chamber slides (LAB-TEK Nalge Nunc International) and transiently transfected with wild type and mutant Cx50. Forty-eight hours post-transfection, cells were rinsed with phosphate buffered saline (PBS) and fixed for 15 min in 4% paraformaldehyde at room temperature and permeabilised in 1% Triton X-100 in PBS. Immunofluorescence was performed using affinity purified rabbit polyclonal anti-Cx50 antibodies and mouse monoclonal antibodies against protein disulphide isomerase (Affinity Bioreagents), ERGIC-53 (Axxora LLC, San Diego, California, USA) or Golgi 58K protein (Sigma Chemical Company) as previously described.27
Immunoblotting
Cells at about 90% confluence were harvested in PBS, 4 mM EDTA, 2 mM PMSF, 1 mg/ml Pefabloc, 10 μg/ml leupeptin, 10 μg/ml pepstatin and 1 μg/ml aprotinin, and sonicated. Aliquots from cell homogenates were resolved on 9% SDS-containing polyacrylamide gels, transferred to Immobilon P membranes (Millipore), and subjected to immunoblotting using anti-Cx50 antibodies.27
Expression of connexins in Xenopus oocytes
Plasmids were linearised with SalI. Capped RNA was synthe-sised using the mMessage mMachine SP6 in vitro transcription kit (Ambion, Austin, Texas, USA). RNA was quantified by measuring absorbance at 260 nm and purity was further assessed by agarose gel electrophoresis.
Adult female Xenopus laevis were anaesthetised with tricaine and a partial ovariectomy was performed. The animals were maintained and treated in accordance with National Institute of Health guidelines. Oocytes were defolliculated and microin-jected with an oligonucleotide antisense to the endogenous Xenopus Cx38 and 14.5 ng of connexin cRNAs (or no RNA for controls) as previously described.28
Electrophysiological measurements
Connexin cRNA-injected oocytes were devitellinised and paired.29 The oocyte pairs were allowed to incubate at room temperature for 24 h before electrophysiological recording. Double two-microelectrode voltage-clamp experiments were performed using Geneclamp 500 and an Axoclamp 2A voltage-clamp amplifier (Axon instruments, Union City, California, USA). The microelectrodes were filled with 3 M KCl and had a resistance between 0.1–0.6 MΩ. To prevent electrode leakage, the tips of the electrodes were backfilled with 1% agar in 3 M KCl. For measurements of gap junctional coupling, both cells of a pair were held initially at −40 mV; 5–10 mV increments were applied to one cell while holding the second cell at −40 mV. The junctional conductance (gj) was calculated as (gj = Ij/Vj), where Vj = Vcell 2−Vcell 1. Pulse generation and data acquisition were performed using a PC computer equipped with PCLAMP6 software and a TL-1 acquisition system (Axon Instruments). Currents were filtered at 20–50 Hz and digitised using PCLAMP6 software and a Digidata 1200 (Axon Instruments). All experiments were performed at room temperature (20–22°C).
RESULTS
Identification of a new Cx50 mutation
Sequence analysis of the GJA8 gene revealed a novel GJA8 heterozygous mutation in one patient in the panel who had nuclear pulverulent opacities (fig 1A,B). The mutated sequence contained a 139G>A nucleotide transition within the coding region that resulted in the replacement of aspartic acid by asparagine (D47N) (fig 1C). Four members of this two generation Caucasian family were affected with pulverulent cataracts that were inherited in an autosomal dominant pattern (fig 1D). The sequence change segregated together with the cataracts in affected family members and was not seen in any of 156 ethnically matched controls.
Figure 1. Cataract phenotype and mutational analysis.
