

Abstract
Purpose
Poly(ADP-ribose) polymerase plays an important role in DNA repair and PARP inhibitors can enhance the activity of DNA damaging agents in vitro and in vivo. AG014699 is a potent PARP inhibitor in Phase II clinical development. However the range of therapeutics with which AG014699 could interact via a DNA-repair based mechanism is limited. We aimed to investigate a novel, vascular-based activity of AG014699, underlying in vivo chemosensitisation, that could widen its clinical application.
Experimental design
Temozolomide response was analysed in vitro and in vivo. Vessel dynamics were monitored using “mismatch” following the administration of perfusion markers and real-time analysis of fluorescently-labeled albumin uptake in to tumours established in dorsal window chambers. Further mechanistic investigations employed ex vivo assays of vascular smooth muscle relaxation, gut motility and myosin light chain kinase inhibition.
Results
AG014699 failed to sensitise SW620 cells to temozolomide in vitro but induced pronounced enhancement in vivo. AG014699 (1mg/kg) improved tumour perfusion comparably with the control agents nicotinamide (1g/kg) and AG14361 (fore-runner to AG014699; 10mg/kg). AG014699 and AG14361 relaxed pre-constricted vascular smooth muscle more potently than the standard agent, hydralazine, with no impact on gut motility. AG014699 inhibited myosin light chain kinase at concentrations that relaxed isolated arteries, whereas AG14361 had no effect.
Conclusion
Increased vessel perfusion elicited by AG014699 could increase tumour drug accumulation and therapeutic response. Vasoactive concentrations of AG014699 do not cause detrimental side-effects to gut motility and may increase the range of therapeutics with which AG014699 could be combined with for clinical benefit.
Keywords: PARP, vasoactivity, AG014699, therapeutic, temozolomide
Introduction
Poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme that signals DNA breaks to repair proteins by synthesising ADP-ribose polymers on nuclear proteins using NAD+ as a substrate. Inhibition of DNA repair using PARP inhibitors following DNA damage caused by chemotherapeutic drugs or radiation is a newly recognised therapeutic manoeuvre to improve cancer therapy. PARP inhibitors have been shown to enhance the anti-tumour activity of DNA methylating agents, topoisomerase I poisons and ionising radiation in advanced preclinical studies (1).
AG14447 is a potent small molecule inhibitor of PARP-1 and 2. These properties were maintained in glucuronate (AG14698) and phosphate (AG014699) salt derivatives that were developed as potential clinical candidates with improved solubility. Pre-clinical studies demonstrated that AG14447 sensitised cells cultured in vitro to DNA-damaging chemo- or radiotherapy, and AG014698 caused profound chemo-potentiation in xenograft models established in nude mice. (2). These promising data led to AG014699 being the first PARP inhibitor to be evaluated in cancer patients. Phase I and II trials have been completed in which AG014699 was combined with the DNA-damaging agent temozolomide (TMZ). These studies have shown excellent PARP inhibition in peripheral blood leucocytes and tumour material at a dose of 12mg/m2 that can be well tolerated in combination with TMZ (100 mg/ m2) and yields encouraging activity in malignant melanoma (3). Importantly, no AG014699-associated toxicity was observed at doses that caused profound and durable suppression of PARP activity. Given the promising clinical profile of AG014699, broadening its potential clinical application to an increased range of anti-cancer therapeutics would be an attractive approach.
One major obstacle to effective chemotherapy is poor drug delivery. It remains challenging to adequately deliver chemotherapeutic agents/drugs directly into the tumour microenvironment due its hostile nature and poor vascular perfusion (4). Fluctuations in tumour blood flow have been observed in patients (5) that could affect micro-regional oxygenation and drug delivery (6). Tumour blood flow has been correlated with poor outcome following chemotherapy, and also radiotherapy and surgery (7). The latter two observations are likely to be associated with fluctuations in oxygen availability causing tumour hypoxia that results in radioresistance and an aggressive disease phenotype.
The early PARP inhibitors, nicotinamide and the benzamides, are potent in vivo radiosensitisers, an effect that was achieved at least in part by virtue of their ability to increase tumour blood flow. Indeed nicotinamide is used clinically in combination with carbogen breathing to improve tumour oxygenation for radiotherapy (8-10). Our initial studies revealed that the PARP inhibitor AG14361, a forerunner of the clinical candidate AG014699, increased the transient perfusion of tumour xenografts by inhibiting spontaneous rhythmic tumour vasoconstriction (11). We postulated that the apparent vasoactivity of AG14361 contributes to its in vivo radio- and chemosensitisation. In keeping with this suggestion, AG14361, coupled with TMZ caused complete regression of SW620 xenografts tumours in vivo whilst having no effect on TMZ sensitivity in SW620 cells cultured in vitro (11). In this present study we investigate the vasoactivity of the clinical lead agent AG014699 in vivo and ex vivo compared with AG14361 and the known vasoactive agents nicotinamide and hydralazine and assess its anti-tumour effects in combination with TMZ.
