

Abstract
G-protein coupled receptors (GPCRs) comprise a large superfamily of proteins that are targets for nearly 50% of drugs in clinical use today. In the past, the use of structure-based drug design strategies to develop better drug candidates has been severely hampered due to the absence of the receptor’s three-dimensional structure. However, with recent advances in molecular modeling techniques and better computing power, atomic level details of these receptors can be derived from computationally derived molecular models. Using information from these models coupled with experimental evidence, it has become feasible to build receptor pharmacophores. In this study, we demonstrate the use of the Hybrid Structure Based (HSB) method that can be used effectively to screen and identify prospective ligands that bind to GPCRs. Essentially; this multi-step method combines ligand-based methods for building enriched libraries of small molecules and structure-based methods for screening molecules against the GPCR target. The HSB method was validated to identify retinal and its analogues from a random dataset of ∼300,000 molecules. The results from this study showed that the 9 top-ranking molecules are indeed analogues of retinal. The method was also tested to identify analogues of dopamine binding to the dopamine D2 receptor. Six of the ten top-ranking molecules are known analogues of dopamine including a prodrug, while the other thirty-four molecules are currently being tested for their activity against all dopamine receptors. The results from both these test cases have proved that the HSB method provides a realistic solution to bridge the gap between the ever-increasing demand for new drugs to treat psychiatric disorders and the lack of efficient screening methods for GPCRs.
Keywords: GPCRs, Virtual screening, Structure-based methods, Shape Signatures, Dopamine receptors, Scoring functions
Introduction
The family of GPCRs represents one of the most important classes of proteins. The sequencing of the human genome has led to the identification of nearly 950 genes coding for GPCRs, of which nearly 450 genes have been implicated as therapeutic targets [1]. There is an exceptional chemical diversity among the endogenous ligands that bind to these receptors. They include biogenic amines, peptides, glycoproteins, nucleotides, metal ions and lipids [2]. They are also involved in the transmission of various stimuli like light, odor and taste [3].
The mechanism of action of GPCRs involves a ligand-induced conformational change that activates the receptor to bind to one or more G proteins leading to the release of GDP followed by binding of GTP [4]. Further, the α-subunit of the G-protein that is bound to GTP dissociates from the receptor and also from the stable βγ-subunits. The GTP bound α-subunit and the βγ-subunits can further participate in various cellular signaling events [5]. Thus GPCRs can be targeted at various stages of cellular signaling process for inhibiting or enhancing their effect in physiological processes. The subfamily of biogenic amine binding GPCRs has provided excellent targets for the treatment of various psychiatric disorders such as schizophrenia [6], depression [7], attention deficit hyperactivity syndrome [8] and migraine [9]. They have also been implicated in other disorders such as allergies, asthma [10], hypertension [11] and gastro intestinal disorders.
Although they are excellent targets for some drugs, they also form good anti-targets for other drugs leading to undesirable side effects. For example, antagonists binding to the α-1A subtype of adrenergic receptors are known to be effective as anti-hypertensive agents, but they also mediate cardiovascular side effects leading to hypotension and dizziness [12, 13]. Similarly, antipsychotic drugs that bind with high affinity to dopamine D2-like receptors and serotonin 5HT2A and 2C receptors reduce the positive symptoms of schizophrenia such as delusions and hallucinations [14, 15] but they also bind to other aminergic GPCRs leading to side effects such as dizziness, depression and tardive dyskinesia [16].
The major factor in the cross reactivities of these essential drugs leading to side effects can be attributed to the common structural folds of all members of the Class-A GPCRs. They all share the characteristic seven transmembrane (TM) spanning α-helical motifs connected by alternating intra and extra cellular loops, with the amino terminus located on the extracellular side and the carboxy terminus on the intracellular side. Although the overall sequence homology among GPCRs is low, the high degree of conservation among a small subset of key residues suggests that they may play an essential role in the structure and function of the receptors leading to cross reactivities among their ligands [17]. However, structure based studies to discover new compounds that have milder side effects have been hampered due to the absence of experimental structural data for GPCRs except for the x-ray crystal structure of bovine rhodopsin. Thus, drug design for GPCR targets has largely comprised either through random screening of compounds using experimental high throughput screening methods and/or virtual screening methods to identify molecules that have a potential to bind to GPCRs. Virtual screening techniques can be broadly classified into two categories: (1) ligand-based methods and (2) structure-based methods. Both these computational methods entail inherent advantages and disadvantages [18-20]. Virtual screening using ligand-based methods such as quantitative structure activity relationship (QSAR) models and (sub)structure searching, although successful in identifying many lead compounds, depend solely on the elements of a pharmacophore that is typically devoid of knowledge about the nature of interaction of the ligand with the protein thus leading to severe receptor mediated side effects. Although structure-based methods represent a potential solution to this problem, they still face technical hurdles pertaining to the conformational flexibility of receptor side chains and ligand molecules, entropic effects due to solvation/desolvation, and other issues associated with docking and scoring based on the complex thermodynamics process of ligand-receptor binding. Also, structure-based methods require detailed knowledge of the three-dimensional (3D) structure of the receptor preferably in complex with the bound ligand.