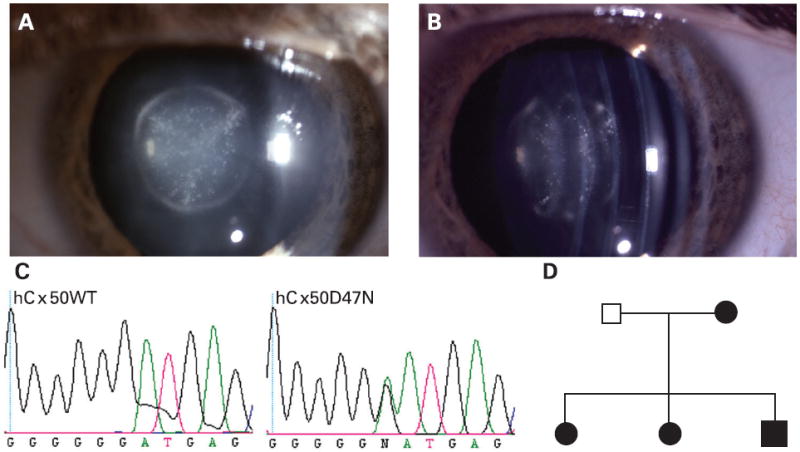
(A) Slit lamp photograph of the lens of an affected individual with a congenital nuclear pulverulent cataract. (B) Oblique view of the same lens demonstrating opacities confined to the fetal and embryonal nucleus. (C) Chromatogram of the DNA sequence from an unaffected individual shows only the wild type Cx50 allele (hCx50WT) which encodes aspartic acid (GAT) at codon 47. The sequence chromatogram from an affected individual (hCx50D47N) shows both G and A at position 139; thus, the mutant allele contains a G to A transition resulting in the substitution of asparagine for aspartate at amino acid residue 47. (D) Pedigree of the family with autosomal dominant nuclear pulverulent cataracts. Black symbols indicate affected individuals and white symbols indicate unaffected individuals.
Altered trafficking/targeting of Cx50D47N
To study the cellular consequences of this mutation, HeLa cells were transiently transfected with wild type Cx50 and Cx50D47N. Protein production was confirmed by immunoblotting. A band of Mr ~62 kDa was detected in homogenates of cells transfected with either wild type Cx50 or Cx50D47N (fig 2A). No immunoreactive Cx50 bands were seen in homogenates of untransfected HeLa cells.3 27
Figure 2. Expression of wild type and mutant Cx50 in transfected cells as detected by immunoblotting and immunofluorescence.

(A) Proteins from homogenates of HeLa cells transiently transfected with wild type Cx50 (lane 1) or Cx50D47N (lane 2) were resolved by SDS-PAGE and subjected to immunoblotting using rabbit polyclonal anti-human Cx50 antibodies. The positions of the molecular mass standards are indicated on the right. (B, C) Photomicrographs show the distribution of immunoreactivity to anti-Cx50 antibodies in HeLa cells transfected with wild type (B) or mutant (C) Cx50. Gap junctions are indicated by arrows. Bar, 12 μm for B and 11 μm for C.
The distribution of Cx50 in transiently transfected HeLa cells was assessed by immunofluorescence (fig 2B, C). Wild type Cx50 localised at appositional membranes and within the perinuclear region as expected.3 27 In contrast, in cells transfected with Cx50D47N, anti-Cx50 immunoreactivity was found within the cytoplasm.
To determine the intracellular compartment in which Cx50D47N localised, we performed double labelling immunofluorescence experiments using antibodies against proteins that reside in different subcellular compartments. Anti-Cx50 immunoreactivity nearly precisely overlapped with the distribution of protein disulfide isomerase (an ER-resident protein) (fig 3A–C). Cx50D47N did not co-localise with immunoreactivities for resident proteins of the ER-Golgi Intermediate Compartment (ERGIC-53) or Golgi (Golgi-58K) (data not shown).
Figure 3. Subcellular localisation of Cx50D47N in transfected cells.
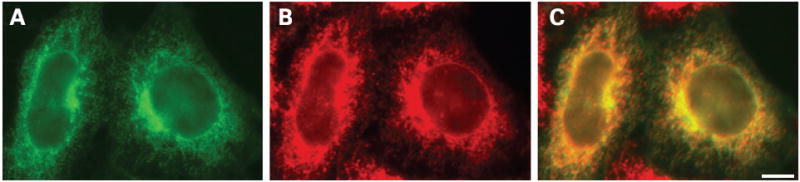
(A–C) Photomicrographs show the distribution of Cx50 (A) and protein disulfide isomerase (B) after double-immunofluorescence staining of HeLa cells transiently transfected with Cx50D47N using rabbit polyclonal anti-Cx50 antibodies and a mouse monoclonal antibody against protein disulfide isomerase. The overlap of the immunofluorescent signals in A and B appears yellow in C. Bar, 7.4 μm.