Materials and Methods
Reagents
For in vitro studies we dissolved temozolomide (TMZ) (gift from Cancer Research UK), and PARP inhibitors AG14361 and AG014699 (Pfizer GDR, La Jolla, USA) in DMSO to allow addition to cell cultures at a final DMSO concentration of 1% (v/v). All other chemicals and reagents were from Sigma (Poole, UK) unless otherwise stated. For in vivo evaluation we prepared all agents immediately before administration. TMZ was suspended and AG014699 and nicotinamide were dissolved in normal saline. AG14361 and AG14447 were prepared as the HCl salt by dissolving in equimolar HCl in saline.
Cell lines and culture
We maintained exponentially growing cultures of SW620 (American Type Culture Collection, Manassas, VA) and HT29 (a kind gift from Professor Caroline Dive, University of Manchester, UK) colorectal cancer cells in RPMI 1640 containing 10% (v/v) foetal calf serum. Cells were verified as mycoplasma-free (MycoAlert™ Cambrex Bioscience, Rockland USA).
In vitro chemosensitisation
We estimated cell growth inhibition in exponentially growing SW620 cells in 96 well plates exposed to increasing concentrations of TMZ, alone or in combination with AG014699 (0.4 μM) for 5 days prior to staining with sulphorhodamine B as previously described (12). Cell growth, determined after subtraction of time 0 values, was expressed as a % of the relevant DMSO or AG014699 alone control, as appropriate. GI50 (concentration of drug that inhibited growth by 50%) values were calculated from the computer-generated curves (GraphPad Software, Inc. San Diego Ca USA).
In vivo TMZ studies
All of the in vivo experiments were reviewed and approved by the relevant institutional animal welfare committees, and performed according to national law (Scientific Procedures Act 1986) and UK Coordinating Committee on Cancer Research Guidelines. Adult female athymic nude mice (nu/nu) used for anti-tumour studies were maintained and handled in isolators under specific pathogen-free conditions. For TMZ studies we implanted 1 × 107 SW620 cells subcutaneously (s.c.) into one flank of each mouse. When tumours were palpable (10–12 days after implantation) we randomised the mice (5 animals/ group) to receive five daily doses of TMZ administered p.o. as a suspension in saline at 68 mg/kg either alone or in combination with five daily i.p. administrations of AG014699 at 1.0 mg/kg, normal saline p.o. and i.p. (controls) or normal saline p.o and AG014699 (10 mg/kg) i.p. Data are presented as median relative tumour volumes (RTV), defined as the calculated tumour volume divided by the calculated tumour volume on the initial day of treatment (day 0). Thus, on day 0 the RTV value is 1 and RTV4 is when the tumour is 4 times as large as its initial value. Tumour growth delays were calculated as follows: Tumour growth delay (TGD) = Median time to RTV4 in treated group − median time to RTV4 control
Vessel mismatch studies
Once SW620 xenografts (established as described above) had reached an approximate diameter of 10 mm, we administered AG14361 (10 mg/kg), AG014699 (1 mg/kg), nicotinamide (1 g/kg) or saline (control) i.p., followed 30 minutes later by Hoechst 33342 (i.v., 15 mg/kg, dissolved in phosphate-buffered saline) and finally carbocyanine (i.v. 1 mg/kg, dissolved in 75% dimethyl sulphoxide) after a further 20 minutes. We excised and rapidly froze the tumours 5 minutes later as described previously (9). Tumour sections (10 μm) were prepared and scanned under a Nikon Eclipse E800 microscope to analyse Hoechst 33342 and carbocyanine positive vessels (excitation wavelengths; 340–380 nm and 450–490 nm; emission wavelengths; 480 nm and 510 nm, for Hoechst and carbocyanine, respectively).