In the case of GPCRs, the only available crystal structure is that of bovine rhodopsin. However, in a recent study [21] the use of rhodopsin-based homology models of GPCRs to discover new lead molecules has been shown to be very reliable. In the present study, we propose a novel HSB method that combines both ligand-based and structure-based techniques to discover a new generation of molecules that have high affinity and selectivity towards a particular receptor family. We previously demonstrated that certain 1,4 disubstituted aromatic piperidines and piperazines have shown extreme selectivity towards dopamine D4 receptors. Using molecular models of dopamine D2 and D4 receptors coupled with site directed mutagenesis studies [22, 23], we identified microdomains formed by residues from TM2 and TM3 to be coding for the selectivity between dopamine D2 and D4 receptors. Inspired by the results from our previous studies, molecular level details were incorporated in the HSB screening method that would lead to identification of receptor specific compounds thereby reducing receptor-mediated side effects.
The HSB method uses ligand-based methods to build enriched libraries of small molecules, and then employs a combined receptor-ligand pharmacophore to screen molecules from the enriched library and to further dock the molecules to their receptor. These docked complexes were scored based on a number of scoring functions, including a customized weighted knowledge-based scoring scheme to mark high ranking molecules. The results from this detailed analysis of the dynamic mode of association between the receptor and ligand was used to list candidate molecules that were suitable for biochemical testing. The method was tested to identify retinal and its analogues that could bind to rhodopsin from a pool of nearly 300,000 randomly selected organic molecules. Results from this study show that the top-ranking molecules are indeed analogues of retinal and have high affinity to rhodopsin. Similarly, the HSB method was also helpful in identifying high affinity analogues of dopamine binding to the dopamine D2 receptor. The HSB method is highly comprehensive and can be extended to identification of highly selective compounds for other class-A GPCRs that could provide a solution to minimizing the side effects that are commonly associated with drugs used to treat psychiatric disorders.
Methodology
The HSB method for screening molecules binding to GPCRs combines ligand-based and structure-based methods within a coherent framework. A flow chart describing various phases of the screening procedure has been described in Fig. 1. Accordingly, the first phase of the procedure involved the development of an enriched database of drug like molecules starting from libraries of molecules randomly selected from the ZINC database [24] (http://blaster.docking.org/zinc/). The newly developed Shape Signatures method was used to identify molecules that had similar shape but varying structural compositions. The Shape Signatures technology has been described in detail elsewhere [25] and only a brief summary will be discussed here.
Fig. 1.

A flowchart describing the workflow for the HSB method is shown. The various interdependent phases and the objectives to be achieved in the workflow are labeled Phase-I—IV and colored blue, cyan, purple and red respectively
The Shape Signatures approach compactly encodes molecular shape information in conjunction with other properties such as the molecular electrostatic potential mapped onto a molecular surface. The procedure employs a customized ray tracing algorithm, in which the volume of the molecule (defined by its solvent accessible surface area) is explored by a single ray, propagated in the interior of the molecule by the rules of optical reflection. The triangulated solvent accessible surface area was generated using the SMART algorithm [26]. A histogram was obtained by binning ∼100,000 ray segments obtained from the ray-tracing procedure inside the triangulated molecular surface area. The histograms, also called one-dimensional (1D) Shape Signatures, are unique and reproducible for each molecule regardless of the point of initiation of the rays and are invariant to the rotation of the molecule. Further, inclusion of a surface property such as the molecular electrostatic potential yielded a 2D signature. The histograms of two or more molecules can be compared rapidly using a simple method such as the L1 norm [27] or the χ2 metric. This measure of the deviation between the histograms provides the dissimilarity score between two molecules under study.
To generate a signature for a molecule, only the atomic coordinates, atomic radii, and a solvent probe radius are required. Once the Shape Signatures of the molecules in the dataset were obtained, the CPU time required to compare signatures was exceedingly fast which is one of the major strengths of the method. Searchable Shape Signatures libraries of molecules belonging to a number of large databases such as the NCI repository and other sources listed under the publicly available ZINC database were developed in our laboratory. In order to build a customized library of aminergic GPCR binding molecules, Shape Signatures were produced for the query set of molecules whose activity against aminergic receptors has been well established. Using this set of GPCR-directed molecules as queries, various databases of small molecules were virtually screened in search of additional molecules whose Shape Signatures matched the query ligands. This highly enriched set of molecules constituted the in-house GPCR ligand database that will be updated with new lead molecules periodically. In cases where the 3D structure of the query small molecule was unavailable, molecular models were constructed using the builder module in SYBYL (Ver 7.1 Tripos Inc., St. Louis, Missouri, USA), with partial charges derived from molecular electrostatic potential calculations using MOPAC [28]. The molecules were geometry optimized using ab initio quantum chemical calculations. Systematic conformational searches for flexible molecules were performed using a Monte Carlo simulated annealing method.
