

Abstract
Evidence for the important role of the potent oxidant peroxynitrite in peripheral diabetic neuropathy and neuropathic pain is emerging. This study evaluated the contribution of neuronal nitric oxide synthase (nNOS) to diabetes-induced nitrosative stress in peripheral nerve and dorsal root ganglia, and peripheral nerve dysfunction and degeneration. Control and nNOS−/− mice were made diabetic with streptozotocin, and maintained for 6 weeks. Peroxynitrite injury was assessed by nitrotyrosine and poly(ADP-ribose) immunoreactivities. Peripheral diabetic neuropathy was evaluated by measurements of sciatic motor and hind-limb digital sensory nerve conduction velocities, thermal algesia, tactile allodynia, and intraepidermal nerve fiber density. Control nNOS−/− mice displayed normal motor nerve conduction velocity and thermal response latency, whereas sensory nerve conduction velocity was slightly lower compared with non-diabetic wild-type mice, and tactile response threshold and intraepidermal nerve fiber density were reduced by 47 and 38%, respectively. Both diabetic wild-type and nNOS−/− mice displayed enhanced nitrosative stress in peripheral nerve. In contrast to diabetic wild-type mice, diabetic nNOS−/− mice had near normal nitrotyrosine and poly(ADP-ribose) immunofluorescence in dorsal root ganglia. Both diabetic wild-type and nNOS−/− mice developed motor and sensory nerve conduction velocity deficits and thermal hypoalgesia although nNOS gene deficiency slightly reduced severity of the three disorders. Tactile response thresholds were similarly decreased in control and diabetic nNOS−/− mice compared with non-diabetic wild-type mice. Intraepidermal nerve fiber density was lower by 27% in diabetic nNOS−/− mice compared with the corresponding non-diabetic group, and by 20% in diabetic nNOS−/− mice compared with diabetic wild-type mice. In conclusion, nNOS is required for maintaining the normal peripheral nerve function and small sensory nerve fibre innervation. nNOS gene deficiency does not protect from development of nerve conduction deficit, sensory neuropathy and intraepidermal nerve fiber loss.
Keywords: nerve conduction, neuronal nitric oxide synthase, nitrosative stress, peripheral diabetic neuropathy, streptozotocin-diabetic mouse
Introduction
Accumulation of the potent oxidant peroxynitrite, a product of superoxide anion radical reaction with nitric oxide, is associated with multiple detrimental consequences including, but not limited by, initiation of lipid peroxidation, DNA breakage and base modifications, metalloproteinase and poly(ADP-ribose) polymerase (PARP) activations, impairment of cell signaling, changes in transcriptional regulation, inflammatory response, and, in extreme cases, cell death by necrosis and apoptosis (1–4). Evidence for the important role of peroxynitrite in diabetic complications including vascular disease (5), cardiomyopathy (2–4), peripheral (PDN; 6–11) and autonomic (12) neuropathy, and retinopathy (13) is emerging. Using a pharmacological approach with structurally different peroxynitrite decomposition catalysts and several animal models of diabetic neuropathy, we demonstrated an important role for peroxynitrite injury (nitrosative stress) in diabetes-associated nerve blood flow and nerve conduction deficits, impaired vascular reactivity of epineurial arterioles, thermal hyper- and hypoalgesia, mechanical hypoalgesia, tactile allodynia, and small sensory nerve fiber degeneration (6–11). Furthermore, nitrosative stress in peripheral nerve, spinal cord, and dorsal root ganglion (DRG) neurons has been identified as an important factor leading to PARP activation, another important mechanism in the pathogenesis of PDN (14–17).