Several mutant transmembrane proteins that traffic poorly to the plasma membrane can reach the plasma membrane more efficiently when they are incubated at reduced temperatures.30 To test whether trafficking of the mutant Cx50 was temperature sensitive, HeLa cells transiently transfected with Cx50D47N were subjected to a temperature shift by incubating the cells at 27°C. Under these conditions, anti-Cx50 immunoreactive gap junction plaques were observed (fig 4).
Figure 4. Distribution of Cx50D47N in transfected cells incubated at normal or reduced temperatures.
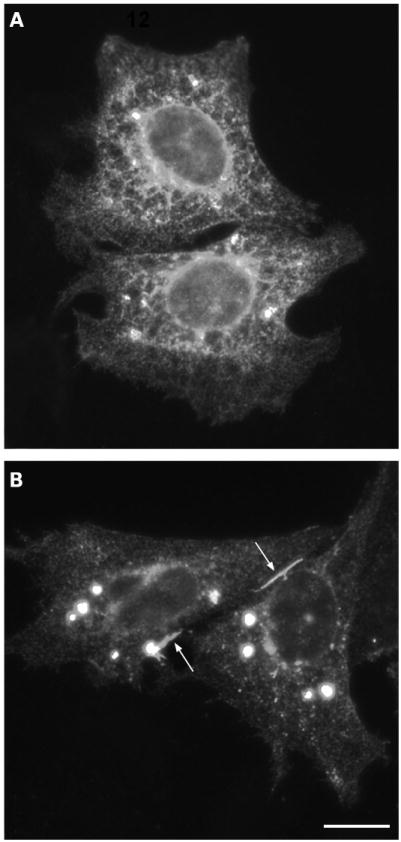
(A, B) Photomicrographs show the distribution of anti-Cx50 immunoreactivity in HeLa cells transiently transfected with Cx50D47N that were incubated at 37°C and kept at 37°C (A) or transferred to 27°C for 24 h (B). Gap junctions are indicated by arrows. Bar, 14.8 μm.
Functional characterisation of wild type and mutant Cx50
The functional behaviour of Cx50D47N was examined by expression in Xenopus oocytes. Oocyte pairs injected with wild type Cx50 cRNA developed large gap junctional conductances when tested 24 h after pairing (table 1). In contrast, oocytes injected with Cx50D47N cRNA and paired for 24 h did not demonstrate significant gap junctional coupling as compared to control oocytes injected only with Xenopus Cx38 antisense oligonucleotides (table 1). Similar results were obtained in two other batches of oocytes from different frogs (data not shown).
Table 1.
Wild type and mutant Cx50 gap junction coupling
| Oocyte injection |
Gap junction coupling |
||
|---|---|---|---|
| Cell 1 | Cell 2 | Mean conductance† (μS) | n |
| Wild type Cx50 | Wild type Cx50 | 15.77 (3.81) | 10 |
| Cx50D47N | Cx50D47N | 0.073 (0.010)* | 10 |
| Wild type Cx50:Cx50D47N (1:1) | Wild type Cx50:Cx50D47N (1:1) | 21.70 (4.00) | 10 |
| No connexin cRNA | No connexin cRNA | 0.073 (0.011)* | 5 |
p<0.01 as compared to wild type Cx50 by Student’s t-test
Values represent mean (SEM); n, number of cell pairs.
To facilitate comparison of the homotypic and heteromeric pair data, the amount of wild type Cx50 cRNA injected into each cell was held constant.
To determine the effect of mutant Cx50D47N upon wild type Cx50 channel activity, gap junctional conductance was recorded from oocyte pairs co-injected with equal amounts of wild type Cx50 and Cx50D47N cRNAs. These pairs showed no reduction in gap junction conductance compared to oocytes injected with wild type Cx50 alone, suggesting that Cx50D47N acts as a loss-of-function mutation.
Subcellular localisation of mouse Cx50D47A
A mouse Cx50 mutant associated with cataracts also has a missense mutation at amino acid residue 47; in these mice, aspartate 47 is replaced with alanine (Cx50D47A).25 Similar to human Cx50D47N, mouse Cx50D47A behaves as a loss-of-function mutant when expressed in Xenopus oocytes.26 To test whether mouse Cx50D47A also showed impaired trafficking, HeLa cells were transiently transfected with wild type mouse Cx50 or mouse Cx50D47A and their subcellular distribution was studied by immunofluorescence. While gap junction plaques were detected in cells transfected with wild type mouse Cx50, no gap junction plaques were observed in cells transfected with mouse Cx50D47A (fig 5A and data not shown). The mutant connexin localised intracellularly and showed abundant overlap with markers for ER (fig 5).