The second phase of the method involved building homology model of the dopamine D2 receptor whose crystal structure was not available. The primary sequence of the dopamine D2 receptor was retrieved from the Swiss-Prot repository [29]. Pairwise sequence alignment of this sequence with the bovine rhodopsin sequence was done using CLUSTALW [30] (see Fig. 2). TM forming regions were identified using multiple sequence alignment of class-A GPCRs from GPCR database (GPCRDB http://www.gpcr.org/7tm/index.html) [31]. Additionally the beginning and ending regions of the transmembrane helices were identified using the procedure previously described [32]. The TM regions of D2 receptors were built using the corresponding TM regions of the bovine rhodopsin crystal structure (pdb code: 1U19) as template for the molecular modeling program MODELER [33]. Care was taken to model regions of inconsistencies such as kinks produced by Prolines [34, 35] and multiple Glycines.
Fig. 2.

Pairwise alignment of dopamine D2 rat sequence with bovine rhodopsin sequence is shown. The corresponding transmembrane forming residues are highlighted in red, while the putative binding site residues are colored blue
The loop forming regions was modeled using an ab initio method for predicting the 3D structures of loop forming segments of GPCRs using only the amino acid sequence [36]. Details of the method as applied to modeling the extra and intra cellular dopamine D2 and D4 receptors have been described elsewhere [Kortagere et al. (under preparation)]. This approach used the Screened Coulomb Potential-Implicit Solvent Model (SCP-ISM) [37] to represent solvent effects implemented in CHARMM (ver 32 [58]) and was validated on a number of known structures [38, 39]. This multi-step loop modeling protocol utilizes a Monte-Carlo simulated annealing method to explore the various conformations available to a loop segment that is tethered at one end. Further, using a stochastic sampling technique based on the Scaled collective variable Monte Carlo method, the loop segments are closed. The procedure is repeated until a native like structural ensemble is found that can represent the final structure of the loop segment. Complete details of the methodology and validation of this method has been described by Mehler et al. [36].
The complete model of the dopamine D2 receptor was refined using standard molecular dynamics procedures with a production run of 2ns. All simulations were performed using the Implicit Membrane Model (IMM1-pore) implemented in CHARMM. The IMM1-pore model, which has been well tested on a number of membrane proteins, can also account for the intra and inter helical hydrogen bonding patterns [40, 41]. Placement of the helices in our structural model for the D2 receptor was based on the bovine rhodopsin crystal structure, which served as the template for homology modeling. Accordingly, the TM4 helix that has been known to be approximately perpendicular to the plane of the membrane was used to adjust the positioning of the other helices with reference to the membrane axis.
The study involved identifying analogues of dopamine, an endogenous agonist to dopamine receptors. Hence, the agonist model of dopamine D2 receptor was built using the complete model of dopamine D2 receptor (described above) as an initial structure. Experimental evidence has suggested that activation of class-A GPCRs by agonists involved the movement of TM3 and an anticlockwise rotation of TM6 on its axis by 30° [42-45]. Thus to derive a functional agonist model, experimental mutational data for dopamine D2 receptor was obtained from GPCRDB (31). These pieces of experimental evidence were incorporated into the D2 receptor model as harmonic distance constraints between the various residues listed as mutants during the refinement protocol. Specifically, residues involved in binding agonists such as D3.32, F6.51, F6.52, S5.42, S5.43, S5.46 were given higher weightage as opposed to residues that were distant from the binding site. These constraints were reduced over cycles of refinement and the final 1ns production run was devoid of any constraints. Dopamine was docked in the binding site and the complex was further refined using AMBER (ver 8.0 [46]).
Combined pharmacophore
For the two case studies described here, the combined pharmacophore was derived using the docked protein—ligand complex of rhodopsin with retinal and D2 receptor with dopamine. The binding site of retinal was extracted from the bovine rhodopsin crystal structure complex (pdb code: 1U19). All residues that were within 15 Å radius from C10 atom of retinal were considered to represent the binding site. All interactions with the hydrophobic ring were mapped onto the hydrophobic regions of the retinal molecule and the covalent linkage of C15 atom with LYS296 (K7.43) was treated with a steric constraint (see Fig. 3). Similarly, for the case of dopamine binding to D2 receptor, hydrophobic interaction of the catechol ring with the aromatic cluster in the binding site of D2 receptor was mapped to the catechol ring, while the catechol hydroxyls and amine interactions with the Ser residues in TM5 and Asp in TM3 respectively were treated as hydrogen bond donor—acceptor complexes. The UNITY search module of SYBYL was used to query the combined pharmacophore against the enriched GPCR ligand database to identify small molecules that had a higher propensity to bind to the D2 receptor.
Fig. 3.