Until now, the sources of superoxide and nitric oxide for diabetes-related peroxynitrite formation in peripheral nervous system (PNS) have been elusive. Two pharmacological studies (18,19) revealing beneficial effects of the NAD(P)H oxidase inhibitor apocynin and the xanthine oxidase inhibitor allopurinol on early PDN, suggest that these two superoxide-generating enzymes contribute to diabetes-associated superoxide production in PNS. Increased superoxide production has been documented in vasa nervorum in rat models of both Type 1 and Type 2 diabetes (16,20,21), and in high glucose-exposed human Schwann cells (22). Superoxide dismutase activity was found to be reduced in the diabetic nerve (23,24). Insufficient information is available on the role of individual isoforms of nitric oxide synthase (NOS), an enzyme producing the second component for peroxynitrite formation, i.e., nitric oxide, in nitrosative stress in PNS and PDN. Endothelial NOS (eNOS) is known to play an important role in maintenance of normal nerve blood flow, and its uncoupling in diabetes causes neurovascular dysfunction and nerve conduction deficits (25,26). Inducible NOS (iNOS) has recently been identified as a major contributor to oxidative-nitrosative stress in peripheral nerve as well as nerve conduction deficit, sensory disorders, and intraepidermal nerve fiber loss characteristic for PDN (12). The present study was aimed at evaluating the role for neuronal NOS (nNOS) in oxidative-nitrosative stress in peripheral nerve and DRG, and in development of neuropathic changes in STZ-diabetic mice, a model of Type 1 diabetic neuropathy. STZ-induced diabetic mice of all genetic strains studied so far, i.e., C57Bl6/J (6,8,9,12,27–30), 129S1/SvImJ (17), Swiss-Webster (31,32), and C57B6:129S7 (33) mice, developed both functional and structural changes characteristic for PDN.
Materials and methods
Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality, and were purchased from Sigma Chemical Co., St. Louis, MO. Rabbit polyclonal anti-nitrotyrosine (NT) antibody was purchased from Upstate, Lake Placid, NY, and mouse monoclonal anti-poly(ADP-ribose) from Trevigen, Inc., Gaithersburg, MD. Secondary Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 488 goat anti-mouse antibodies as well as Prolong Gold Antifade Reagent were purchased from Invitrogen, Eugene, OR. Avidin/Biotin Blocking Kit, M.O.M. Basic Kit, Vectastain Elite ABC Kit (Standard*), DAB Substrate Kit, and 3,3′-diaminobenzidine were obtained from Vector Laboratories, Burlingame, CA. Rabbit polyclonal anti-protein gene product 9.5 (ubiquitin c-terminal hydrolase) antibody was purchased from Chemicon International, Inc., Temecula, CA. Other reagents for immunohistochemistry were purchased from Dako Laboratories, Inc., Santa Barbara, CA.
Animals and limitations of the nNOS−/− mouse model
The experiments were performed in accordance with regulations specified by the National Institutes of Health ‘Principles of Laboratory Animal Care, 1985 Revised Version’ and Pennington Biomedical Research Center Protocol for Animal Studies. Several breeding pairs of B6.129S4-Nos1tm1Plh/J (nNOS−/−) mice, C57Bl6/J background, were obtained from Jackson Laboratories (Bar Harbor, ME, stock number 002986). The use of nNOS−/− mice for diabetic neuropathy-related studies is associated with some limitations. nNOS-deficient mice have enlarged stomachs, and a significant proportion develops a gastroparesis-like condition in both non-diabetic and diabetic states. Furthermore, hyperglycemic-euglycemic clamp studies demonstrate that homozygotes exhibit insulin resistance at the level of peripheral tissues (http://jaxmice.jax.org/strain/002986.html).
For the main experiment, a colony of nNOS−/− mice was established at Pennington Biomedical Research Center. Mature male C57Bl6/J mice were purchased from Jackson Laboratories and served as controls. All the mice were fed standard mouse chow (PMI Nutrition International, Brentwood, MO) and had access to water ad libitum. Diabetes was induced by a single injection of streptozotocin (STZ), 100 mgkg−1d−1, i.p., to non-fasted animals. Blood samples for glucose measurements were taken from the tail vein three days after STZ injection and the day before the animals were sacrificed. The mice with blood glucose ≥13.8 mM were considered diabetic. The injected mice that had blood glucose concentration in non-diabetic range have been given low-dose STZ injections (40 mgkg−1d−1, i.p.) until they developed hyper-glycemia (typically, one-three additional injections). At the end of the study (6-week duration of diabetes), the physiological and behavioral tests were performed in the following order: tactile responses to flexible von Frey filaments (first day), thermal algesia by tail-flick test (second day), thermal algesia by paw withdrawal test (third day), SNCV and MNCV (fourth day). Measurements of MNCV and SNCV were performed in mice anaesthetized with a mixture of ketamine and xylazine (45 mgkg−1 body weight and 15 mgkg−1 body weight, respectively, i.p.).