Real time analysis of tumours established in the Dorsal Window Chambers
We implanted dorsal window chambers (DWC), attached to the back of female nude mice as described previously (13), with SW620 or HT29 cells (approximately 50μl of a 1×107/ml stock prepared in serum-free RPMI). At tumour volume of approximately 100mm3, mice were anaesthetised and prepared for intra-vital microscopy. Tumour vasculature images were taken using bright-field microscopy (Nikon Eclipse E800) as well as background fluorescence readings prior to the i.v. administration of bovine serum albumin labelled with Alexa fluorochrome (BSA-647 excitation wavelength 647nm, prepared at1mg/ml in sterile saline; 0.1ml/mouse; Molecular Probes, Invitrogen, Paisley, UK). Changes in fluorescence (emission 668nm) were monitored in real-time (Metamorph analysis system) continuously pre- and post- injection of AG014699 (1-10 mg/kg i.p.).
Ex vivo assay: Rat-tail artery
Male albino Wistar rats (8-12 weeks old; Harlan UK Ltd., Oxon, England) were killed by CO2-asphyxiation followed by cervical dislocation. We removed the tail from the animal at its most proximal point and peeled back the skin on the ventral surface to reveal the vascular bed containing the tail artery. We removed the membrane over-lying the artery in the same manner and bathed the exposed artery in ice-cold Krebs’ solution (118 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 1.15 mM NaH2PO4, 2.5 mM CaCl2, 1.1 mM MgCl2, 5.6 mM C6H12O6) to prevent dehydration. We then introduced a cannula of 1 mm polythene gas chromatography tubing into the artery, and slid the artery over it until an insertion overlap of approximately 5 mm was achieved before securing it to the cannula using double-knotted thread. A length of artery approximately 10 mm beyond the end of the cannula was freed from its vascular bed and cut.
We connected the cannulated artery to an internal/external perfusion apparatus and perfused it with oxygenated Krebs’ (95% O2/5% CO2, BOC, Manchester, UK) at 37°C, increasing the rate of perfusion from 0.25 ml/min to 2 ml/min in 0.25 ml/min increments over a period of 1 h. After equilibration of the artery segments for 1 hour we elicited sub-maximal constriction by perfusing the arterial segments with perfusate that contained 10 μM phenylephrine (PE). We determined the dilatory properties of AG14361/AG014699 by replacing the constricting perfusate with one that contained 10 μM PE plus the relevant concentration of either AG14361 or AG014699. Arterial constriction or dilation was detected by an increase or decrease in pressure detected by transducers (by changes in pressure, monitored by water displacement) connected to a MacLab system (AD Instruments Pty Ltd., Australia), and was visualised on a PC. The degree of relaxation elicited by AG14361/AG014699 treatment was expressed as a percentage of the magnitude of constriction that was observed following 10 μM PE treatment.
Rat ileum
We removed a segment of ileum from rats killed as described above. After flushing the faecal matter from the lumen using ice-cold Krebs’ solution we placed the tissue in fresh, ice-cold oxygenated Krebs’. We mounted 5 mm thick rings of ileum on the pins of isometric force transducers and lowered them into a tissue bath containing Krebs’ at 37°C, with oxygenated Krebs’ flowing through at a rate of 2 ml per minute. In this system, one pin is stationary, while the second pin is free to move vertically and is used to apply tension to the piece of tissue. With increasing application of tension, the rings begin contracting spontaneously. When the average tension on each ring reached 0.5 g, we allowed the tissue to equilibrate (average 15 min) before applying of the test agent by perfusing the tissue bath with the relevant agent. Intestinal contraction was detected by the transducers connected to a MacLab system (AD Instruments Pty Ltd., Australia) and was visualised on a Macintosh computer. Differences in the magnitude of contractions 15 min following application of the agent were compared with the magnitude of contraction in the lead up to treatment and were expressed as a percentage of the magnitude of contraction before treatment.
In vitro MLCK activity analysis
Kinase activity analysis was performed using Upstate’s IC50 Profiler Express service. 5 mM samples of AG14361 and AG014699 were sent to Upstate’s Drug Discovery Service department (Dundee, Scotland). From this sample, a 10-point, ½ logarithmic dilution series was generated, resulting in an agent test concentration range of 100 nM – 100 μM, plus 0 μM as control (1% (v/v) DMSO). The kinase activity analysis protocol employed by Upstate is available on their website1. Briefly, in a final reaction volume of 25 μL, MLCK (h) (5-10 mU) was incubated with 8 mM MOPS pH 7.0, 0.2 mM EDTA, 0.5 mM CaCl2, 16 μg/mL calmodulin, 250 μM KKLNRTLSFAEPG, 10 mM MgAcetate and [γ-33P-ATP]. The reaction was initiated by the addition of the MgATP mix. After incubation for 40 min at room temperature, the reaction was stopped by the addition of 5 μL 3% phosphoric acid solution. 10 μL of the reaction was then spotted onto a P30 filtermat and washed three times for 5 min in 75 mM phosphoric acid and once in methanol prior to drying and scintillation counting. The relative activity of MLCK following treatment with the relevant agent was then expressed as a percentage of the activity of the kinase in the absence of AG14361 or AG014699. MLCK activity analysis was performed in duplicate.