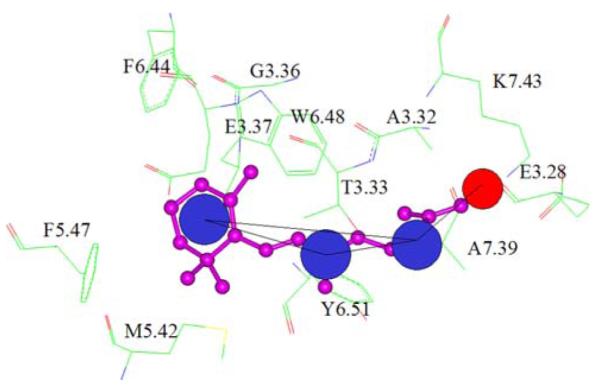
The binding site of retinal in bovine rhodopsin crystal structure is shown along with the combined pharmacophore mapped onto retinal. The residues in the binding site are displayed as sticks and colored atom type with Carbon in green, oxygen in red and nitrogen in blue and labeled according to the generic numbering scheme for GPCRs [59]. Retinal is rendered as ball and stick and colored purple. Hydrophobic features are depicted as blue spheres and steric constraint is shown as red sphere on C15
Docking methodology
The GOLD program (ver 3.0 [47]) was used for docking ligands to the binding sites of the receptor. The “library screening mode” option was used for fast docking. Further, given the non-deterministic nature of genetic algorithms, 20 independent docking runs were performed for each ligand. The “early-termination” option was applied when the top three solutions were within 0.5 Å root-mean-square deviation (RMSD). The docked protein-ligand complexes were energy minimized using SYBYL.
Scoring methodology
The docked receptor—ligand complexes were scored using a customizable knowledge based scoring function that was developed based on the nature of interaction of ligand atoms with amino acids in known protein ligand complexes. Normalized interaction propensities of various functional groups such as halogens, keto, hydroxyl and sulfonamide with the side chains of all the amino acids were derived from these contact maps. The propensity of interaction of atoms of the functional group X with 19 amino acids in the entire data set was recorded as the observed frequency (OBS-Freq). Also, the expected frequency (EXP-Freq) for each such interacting pair was determined as follows:
| (1) |
where, and are the probabilities of occurrence of the interacting pairs A and B in the pool of N(X,X) interactions. The observed propensities were normalized according to
| (2) |
where Xi is the propensity of interaction of the functional group of the ligand with the ith amino acid side chain atom. These interaction propensities were used to rank and score the docked protein—ligand complexes. Accordingly, an in-house program was used to scan the docked complexes for contacts between the ligand and protein atoms. These contacts were then scored based on the normalized contact propensities. To avoid a bias towards scoring based on contact propensities, a consensus-scoring scheme was developed. A contact score weight wi was assigned to each of the ligand i docked into the protein such that:
| (3) |
The weighted docking score of an active compound j with i conformations was described as
| (4) |
where sij was the original GOLD docking score for the compound j in its ith conformation. The best docking score of the compound j with N conformations was calculated as:
| (5) |
In general, the GOLDscore could be replaced by C-score that comprises of various scoring functions such as D-score [48], PMF-score [49], Chemscore [50], G-score and F-score. This scoring scheme has been customized to every receptor by computing only the required contacts for the activity of the receptor—ligand complex, thereby incorporating key experimental information that was available for a particular ligand class interacting with the receptor.
Results and discussion
The HSB method was developed as a screening tool to identify new lead compounds that could bind to GPCRs with high affinity and selectivity. The goal was to effectively combine ligand-based and structure-based methods in order to circumvent the inherent technical problems involved in virtual screening of GPCRs. Thus a multi-step protocol (Fig. 1) was developed that used ligand-based methods such as the Shape Signatures to build a customized, enriched library of small molecules from databases of random drug-like molecules (such as the ZINC database). Beginning with a database of ∼300,000 molecules, the enrichment led to identification of nearly 383 molecules that had a better propensity to bind to rhodopsin and 500 molecules to bind to dopamine D2 receptor. The protocol was validated based on two case studies detailed below:
Identification of retinal like molecules from a random set of compounds using the HSB method
Bovine rhodopsin, closely related to biogenic amine binding GPCRs, is the only GPCR for which a high-resolution x-ray crystal structure with its cognate ligand retinal is available in the Protein Data Bank (PDB [51]). Ever since this first GPCR crystal structure was determined [52], molecular level details of the interaction of retinal with rhodopsin has been well studied by many researchers. The 11-cis isomer of retinal mediates the detection of a light photon in the dark state of rhodopsin, followed by conversion of 11-cis retinal to its trans isomeric form, leading to activation of the receptor and initiation of the visual cascade [53-56]. Crystallographic studies have provided a snapshot of the binding of the chromophore retinal to rhodopsin (Fig. 3). Several analogues of retinal have also been co-crystallized and their mode of binding has been found to be very similar to that of retinal. In general the binding mode of retinal has been defined by a covalent linkage of the C15 carbon with LYS296 (K7.43), the beta-ionone ring with a hydrophobic core formed by residues PHE212 (F5.47), PHE261 (F6.44), TRP265 (W6.48) and other residues such as MET207 (M5.42), GLY121 (G3.36), HIS211 (H5.46) and GLU122 (E3.37). Apart from the covalent interaction with LYS296 (K7.43), the hydrophobic tail also interacts with TYR268 (Y6.51), THR118 (T3.33), ALA117 (A3.32), ALA292 (A7.39) and GLU113 (E3.28). In this study, we tested the ability of the HSB method to identify retinal and its analogues from a random data set of 300,000 molecules. The dataset randomly selected from the ZINC database, included retinal and various other analogues apart from numerous inactive and unrelated compounds.