Figure 5. Distribution of mouse Cx50D47A in transfected cells.

(A–C) Photomicrographs show the distribution of anti-Cx50 (A) and anti-protein disulfide isomerase (B) immunoreactivities in HeLa cells transiently transfected with mouse Cx50D47A after double immunofluorescence. The overlap of the immunofluorescent signals in A and B appears yellow in C. Bar, 13.8 μm.
DISCUSSION
We have identified a novel mutation in the GJA8 gene encoding Cx50 in affected individuals within a family with autosomal dominant nuclear pulverulent cataract. Cx50 is a gap junction protein expressed in mature lens fibres23 and lenticular epithelial cells.31 Gap junction channels facilitate lens homeostasis by contributing an intercellular pathway that is integral to the internal microcirculation of the lens32; gap junctions are essential for maintaining lens transparency.33 Connexin50 has also been implicated in cell proliferation and lens size.34 35 The central role of connexins in maintaining lens transparency in man has been confirmed by the identification of several mutations in Cx46 and Cx50 in individuals with congenital cataract.2-18
Connexins are integral membrane proteins containing four transmembrane domains, two extracellular loops, an intracellular loop, and cytoplasmic amino and carboxyl termini. The 139G>A transition in Cx50 results in the substitution of a polar, uncharged asparagine for a negatively charged aspartic acid at position 47. The aspartic acid at position 47 is conserved in all connexins (position 46 in β-connexins). Structural analysis of connexins has not been performed at a sufficiently high resolution to determine the localisation of specific amino acids in relation to the membrane or other parts of the protein. However, hydropathy plots of Cx50 and other connexins place aspartate 47 at the interface between the first transmembrane domain and the first extracellular loop (E1) or within the first few amino acids of E1. Modification of charge at this position may result in altered protein folding/conformation. The intracellular localisation of the mutant protein and the lack of gap junction plaques in transfected cells support the hypothesis that this amino acid substitution caused a conformational change that impaired trafficking. Moreover, Cx50D47N was able to form gap junction plaques when the cells were grown under a condition that facilitates protein folding (reduced temperature), further suggesting that a conformational change was responsible for the intracellular localisation.
Cx50D47N did not form functional gap junction channels when expressed in Xenopus oocyte pairs. Furthermore, co-expression of Cx50D47N with wild type Cx50 did not inhibit the activity of wild type Cx50. This behaviour is very similar to that of mouse Cx50D47A,26 a mutation underlying the cataracts in the No2 mouse.25 This observation contrasts with the behaviour of other Cx50 mutants (that is, Cx50P88S or Cx50P88Q) that act as dominant negatives by significantly decreasing the activity of co-expressed wild type Cx50.3 36 The simplest explanation for this behaviour is that Cx50D47N does not oligomerise with wild type Cx50 to form hexamers.
Several amino acid substitutions of D47 or nearby amino acids have been identified in Cx50 in association with congenital cataracts in mice and humans including Cx50D47A, Cx50E48K, and Cx50V44E.5 8 25 Mutations of residues in this region of other connexins have been identified in families with deafness,37-39 keratitis–ichtyosis–deafness syndrome,40 41 and X-linked Charcot–Marie–Tooth disease42 and include Cx26W44C,37 Cx26W44S,38 Cx26V37I,39 Cx26D50N40 41 and Cx32C53S,42 respectively.
Reduced intercellular communication due to the presence of only one functional Cx50 gene likely contributes but does not fully explain the cataract observed in individuals with the Cx50D47N mutation. Mice that are heterozygous for a targeted deletion of Cx50 do not develop cataracts34 even though the 50% reduction in Cx50 is associated with a 28% reduction in normalised coupling conductance in differentiating fibres.43 Mice heterozygous for the Cx50D47A mutation have cataractous lenses25 44 as do affected members of the family reported here who carry one copy of the Cx50D47N mutation. There are several possible explanations for the difference in phenotypes between heterozygous Cx50-null mice and individuals carrying non-functional Cx50 mutations: (1) cytoplasmic retention of the mutant protein (human Cx50D47N or mouse Cx50D47A) may have additional adverse effects on the lens; (2) the mutant protein may interact and alter the function of other lens fibre cell components including other connexins; and (3) other modifying genes may be present, since the severity of cataracts in the Cx46- and Cx50-null mice has been previously shown to vary with genetic background.45 46
In summary, Cx50D47N is a novel mutation in the GJA8 gene that was identified in an English family with autosomal dominant nuclear pulverulent congenital cataracts. The Cx50D47N protein localised within the ER and did not form gap junction plaques at 37°C, suggesting that it has a trafficking impairment. Furthermore, the mutant Cx50 acted as a loss-of-channel-function mutation without dominant negative inhibition of wild type Cx50 when expressed in Xenopus oocyte pairs. These results suggest that the cataracts observed in these patients are due to changes secondary to retention of the mutant connexin within the synthetic/secretory pathway, including reduced intercellular communication.