Results
AG014699 does not enhance TMZ-induced growth inhibition in vitro but profoundly increases the activity of TMZ against SW620 xenografts
SW620 cells express low levels of methylguanine methyl transferase and have functional mismatch repair and are therefore profoundly sensitive to TMZ (Arris, C.E.A and Curtin, N. J., unpublished observations; 14). We had previously found that AG14361 did not enhance TMZ-induced growth inhibition in these cells (11) and we confirmed in the current study that there was no statistically significant difference in growth inhibition by TMZ alone or in combination with AG014699 (p = 0.596 from 5 independent experiments; Figure 1A). Furthermore, co-administration of a PARP inhibitor did not enhance the cytotoxicity of TMZ in SW620 cells by clonogenic survival assay (supplementary figure S1). However, when AG014699 was combined with TMZ in vivo a completely different picture emerged. TMZ alone caused transient regression followed by regrowth resulting in a 25-day tumour growth delay (time to RTV4) in tumour growth. AG014699 alone had no impact on tumour growth but the combination of TMZ and AG014699 resulted in prolonged regression lasting at least 53 days (time to RTV 4 = 60, 63 and 100 days in 3 mice that relapsed) with 2/5 mice remaining tumour free at day 100 (Figure 1B). The difference in in vivo and in vitro chemosensitisation by AG014699 was not thought to be due to divergent concentrations of the drug in the two systems as we have shown previously that concentrations of the parent drug, AG14447, are between 1.5 and 0.6 μM for 24 hr after a single i.p. injection of AG014699 at a concentration of 1 mg/kg (15).
Figure 1. The different effects of AG014699 on TMZ-induced growth inhibition of SW620 cells in vitro and xenografts in vivo.

(A) TMZ alone (filled circles, solid line) caused a concentration-dependent inhibition of cell growth that was not significantly increased by the combination with 0.4 μM AG014699 (open circles, broken line). Data are mean ± standard deviation of 5 independent experiments. (B) Growth of SW620 xenografts treated with control vehicle (filled circles, solid line), AG014699 (10 mg/kg daily ×5) alone (open circles broken line), TMZ (68 mg/kg daily ×5) alone (filled triangles, solid line) or the combination of TMZ with AG014699 at 1 mg/kg (open triangles, broken line). Data are median RTV from 5 animals/group.
PARP inhibitors reduce vessel mismatch in SW620 tumour xenografts in vivo
To determine why there was such a profound in vivo chemosensitisation in the absence of in vitro activity, we investigated whether AG014699 (in comparison AG14361 and nicotinamide) might be increasing the area of tissue that received TMZ through an effect on tumour perfusion. Hoechst 33342 and carbocyanine are routinely used as markers of tumour perfusion. As they have distinct emission wavelengths they can be analysed simultaneously in the same tumour. We administered Hoechst 33342 20 min before carbocyanine and observed the staining pattern of the tumour blood vessels. Vessels staining with one dye only are denoted as mismatched and have either closed (Hoechst 33342 only) or opened (carbocyanine only) in the period between administration of the dyes. Vessels open throughout the 20-minute interval stain with both dyes. In keeping with our previous findings (11), vessel mismatch was markedly reduced in the AG14361 (10 mg/kg) pre-treated SW620 xenografts compared to control (18 versus 51%; Figure 2A). Vessel mismatch was also reduced following AG014699 (1 mg/kg; 28%) and nicotinamide (1 g/kg; 19%) pre-treatment (Figure 2A). Thus, AG014699 has a similar effect to a 10x higher concentration of AG14361 and a 1,000x higher concentration of nicotinamide. Analysis of the proportion of vessels stained with only one dye revealed subtle differences between the agents used (Figure 2B). Whereas in saline control treated animals, 30% of vessels closed between Hoechst and carbocyanine administration, this was reduced to 12% in AG14361 and nicotinamide treated tumours. Similarly the number of vessels stained with carbocyanine only was also reduced in tumours treated with AG14361 and nicotinamide suggesting that for these agents vessels that were open at the point of Hoechst 33342 injection remained so, such that the proportion of Hoechst 33342 only (closed) and carbocyanine only (opened) vessels was low. For AG014699 the amount of vessel closure was further reduced compared with AG14361 and nicotinamide to only 5% in mice treated with AG014699 (Figure 2B). Furthermore the proportion of vessels open at the time of carbocyanine injection was greater in AG014699 treated mice (21%) compared with AG14361 and nicotinamide (7% for both; Figure 2B) and was comparable to controls. Furthermore the proportion of vessels open at the time of carbocyanine injection was greater in AG014699 treated mice (21%) compared with AG14361 and nicotinamide (7% for both Figure 2B), indicating that a substantial amount of the vessel mismatch was a consequence of vessel opening.