The 11-cis-retinal molecule devoid of the carbonyl group was used as the query for retrieving molecules using Shape Signatures. A number of top-scoring molecules ranked according to their dissimilarity score were retrieved that formed the enriched dataset (Fig. 4). This enriched small molecule library consisted of 383 molecules (i.e., roughly 0.1% of the total data set). The crystal structure of bovine rhodopsin (pdb code 1U19-chain A) was used as the target for docking studies. The binding site of rhodopsin was defined as all those residues that were found within a sphere of radius 10 Å, with the atom C9 of retinal molecule as the center. In order to simplify the system, all hetero-atoms (such as ions, water molecules) other than retinal molecule were removed for docking studies, although two water molecules around the chromophore were known to be involved in hydrogen bonding network. The protein was treated as a rigid body, while torsional flexibility was allowed for the ligands. Our aim in this study was to validate our method in identifying retinal like molecules that could bind to the receptor and not to understand the dynamic nature of binding of the ligand, which justified the simplifications assumed in this protocol.
Fig. 4.

a list of retinal analogues derived from shape signature search method that form the enriched retinal database is shown. The molecule along with its 1D and 2D scores and their parent database referencing number are shown
In the second phase of the study, a combined receptor—ligand pharmacophore was built (Fig. 5a) to screen the enriched library of 383 molecules to filter out molecules that lacked the essential functional features to bind to rhodopsin. The combined pharmacophore screening included key information from the binding site such as a steric constraint at C15 (representing the covalent linkage with LYS296 of the protein) and the fixed hydrophobic centers denoting the hydrophobic core surrounding the retinal molecule. However, the pharmacophore also included flexible regions in order to choose molecules that mimicked retinal but did not essentially have the same structural components. The screening was divided into two stages, the first involved screening based on the rigid pharmacophore which resulted in molecules shown in Fig. 5b; the second involved a search based on a flexible pharmacophore (Fig. 6a) that resulted in additional molecules (Fig. 6b). The only difference in the rigid vs. flexible pharmacophore was the change in the distance constraints that led to the identification of not only retinal but also other structural analogues.
Fig. 5.

(a) Schematic representation of the combined pharmacophore for retinal in the binding pocket of bovine rhodopsin. The ligand retinal is depicted as ball and stick and colored grey. Blue spheres indicate regions of the ligand strictly interacting with hydrophobic residues and the red sphere indicates a steric interaction signifying the covalent linkage between C15 of the ligand and LYS296 of the receptor. The pharmacophore is shown along with the distance constraints. (b) 2D representations of various retinal analogues that were retrieved from the enriched database using the combined pharmacophore. Note that all the molecules shown here strictly follow the pharmacophore and skeletal structure of the query ligand retinal
Fig. 6.

(a) Schematic representation of the relaxed combined pharmacophore for retinal is shown. Note the change in shape of the pharmacophore and relaxing the distance constraints leads to a selection of a larger number of molecules that have more variations in the skeletal structure. (b) 2D representations of some of the molecules that were retrieved based on the relaxed combined pharmacophore. A total of 110 molecules were retrieved from the enriched database
This second phase of screening led to the selection of 110 molecules that were then docked using the program GOLD. Ten random conformations for each of the ligands were sampled for docking to the protein. The docked poses were scored using Gold score, Chemscore, and our customized in-house scoring function. The customized scoring scheme consisted of scanning all the rhodopsin—ligand contacts using an in-house script; all ligands that docked in a manner similar to the retinal binding mode in the crystal structure complex were scored positively and ligands that docked in other modes were penalized. A comparison of the 10 top-ranking molecules based on Chemscore and customized score is summarized in Table 1.
Table 1.
Ten best ranking analogues of retinal and their ranking based on Chemscore and Customized scoring scheme are shown. The ligand names relate to the database from which they were chosen and their 2D structure is depicted for clarity
| Ligand name |
Ligand structure | Chemscore | Custom score |
|---|---|---|---|
| Aldrich- 294332 |
12 | 1 | |
| Aldrich- 223018 |
31 | 2 | |
| Retinal | 11 | 3 | |
| Aldrich- 294314 |
18 | 4 | |
| Retinoic acid |
36 | 5 | |
| All-trans- 4- oxoretinol |
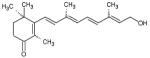 |
4 | 6 |
| N-ethyl- retinamide |
5 | 7 | |
| Aldrich- 223026 |
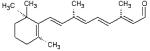 |
23 | 8 |
| All-trans- axerophthe ne |
10 | 9 | |
| Tripos- 611983 |
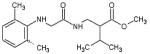 |
85 | 10 |
The results from this study indicated that the customized scoring scheme outperformed Chemscore in correctly identifying top-ranking molecules in seven of ten cases. There was consensus between the two scoring schemes in identifying three molecules. Thus using this protocol, we were able to identify retinal like molecules as the best set of molecules that can bind to rhodopsin binding site. The final scoring was obtained using the consensus-scoring scheme with the contact map scores as the weighting factor.