Acknowledgments
We would like to thank the family for participating in this study. Anita Arora is supported by the Wellcome Trust (Grant 068083/Z/02/Z). Eric Beyer is supported by the National Institute of Health (Grant EY08368). Lisa Ebihara is supported by the National Institute of Health (Grant EY10589).
Footnotes
Competing interests: None declared.
Ethics approval: Ethical approval for this study was obtained from the local research ethics committee
Patient consent: Informed consent was obtained from all adult individuals and from the parents of children under 16 years of age for publication of this report
References
- 1.Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT. Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol. 2004;49:300–15. doi: 10.1016/j.survophthal.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Addison PK, Berry V, Holden KR, Espinal D, Rivera B, Su H, Srivastava AK, Bhattacharya SS. A novel mutation in the connexin 46 gene (GJA3) causes autosomal dominant zonular pulverulent cataract in a Hispanic family. Mol Vis. 2006;12:791–5. [PubMed] [Google Scholar]
- 3.Arora A, Minogue PJ, Liu X, Reddy MA, Ainsworth JR, Bhattacharya SS, Webster AR, Hunt DM, Ebihara L, Moore AT, Beyer EC, Berthoud VM. A novel GJA8 mutation is associated with autosomal dominant lamellar pulverulent cataract: further evidence for gap junction dysfunction in human cataract. J Med Genet. 2006;43:e2. doi: 10.1136/jmg.2005.034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett TM, Mackay DS, Knopf HL, Shiels A. A novel missense mutation in the gene for gap-junction protein α3 (GJA3) associated with autosomal dominant “nuclear punctuate” cataracts linked to chromosome 13q. Mol Vis. 2004;10:376–82. [PubMed] [Google Scholar]
- 5.Berry V, Mackay D, Khaliq S, Francis PJ, Hameed A, Anwar K, Mehdi SQ, Newbold RJ, Ionides A, Shiels A, Moore T, Bhattacharya SS. Connexin 50 mutation in a family with congenital “zonular nuclear” pulverulent cataract of Pakistani origin. Hum Genet. 1999;105:168–70. doi: 10.1007/s004399900094. [DOI] [PubMed] [Google Scholar]
- 6.Burdon KP, Wirth MG, Mackey DA, Russell-Eggitt IM, Craig JE, Elder JE, Dickinson JL, Sale MM. A novel mutation in the Connexin 46 gene causes autosomal dominant congenital cataract with incomplete penetrance. J Med Genet. 2004;41:e106. doi: 10.1136/jmg.2004.018333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devi RR, Reena C, Vijayalakshmi P. Novel mutations in GJA3 associated with autosomal dominant congenital cataract in the Indian population. Mol Vis. 2005;11:846–52. [PubMed] [Google Scholar]
- 8.Devi RR, Vijayalakshmi P. Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea. Mol Vis. 2006;12:190–5. [PubMed] [Google Scholar]
- 9.Hansen L, Yao W, Eiberg H, Funding M, Riise R, Kjaer KW, Hejtmancik JF, Rosenberg T. The congenital “ant-egg” cataract phenotype is caused by a missense mutation in connexin46. Mol Vis. 2006;12:1033–9. [PubMed] [Google Scholar]
- 10.Jiang H, Jin Y, Bu L, Zhang W, Liu J, Cui B, Kong X, Hu L. A novel mutation in GJA3 (connexin46) for autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2003;9:579–83. [PubMed] [Google Scholar]
- 11.Li Y, Wang J, Dong B, Man H. A novel connexin46 (GJA3) mutation in autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2004;10:668–71. [PubMed] [Google Scholar]
- 12.Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet. 1999;64:1357–64. doi: 10.1086/302383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Polyakov AV, Shagina IA, Khlebnikova OV, Evgrafov OV. Mutation in the connexin 50 gene (GJA8) in a Russian family with zonular pulverulent cataract. Clin Genet. 2001;60:476–8. doi: 10.1034/j.1399-0004.2001.600614.x. [DOI] [PubMed] [Google Scholar]