Figure 2. AG014699 and AG14361 are more potent inhibitors of vessel mismatch than the positive control agent nicotinamide.
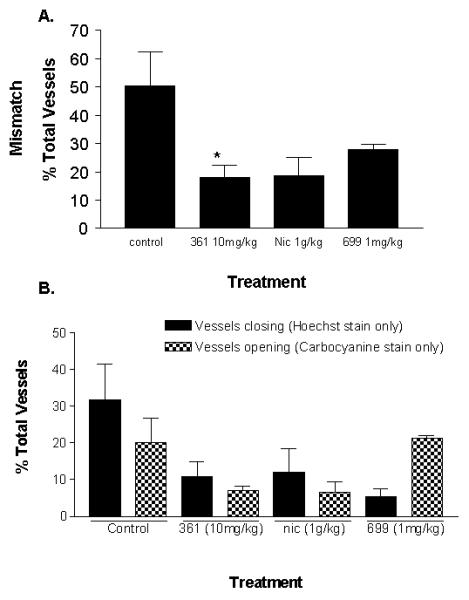
The perfusion markers Hoechst and carbocyanine were administered i.v. 20 minutes apart to SW620 tumour bearing mice that had been pre-treated with AG014699 (1mg/kg), AG14361 (10mg/kg), nicotinamide (1g/kg) or saline (control). Data presented in (A) are average vessel mismatch values (n=4 per group, ± se). *p=0.03 versus saline treated control. (B) shows the impact of the agents on percentage of vessels closing (Hoechst positive only) or opening (carbocyanine positive only; average values ± se).
The effects of AG014699, AG14361 and nicotinamide on vascular perfusion in tumours xenografts established in dorsal window chambers (DWC)
To further investigate the effects of the PARP inhibitors on tumour haemodynamics in real time we measured Alexa-fluor-647-conjugated serum albumin (BSA-647) accumulation as a marker of tumour perfusion in SW620 tumours established in DWC. We recorded background fluorescence prior to BSA-647 injection and when BSA-647 fluorescence had reached a plateau we administered saline or AG014699. The arbitrary fluorescence units (FU) remained unchanged following the administration of saline i.p. or i.v. (Figure 3). In contrast, 20 min after the injection of AG014699 (1 mg/kg i.p) the BSA-647 accumulation rose steadily reaching a plateau at 70 minutes yielding a 1.3 fold enhancement in fluorescence compared with the base-line plateau (Figure 3). This suggested that AG014699 enhances tumour perfusion in SW620 tumours established in DWC. We then undertook comparative studies using nicotinamide (1 g/kg) and AG14361 (10 mg/kg) and the fold enhancements in BSA-647 accumulation following treatment were 1.4 and 1.5 respectively (data not shown). The studies were repeated using the HT29 colorectal model established in DWC (Figure 3). Again AG014699 (1 mg/kg) caused an increase in BSA-647 accumulation (1.3-fold) that compared favorably with AG14361 (1.2-fold) and nicotinamide (1.7-fold) administered at higher doses (10 mg/kg and 1 g/kg respectively; latter data not shown).
Figure 3. Real-time analysis of the effects of AG014699 on the accumulation of BSA-labelled with Alexa-647 in SW620 and HT29 tumours established in DWC.