Identification of dopamine analogues to bind to dopamine D2 receptor
Dopamine receptors, which typify the aminergic GPCR family, share the rhodopsin-like structure. In our previous studies we have demonstrated that using homology models of the TM regions of the receptors, we were able to explain the molecular mechanisms involved in the selectivity of certain 1,4-disubstituted piperizines and piperidines towards the D4 receptor. In the absence of detailed 3D structural information for these proteins, the structure of the only crystallized GPCR rhodopsin has enabled homology modeling of the TM regions, but not the loop segments due to the generally low homology between corresponding loops in GPCRs. Ab initio methods have been used to model the loop forming regions of dopamine D2 receptor.
Using the Shape Signatures method and with dopamine molecule as a query compound, nearly 500 molecules were retrieved from various databases of small molecules. These molecules formed the enriched database for further screening experiments. Dopamine was docked into the agonist model of the dopamine D2 receptor. The binding site for most of the biogenic amine binding GPCRs has been well characterized using site-directed mutagenesis and spectroscopic experiments [57]. The binding mode of dopamine in the D2 receptor derived from various biochemical experiments suggested favorable hydrogen bonded interactions of the catechol hydroxyls with SER residues in TM5 (S5.42 and S5.43) and aromatic interactions of the catechol ring with residues in TM6 and TM7 (F6.51, F6.52 and H6.55). Similar to other biogenic amines, the protonated amine group of dopamine engaged in favorable ionic interaction with ASP in TM3 (D3.32) (see Fig. 7a). Based on these interactions, a combined receptor—ligand pharmacophore was built as shown in Fig. 7b. A flexible combined pharmacophore was used to screen the enriched dopamine ligand database to identify 183 molecules that could potentially bind to dopamine D2 receptor (Fig. 7c). These molecules were then docked to the D2 receptor binding site using GOLD program with 20 random starting conformations for each ligand. The docked complexes were scored using GOLD score and were then re-scored with our customized scoring scheme that ranked the complexes based on the nature of interaction of the ligands with important residues in the binding site. All those molecules that had an interaction profile similar to dopamine in the D2 binding site were scored positively and other docking poses were penalized. Finally, the molecules were ranked based on the consensus-scoring scheme. Table 2 shows the set of six top-ranking molecules and their 2D structure, that are known analogues of dopamine, while, experiments are being conducted to evaluate the affinity of molecules ranked 7–40 (see supplementary information), the results of which will be discussed elsewhere.
Fig. 7.

(a) The binding site of dopamine in the modeled dopamine D2 receptor is shown along with the combined pharmacophore mapped onto dopamine. The residues in the binding site are displayed as sticks, numbered according to the generic numbering scheme and colored pink. Dopamine is rendered as ball and stick and colored atom type with carbon in green, oxygen in red and nitrogen in blue. Hydrophobic feature is depicted as cyan spheres. Hydrogen bonding interactions of dopamine with the binding site residues are shown as dotted black lines. For sake of clarity the hydrogen atoms from both dopamine and the binding site residues are not shown. (b) Schematic representation of the combined pharmacophore mapped onto dopamine is shown. Dopamine is represented as ball and stick and colored atom type with Carbon in green, Nitrogen in blue and oxygen in red. The catechol ring interacts favorably with the aromatic residues in the binding site and hence is mapped to a hydrophobic feature and the catechol hydroxyls and amine group are mapped as hydrogen bond donors. (c) Molecules that were retrieved from combined pharmacophore search are listed in their 2D form. A total of 183 molecules were chosen that obeyed the pharmacophore
Table 2.
Final results from the HSB method for the query ligand dopamine. Six best ranking compounds that are analogues of dopamine and identified using Consensus scoring scheme are shown along with their 2D structure
| Ligand name | Ligand structure | Consensus rank |
|---|---|---|
| L-Dopamine | 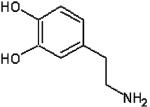 |
1 |
| Dimethoxydopamine | 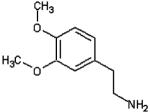 |
2 |
| Dopaminequinone | 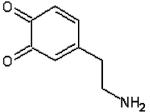 |
3 |
| 3-benzyl dopamine | 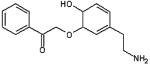 |
4 |
| Fluorodopamine |  |
5 |
| (2S)-N-[2-(3,4- dihydroxyphenyl)ethyl]- 2-[3-(2,5- dimethoxyphenyl)prop- 2-enoy lamino]-3-hydroxy- propanamide |
 |
6 |
Conclusions
In the past, drug discovery for GPCRs has been primarily ligand-based due to the absence of experimental structural information. However, advances in homology-based and ab initio modeling methods, together with the availability of the crystal structure of the rhodopsin—retinal complex have led to an increase in the use of structure-based methods for virtual screening. A suitable combination of both the ligand- and structure-based methods may guide the discovery of new lead compounds that possess high affinity and specificity for their targets leading to minimizing receptor mediated side effects. Further, applying the ligand based shape signature technology to a large database not only reduces the number of compounds to ∼0.1% of its original size, but also enriches the nature of the dataset by including only those compounds that either share a similar scaffold or a similar shape as that of the query molecule. This reductionist approach provides a major savings in computational time and effort as opposed to screening the entire database of 300,000 molecules using the combined pharmacophore. Based on this hypothesis, the HSB method was developed to identify new lead compounds binding to GPCRs. The method was validated for two well-studied cases. In the case of rhodopsin, the HSB method was employed to identify retinal and its analogues from a pool of ∼300,000 molecules comprising of retinal and its analogues and other random molecules. The results from this study showed that all the 10 top-ranking molecules were known binders to rhodopsin with high affinity and are, indeed, analogues of retinal. In a study on dopamine binding to dopamine D2 receptor, the HSB method was adept in identifying dopamine and five other analogues as the best six compounds including a known prodrug. In addition, thirty-four new molecules were predicted to exhibit high affinity for the D2 receptor. These newly identified molecules will be tested for their affinity against all members of the dopamine receptor family (D1—D5).