- 14.Rees MI, Watts P, Fenton I, Clarke A, Snell RG, Owen MJ, Gray J. Further evidence of autosomal dominant congenital zonular pulverulent cataracts linked to 13q11 (CZP3) and a novel mutation in connexin 46 (GJA3) Hum Genet. 2000;106:206–9. doi: 10.1007/s004390051029. [DOI] [PubMed] [Google Scholar]
- 15.Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet. 1998;62:526–32. doi: 10.1086/301762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanita V, Hennies HC, Singh D, Nurnberg P, Sperling K, Singh JR. A novel mutation in GJA8 associated with autosomal dominant congenital cataract in a family of Indian origin. Mol Vis. 2006;12:1217–22. [PubMed] [Google Scholar]
- 17.Willoughby CE, Arab S, Gandhi R, Zeinali S, Arab S, Luk D, Billingsley G, Munier FL, Héon E. A novel GJA8 mutation in an Iranian family with progressive autosomal dominant congenital nuclear cataract. J Med Genet. 2003;40:e124. doi: 10.1136/jmg.40.11.e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng JQ, Ma ZW, Sun HM. A heterozygous transversion of connexin 50 in a family with congenital nuclear cataract in the northeast of China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2005;22:76–8. [PubMed] [Google Scholar]
- 19.Sáez JC, Berthoud VM, Braňes MC, Martínez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- 20.Beyer EC, Kistler J, Paul DL, Goodenough DA. Antisera directed against connexin43 peptides react with a 43-kD protein localized to gap junctions in myocardium and other tissues. J Cell Biol. 1989;108:595–605. doi: 10.1083/jcb.108.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musil LS, Beyer EC, Goodenough DA. Expression of the gap junction protein connexin43 in embryonic chick lens: molecular cloning, ultrastructural localization, and post-translational phosphorylation. J Membr Biol. 1990;116:163–75. doi: 10.1007/BF01868674. [DOI] [PubMed] [Google Scholar]
- 22.Paul DL, Ebihara L, Takemoto LJ, Swenson KI, Goodenough DA. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J Cell Biol. 1991;115:1077–89. doi: 10.1083/jcb.115.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White TW, Bruzzone R, Goodenough DA, Paul DL. Mouse Cx50, a functional member of the connexin family of gap junction proteins, is the lens fiber protein MP70. Mol Biol Cell. 1992;3:711–20. doi: 10.1091/mbc.3.7.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebihara L, Berthoud VM, Beyer EC. Distinct behavior of connexin56 and connexin46 gap junctional channels can be predicted from the behavior of their hemi-gap-junctional channels. Biophys J. 1995;68:1796–803. doi: 10.1016/S0006-3495(95)80356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steele EC, Jr, Lyon MF, Favor J, Guillot PV, Boyd Y, Church RL. A mutation in the connexin 50 (Cx50) gene is a candidate for the No2 mouse cataract. Curr Eye Res. 1998;17:883–9. doi: 10.1076/ceyr.17.9.883.5144. [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Ebihara L. Characterization of a mouse Cx50 mutation associated with the No2 mouse cataract. Invest Ophthalmol Vis Sci. 1999;40:1844–50. [PubMed] [Google Scholar]
- 27.Berthoud VM, Minogue PJ, Guo J, Williamson EK, Xu X, Ebihara L, Beyer EC. Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur J Cell Biol. 2003;82:209–21. doi: 10.1078/0171-9335-00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong JJ, Liu X, Dong L, Ebihara L. Exchange of gating properties between rat cx46 and chicken cx45.6. Biophys J. 2004;87:2397–406. doi: 10.1529/biophysj.104.039594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebihara L. Expression of gap junction proteins in Xenopus oocyte pairs. In: Rudy Y, Iverson LE, editors. Ion channels. San Diego: Academic Press; 1992. pp. 376–80. [DOI] [PubMed] [Google Scholar]