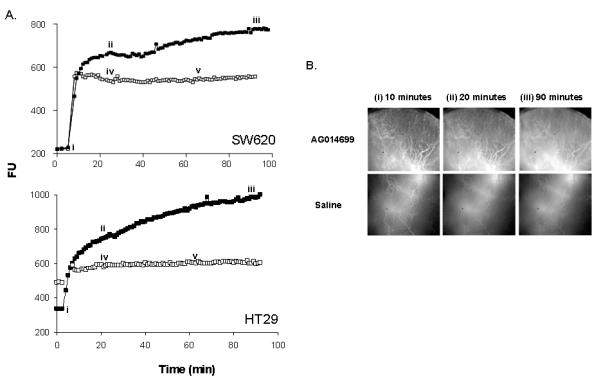
(A) Fluorescence (arbitrary units, FU) was monitored every minute in control (saline; open symbols) and AG014699 (1mg/kg, solid symbols) treated tumours. (B) Images correspond to the distribution of BSA-647 immediately after injection (i), immediately before AG014699 or saline administration (ii) and at the end of the experiment (iii) as illustrated on the uptake curves in (A). Bolus injections of saline via i.p. (iv) or i.v. (v) routes do not alter BSA-647 fluorescence.
Relaxation of pre-constricted rat arteries using PARP inhibitors
AG014699 and AG14361 dose-dependently inhibited PE-induced constriction in rat-tail artery segments (Figure 4 A, and B). The doses at which the agents achieved 50% of their relaxant activity (EC50) were 23 μM (AG014699) and 140 μM (AG14361). The vasodilator hydralazine, included as a positive control, elicited dilation of PE-constricted artery segments with an EC50 of 617 μM. Neither AG14361 nor AG014699 affected the contractility of ileum smooth muscle (Figure 4 C and D).
Figure 4. The PARP inhibitors AG014699 and AG14361 cause relaxation in pre-constricted rat arteries ex vivo.
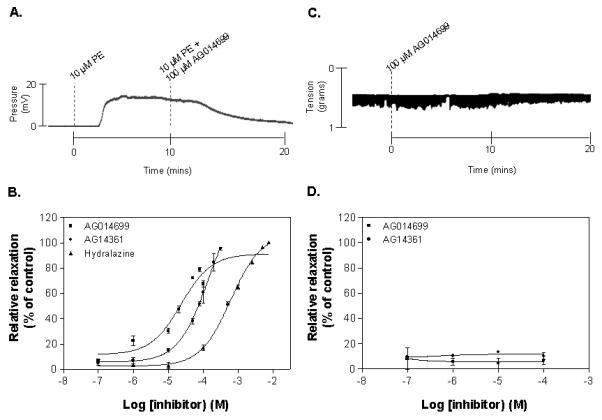
(A) shows a representative trace of the dilatory effect of 100 μM AG014699 in 10 μM PE-constricted rat-tail artery. Evident are the constriction elicited by PE and the relaxant effect of AG014699. Note also the time delay between commencement of perfusion with a particular agent and visualisation of the response, resulting from the transit time of the perfusate through the apparatus. (B) illustrates the dose responses of AG014699, AG14361 and hydralazine in constricted rat-tail artery. (C) shows a representative trace illustrating the lack of an inhibitory effect of 100 μM AG014699 in contracting rat ileum and (D) illustrates the lack of inhibitory effects of AG14361 and AG014699 on the contraction in rat ileum.
AG014699 can inhibit myosin light chain kinase (MLCK) activity
To explore the mechanism of action of AG14361 and AG014699, their inhibitory action upon MLCK, a key regulator of smooth muscle contraction was assessed. AG014699 dose-dependently inhibited phosphorylation of the in vitro substrate peptide (KKLNRTLSFAEPG) by MLCK, with an IC50 of 55 μM. AG14361 did not have any inhibitory effect on the phosphorylation of the peptide substrate by MLCK in the concentration range tested (Figure 5).
Figure 5. AG014699 inhibits myosin light chain kinase activity, but AG14361 does not.

Millipore IC50 Profiler Express.
Discussion
Inhibitors of the DNA-repair enzyme PARP have received much attention recently as a consequence of their potential ability to enhance the effects of a number of DNA-damaging anti-cancer therapeutics (1). However although this application holds much promise, our data suggest that the utility of PARP inhibitors could exceed that initially anticipated through an in vivo mechanism influencing tumour perfusion.
AG014699 was the first PARP inhibitor to be used in cancer patients and is currently in Phase II trials. Analysing the chemopotentiating effect of AG014699 in pre-clinical studies we observed profound differences between in vitro and in vivo models. SW620 cells are exquisitely sensitive to the chemotherapy agent temozolomide. In vitro, AG014699 is unable to enhance this sensitivity, yet in vivo, combination of AG014699 and temozolomide yielded pronounced growth delay or tumour cures at doses of temozolomide that caused only transient growth delay when administered alone. This suggested that mechanisms other than inhibition of DNA-repair were contributing to the observed effects of AG014699 in vivo.