The proposed HSB method can be used for screening and identification of new lead compounds binding to class-A GPCRs. The method is being currently tested for applicability against other GPCRs and globular proteins, and the results will be discussed in future publications.
Supplementary Material
Acknowledgments
The authors acknowledge access to the computational facilities at UMDNJ-Informatics Institute and the Academic computing services. Dr. Peng Zhang is also acknowledged for providing programming expertise. Finally, this study was partially supported by a grant from the National Library of Medicine, National Institutes of Health (G08 LM6230-07).
Footnotes
Electronic supplementary material Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s10822-006-9077-8 and is accessible for authorized users.
References
- 1.Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. FEBS Lett. 2002;520(1–3):97. doi: 10.1016/s0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- 2.Birnbaumer L, Brown AM. Am Rev Respir Dis. 1990;141(3 Pt 2):S106. doi: 10.1164/ajrccm/141.3_Pt_2.S106. [DOI] [PubMed] [Google Scholar]
- 3.Hoon MA, Adler E, Lindemeier J, Battey JF, Ryba NJ, Zuker CS. Cell. 1999;96(4):541. doi: 10.1016/s0092-8674(00)80658-3. [DOI] [PubMed] [Google Scholar]
- 4.Bourne HR, Sanders DA, McCormick F. Nature. 1991;349(6305):117. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 5.Hamm HE. J Biol Chem. 1998;273(2):669. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- 6.Garver DL, Schlemmer RF, Jr, Maas JW, Davis JM. Am J Psychiatry. 1975;132(1):33. doi: 10.1176/ajp.132.1.33. [DOI] [PubMed] [Google Scholar]
- 7.Peroutka SJ, Snyder SH. Science. 1980;210:88. doi: 10.1126/science.6251550. [DOI] [PubMed] [Google Scholar]
- 8.Comings DE. Ann N Y Acad Sci. 2001;931:50. doi: 10.1111/j.1749-6632.2001.tb05773.x. [DOI] [PubMed] [Google Scholar]
- 9.Saxena PR, Ferrari MD. Trends Pharmacol Sci. 1989;10(5):200. doi: 10.1016/0165-6147(89)90238-1. [DOI] [PubMed] [Google Scholar]
- 10.Hallsworth MP, Twort CH, Lee TH, Hirst SJ. Br J Pharmacol. 2001;132:729. doi: 10.1038/sj.bjp.0703866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amer MS. Biochem Pharmacol. 1977;26(3):171. doi: 10.1016/0006-2952(77)90298-2. [DOI] [PubMed] [Google Scholar]
- 12.Kehne JH, Baron BM, Carr AA, Chaney SF, Elands J, Feldman DJ, Frank RA, van Giersbergen PL, McCloskey TC, Johnson MP, McCarty DR, Poirot M, Senyah Y, Siegel BW, Widmaier C. J Pharmacol Exp Ther. 1996;277(2):968. [PubMed] [Google Scholar]
- 13.Peroutka SJ, U’Prichard DC, Greenberg DA, Snyder SH. Neuropharmacology. 1977;16(9):549. doi: 10.1016/0028-3908(77)90023-5. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter WT, Jr, Heinrichs DW, Wagman AM. Am J Psychiatry. 1988;145(5):578. doi: 10.1176/ajp.145.5.578. [DOI] [PubMed] [Google Scholar]
- 15.Worrel JA, Marken PA, Beckman SE, Ruehter VL. Am J Health Syst Pharm. 2000;57(3):238. doi: 10.1093/ajhp/57.3.238. [DOI] [PubMed] [Google Scholar]
- 16.Fann WE, Sullivan JL, Richman BW. Br J Psychiatry. 1976;128:490. doi: 10.1192/bjp.128.5.490. [DOI] [PubMed] [Google Scholar]
- 17.Attwood TK. Trends Pharmacol Sci. 2001;22(4):162. doi: 10.1016/s0165-6147(00)01658-8. [DOI] [PubMed] [Google Scholar]
- 18.Rognan D. J Physiol Paris. 2006;99(2–3):232. doi: 10.1016/j.jphysparis.2005.12.084. [DOI] [PubMed] [Google Scholar]
- 19.Shoichet BK. Nature. 2004;432(7019):862. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorgensen WL. Science. 2004;303(5665):1813. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 21.Bissantz C, Bernard P, Hibert M, Rognan D. Proteins. 2003;50(1):5. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 22.Kortagere S, Gmeiner P, Weinstein H, Schetz JA. Mol Pharmacol. 2004;66(6):1491. doi: 10.1124/mol.104.001321. [DOI] [PubMed] [Google Scholar]
- 23.Floresca CZ, Chen S, Kortagere S, Schetz JA. Arch Pharm (Weinheim) 2005;338(5–6):268. doi: 10.1002/ardp.200400993. [DOI] [PubMed] [Google Scholar]
- 24.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45(1):177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ. J Med Chem. 2003;46(26):5674. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 26.Zauhar RJ. J Comput Aided Mol Des. 1995;9(2):149. doi: 10.1007/BF00124405. [DOI] [PubMed] [Google Scholar]