- 30.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature sensitive. Nature. 1992;358:761–4. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 31.Rong P, Wang X, Niesman I, Wu Y, Benedetti LE, Dunia I, Levy E, Gong X. Disruption of GJA8 (α8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development. 2002;129:167–74. doi: 10.1242/dev.129.1.167. [DOI] [PubMed] [Google Scholar]
- 32.Mathias RT, Rae JL, Baldo GJ. Physiological properties of the normal lens. Physiol Rev. 1997;77:21–50. doi: 10.1152/physrev.1997.77.1.21. [DOI] [PubMed] [Google Scholar]
- 33.Goodenough DA. The crystalline lens. A system networked by gap junctional intercellular communication. Semin Cell Biol. 1992;3:49–58. doi: 10.1016/s1043-4682(10)80007-8. [DOI] [PubMed] [Google Scholar]
- 34.White TW, Goodenough DA, Paul DL. Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol. 1998;143:815–25. doi: 10.1083/jcb.143.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sellitto C, Li L, White TW. Connexin50 is essential for normal postnatal lens cell proliferation. Invest Ophthalmol Vis Sci. 2004;45:3196–202. doi: 10.1167/iovs.04-0194. [DOI] [PubMed] [Google Scholar]
- 36.Pal JD, Berthoud VM, Beyer EC, Mackay D, Shiels A, Ebihara L. Molecular mechanism underlying a Cx50-linked congenital cataract. Am J Physiol. 1999;276:C1443–6. doi: 10.1152/ajpcell.1999.276.6.C1443. [DOI] [PubMed] [Google Scholar]
- 37.Martin PE, Coleman L, Casalotti SO, Forge A, Evans WH. Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal hereditary deafness. Hum Mol Genet. 1999;8:2369–76. doi: 10.1093/hmg/8.13.2369. [DOI] [PubMed] [Google Scholar]
- 38.Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805–12. doi: 10.1093/hmg/ddg076. [DOI] [PubMed] [Google Scholar]
- 39.Oguchi T, Ohtsuka A, Hashimoto S, Oshima A, Abe S, Kobayashi Y, Nagai K, Matsunaga T, Iwasaki S, Nakagawa T, Usami S-i. Clinical features of patients with GJB2 (connexin 26) mutations: severity of hearing loss is correlated with genotypes and protein expression patterns. J Hum Genet. 2005;50:76–83. doi: 10.1007/s10038-004-0223-7. [DOI] [PubMed] [Google Scholar]
- 40.Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynänen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70:1341–8. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Steensel MAM, van Geel M, Nahuys M, Smitt JHS, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Dermatol. 2002;118:724–7. doi: 10.1046/j.1523-1747.2002.01735.x. [DOI] [PubMed] [Google Scholar]
- 42.Yoshimura T, Satake M, Ohnishi A, Tsutsumi Y, Fujikura Y. Mutations of connexin32 in Charcot-Marie-Tooth disease type X interfere with cell-to-cell communication but not cell proliferation and myelin-specific gene expression. J Neurosci Res. 1998;51:154–61. doi: 10.1002/(SICI)1097-4547(19980115)51:2<154::AID-JNR4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 43.Baldo GJ, Gong X, Martinez-Wittinghan FJ, Kumar NM, Gilula NB, Mathias RT. Gap junctional coupling in lenses from α8 connexin knockout mice. J Gen Physiol. 2001;118:447–56. doi: 10.1085/jgp.118.5.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Favor J. A comparison of the dominant cataract and recessive specific-locus mutation rates induced by treatment of male mice with ethylnitrosourea. Mutant Res. 1983;110:367–82. doi: 10.1016/0027-5107(83)90153-7. [DOI] [PubMed] [Google Scholar]
- 45.Gong X, Agopian K, Kumar NM, Gilula NB. Genetic factors influence cataract formation in a3 connexin knockout mice. Dev Genet. 1999;24:27–32. doi: 10.1002/(SICI)1520-6408(1999)24:1/2<27::AID-DVG4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 46.Gerido DA, Sellitto C, Li L, White TW. Genetic background influences cataractogenesis, but not lens growth deficiency, in Cx50-knockout mice. Invest Ophthalmol Vis Sci. 2003;44:2669–74. doi: 10.1167/iovs.02-1311. [DOI] [PubMed] [Google Scholar]