Given the reported vasoactive effects of early PARP inhibitors, we investigated the influence of AG014699 on vascular dynamics using both in vivo and ex vivo models. Comparisons were made with AG14361, the effective forerunner to AG014699 in the development of a clinical lead candidate, and also nicotinamide and hydrazaline that acted as positive controls in the model systems used. Tumour vascular effects were confirmed by both vessel mismatch studies and real-time observations of perfusion using tumours grown in dorsal window chambers. Compellingly, the relative efficacies of agents showed excellent concordance between the mismatch and dorsal window chamber studies using SW620 tumours, and have been replicated using other tumour systems and mouse backgrounds (HT29 data here within and data not shown). Furthermore, vasoactivity was proven in excised rat arteries and both AG014699 and AG14361 were more potent than the control agent hydrazaline at causing relaxation of pre-constricted arteries. The effect was specific to vascular smooth muscle as neither agent had any significant activity on contraction of rat ileum. Preliminary investigations into the mechanisms of the effect of the two agents revealed that AG014699 inhibited the activity of MLCK at concentrations commensurate with its effects on pre-constricted arteries. AG14361 had no effect on MLCK and so its mechanism at this point remains elusive. However the differential impact of the two agents could contribute to the subtle differences observed in the mismatch studies whereby AG14361 appeared to maintain vessels that were already open in an open state whereas significant vessel opening continued during AG014699 treatment.
Relating these data to the observed chemosensitisation effects, they would strongly support a model whereby AG014699 and AG14361 treatment can increase the extent of tumour exposure to drug by enhancing vascular perfusion. As the efficacy of a number of clinically used chemotherapy agents is limited by their very poor distribution in vivo (4, 6), the overall consequence would be to expose a greater number of tumour cells to drug thereby increasing the overall anti-tumour effect. Interestingly previous studies have reported that PARP inhibitors can sensitise tumour xenografts to doxorubicin (16) and cisplatin (17). As neither agent would be anticipated to be enhanced by PARP inhibition through a classical DNA-repair based mechanism, vasoactivity could have been the basis for the enhancement, exemplifying the application of PARP inhibitors with different classes of agent.
In addition, the ex vivo assays performed suggest that the positive effects of AG014699 treatment on drug delivery would not be confounded by negative effects on gut motility. Gastric toxicity limits the therapeutic benefit of nicotinamide (18), so the absence of an inhibitory effect in intestinal tissues in the cases of AG14361 and AG014699 is an attractive finding. It is also important to note is that AG014699 and AG14361 begin exhibiting vasoactivity at a concentration that is two orders of magnitude lower than the concentration at which NA elicits these effects.
AG014699 is an attractive drug for anticancer therapy but the range of drug classes with which it synergises through a DNA-repair based mechanism is quite restricted. However, our data suggest it may have broader application on the basis of improved tumour perfusion and hence drug delivery. In addition enhanced perfusion could also contribute to the in vivo radiosensitisation previously reported for AG14361 (11) and a focus of our ongoing studies with AG014699. The fact that AG014699 appears to have very little clinical toxicity supports its wider application in the clinical setting. We believe our demonstration of the vasoactive effect of AG014699 justifies further in vivo studies with a range of cytotoxic drugs as a prelude to using potential improvements in drug delivery as a therapeutic manoeuvre in the clinical setting.
Supplementary Material
Acknowledgements
This work was predominantly funded by Cancer Research UK with contribution from the Medical Research Council (B. A. Telfer), for which we acknowledge the support of Ian Stratford. We acknowledge the continued support of Pfizer, and in particular Zdenek Hostomsky for his enthusiasm and critical guidance. We are grateful to Ms Lan-Zhen Wang for the data shown in the supplementary figure
Financial support: This work was supported by two grants from Cancer Research UK one awarded jointly to K.J.W. and N.J.C. and one to D.G.H, TR and CS.
Footnotes
Statement of translational relevance
AG014699, a phosphate salt of AG14447, is a potent inhibitor of the DNA-repair enzyme, poly(ADP-ribose) polymerase (PARP). Promising pre-clinical data demonstrating sensitisation of tumour cells to DNA-damaging chemotherapy in vitro and in vivo led to the clinical development of AG014699. Phase I and II trials have reported profound and durable suppression of PARP activity with no appreciable toxicity. AG014699 is well tolerated in combination with temozolomide and yields encouraging activity in malignant melanoma. Although these data are promising, the range of agents that AG014699 could enhance through a DNA-repair based mechanism is limited. We report a novel vascular effect of AG014699 that could contribute to the chemosensitisation achieved with this compound. By increasing tumour perfusion, AG014699 could improve drug delivery and be beneficial in combination with agents whose clinical efficacy is limited by poor distribution.