- 27.Keener JP. Principles of applied mathematics. Addison-Wesley; 1988. [Google Scholar]
- 28.Stewart JJ. J Comput-Aided Mol Des. 1990;4:1. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 29.Bairoch A, Apweiler R. J Mol Med. 1997;75:312. [PubMed] [Google Scholar]
- 30.Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ. Nucl Acid Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G. Nucl Acid Res. 2003;31:294. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Visiers I, Ballesteros JA, Weinstein H. Methods Enzymol. 2002;343:329. doi: 10.1016/s0076-6879(02)43145-x. [DOI] [PubMed] [Google Scholar]
- 33.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Proteins Struct Funct Genet. 1995;23:318. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 34.Visiers I, Braunheim BB, Weinstein H. Protein Eng. 2000;13(9):603. doi: 10.1093/protein/13.9.603. [DOI] [PubMed] [Google Scholar]
- 35.Sansom MS, Weinstein H. Trends Pharmacol Sci. 2000;21(11):445. doi: 10.1016/s0165-6147(00)01553-4. [DOI] [PubMed] [Google Scholar]
- 36.Mehler El, Hassan SA, Kortagere S, Weinstein H. Ab initio computational modeling of loops in G-protein coupled receptors: Lessons from the crystal structure of rhodopsin. Proteins. 2006;64(3):673. doi: 10.1002/prot.21022. [DOI] [PubMed] [Google Scholar]
- 37.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187. [Google Scholar]
- 38.Li X, Hassan SA, Mehler EL. Proteins. 2005;60(3):464. doi: 10.1002/prot.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassan SA, Mehler EL, Zhang D, Weinstein H. Proteins. 2003;51(1):109. doi: 10.1002/prot.10330. [DOI] [PubMed] [Google Scholar]
- 40.Lazaridis T. Proteins. 2005;58(3):518. doi: 10.1002/prot.20358. [DOI] [PubMed] [Google Scholar]
- 41.Mottamal M, Lazaridis T. Biochemistry. 2005;44(5):1607. doi: 10.1021/bi048065s. [DOI] [PubMed] [Google Scholar]
- 42.Javitch JA. Adv Pharmacol. 1998;42:412. doi: 10.1016/s1054-3589(08)60776-0. [DOI] [PubMed] [Google Scholar]
- 43.Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. J Biol Chem. 2001;276(12):9279. doi: 10.1074/jbc.M004871200. [DOI] [PubMed] [Google Scholar]
- 44.Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK. EMBO J. 1997;16(22):6737. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rasmussen SG, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U. Mol Pharmacol. 1999;56(1):175. doi: 10.1124/mol.56.1.175. [DOI] [PubMed] [Google Scholar]
- 46.Pearlman DA, Case DA, Caldwell JW, Ross WR, Cheatham TE, III, DeBolt S, Ferguson D, Seibel G, Kollman P. Comp Phys Commun. 1995;91:1. [Google Scholar]
- 47.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 48.Gohlke H, Klebe G. Curr Opin Struct Biol. 2001;11(2):231. doi: 10.1016/s0959-440x(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 49.Muegge I, Martin YC. J Med Chem. 1999;42:791. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 50.Eldridge MD, Murray CW, Auton TR, Paolinine GV, Mee RP. J Comput-Aided Mol Des. 1997;11:425. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 51.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucl Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289(5480):739. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 53.Fanelli F, Dell’Orco D. Biochemistry. 2005;44(45):14695. doi: 10.1021/bi051537y. [DOI] [PubMed] [Google Scholar]
- 54.Ludeke S, Beck M, Yan EC, Sakmar TP, Sibert F, Vogel R. J Mol Biol. 2005;353(2):345. doi: 10.1016/j.jmb.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 55.Patel AB, Crocker E, Reeves PJ, Getmanova EV, Eilers M, Khorana HG, Smith SO. J Mol Biol. 2005;347(4):803. doi: 10.1016/j.jmb.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 56.Perez DM, Karnik SS. Pharmacol Rev. 2005;57(2):147. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- 57.Tota MR, Strader CD. J Biol Chem. 1990;265(28):16891. [PubMed] [Google Scholar]
- 58.Mackerell AD, Jr, Bashford D, Bellot M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 59.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.