References
- 1.Curtin NJ. PARP inhibitors for cancer therapy. Expert Rev Mol Med. 2005;7:1–20. doi: 10.1017/S146239940500904X. [DOI] [PubMed] [Google Scholar]
- 2.Thomas HD, Calabrese CR, Batey MA, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase (PARP) inhibitor for clinical trial. Mol Cancer Ther. 2007;6:945–56. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- 3.Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-Ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clinical Cancer Res. 2008;14:7917–23. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 5.Pigott KH, Hill SA, Chaplin DJ, Saunders MI. Microregional fluctuations in perfusion within human tumours detected using laser Doppler flowmetry. Radiotherapy and Oncology. 1996;40:45–50. doi: 10.1016/0167-8140(96)01730-6. [DOI] [PubMed] [Google Scholar]
- 6.Tannock IF, Lee CM, Tunggal JK, Cowan DSM, Egorin MJ. Limited penetration of anticancer drugs through tumour tissue: A potential cause of resistance of solid tumours to chemotherapy. Clin Cancer Res. 2002;8:878–84. [PubMed] [Google Scholar]
- 7.Durand RE, Aquino-Parsons C. Clinical relevance of intermittent tumour blood flow. Acta Oncologica. 2001;40:929–36. doi: 10.1080/02841860152708206. [DOI] [PubMed] [Google Scholar]
- 8.Horsman MR. Nicotinamide and other benzamide analogues as agents for overcoming hypoxic cell radiation resistance in tumours. Acta Oncologica. 1995;34:571–87. doi: 10.3109/02841869509094031. [DOI] [PubMed] [Google Scholar]
- 9.Thomas CD, Stern S, Chaplin DJ, Guichard M. Transient perfusion and radiosensitizing effect after nicotinamide, carbogen, and perflubron emulsion administration. Radiotherapy and Oncology. 1996;39:235–41. doi: 10.1016/0167-8140(96)01734-3. [DOI] [PubMed] [Google Scholar]
- 10.Kaanders JHAM, Bussink J, van der Kogel AJ. ARCON: a novel biology-based approach in radiotherapy. Lancet Oncol. 2002;3:728–37. doi: 10.1016/s1470-2045(02)00929-4. [DOI] [PubMed] [Google Scholar]
- 11.Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 12.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 13.Williams KJ, Telfer BA, Shannon AM, Babur M, Stratford IJ, Wedge SR. Combining radiotherapy with AZD2171, a potent inhibitor of vascular endothelial growth factor signaling: pathophysiologic effects and therapeutic benefit. Mol Cancer Ther. 2007;6:599–606. doi: 10.1158/1535-7163.MCT-06-0508. [DOI] [PubMed] [Google Scholar]
- 14.Aquilina , Ceccotti S, Martinelli S, Hampson R, Bignami M. N-(2-chloroethyl)-N’-cyclohexyl-N-nitrosourea sensitivity in mismatch repair-defective human cells. Cancer Res. 1998;58:135–41. [PubMed] [Google Scholar]
- 15.Daniel RA, Rozanska AL, Thomas HD, et al. Inhibition of Poly(ADP-Ribose) Polymerase-1 Enhances Temozolomide and Topotecan Activity against Childhood Neuroblastoma. Clin Cancer Res. 2009;15:1241–1249. doi: 10.1158/1078-0432.CCR-08-1095. [DOI] [PubMed] [Google Scholar]
- 16.Joseph I, Ferguson D, Palma J, et al. Poly(ADP-ribose) polymerase inhibitor, ABT-472 enhances antitumour activity of doxorubicin in human xenograft models and protects against drug-induced cardiac toxicity. Eur J Cancer. 2004;(Suppl2) #8 Abs 473. [Google Scholar]
- 17.Miknyoczki SJ, Jones-Bolin S, Pritchard S, et al. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther. 2003;2:371–82. [PubMed] [Google Scholar]
- 18.Ruddock MW, Burns DM, Murphy LE, O’Rourke MG, Hirst DG. The effect of nicotinamide on spontaneous and induced activity in smooth and skeletal muscle. Radiother Oncol. 2000;56:253–7. doi: 10.1016/s0167-8140(00)00194-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.