Anesthesia, euthanasia and tissue sampling
The animals were sedated by CO2, and immediately sacrificed by cervical dislocation. Sciatic nerves, dorsal root ganglia (DRG), and foot pads were fixed in normal buffered 4% formalin for assessment of nitrotyrosine and poly(ADP-ribose) by immunofluorescent histochemistry and intraepidermal nerve fiber density by conventional immunohistochemistry. Poly(ADP-ribose) abundance is a measure of poly(ADP-ribose) polymerase activity (14–17,34).
Specific methods
Physiological tests
Sciatic MNCV and hind-limb digital SNCV were measured as we have described elsewhere (11,14,17).
Behavioral tests
i) Tactile response threshold
Tactile responses were evaluated by quantifying the withdrawal threshold of the hindpaw in response to stimulation with flexible von Frey filaments as we have described (35).
ii) Thermal algesia
The paw withdrawal latency in response to the radiant heat (15% intensity which produced a heating rate of ~1.3°C per sec, cut-off time 30 sec) was determined as we have described (7–9,17,35) using the IITC model 336 TG combination tail-flick and paw algesia meter (IITC Life Science) with a floor temperature ~32–33°C (manufacturer’s set up). For assessment of tail flick response latencies, the device was set at 40% heating intensity (heating rate ~2.5°C per sec) with a cut-off at 10 sec. In both tests, at least three readings per animal were taken at 15 min interval, and the average was calculated.
Immunohistochemical studies
All sections were processed by a single investigator and evaluated blindly. Low power observations of skin sections stained for PGP 9.5 were made using a Zeiss Axioskop microscope. Color images were captured with a Zeiss Axiocam HRc CCD camera at 1300×1030 resolution. Low power images were generated with a 40x acroplan objective using the automatic capturing feature of the Zeiss Axiovision software (Ver. 3.1.2.1). Low power observations of sciatic nerve sections stained for NT and DRG sections stained for NT and poly(ADP-ribose) were made using a Zeiss Axioplan 2 imaging microscope. Color images were captured with a Photometric CoolSNAP™ HQ CCD camera at 1392×1040 resolution. Low power images were generated with a 40x acroplan objective using the RS Image™ 1.9.2 software.
i) NT immunoreactivity in sciatic nerve and DRG neurons
NT immunoreactivities in the sciatic nerve and DRG were assessed by immunofluorescent histochemistry. In brief, sections were deparaffinized in xylene, rehydrated in decreasing concentrations of ethanol, and washed in water. For immunofluorescent histochemistry, rabbit polyclonal anti-NT antibody was used in a working dilution 1:400. Primary antibody was omitted in negative controls. Secondary Alexa Fluor 488 goat anti-rabbit antibody was applied in a working dilution 1:200. Sections were mounted in Prolong Gold Antifade Reagent. The intensity of fluorescence was quantified using the ImageJ 1.32 software (National Institutes of Health, Bethesda, MD) and expressed as mean ± SEM for each experimental group.
ii) Poly(ADP-ribose) (PAR) immunoreactivity in DRG
PAR immunoreactivity was assessed as described (14) with minor modifications. In brief, sections of DRG were deparaffinized in xylene, rehydrated in decreasing concentrations of ethanol and washed in water. Non-specific binding was blocked with the mouse IG blocking reagent supplied with the Vector M.O.M. Basic Immunodetection Kit. Then mouse monoclonal anti-PAR antibody was diluted 1:100 in 1% BSA in TBS, and applied overnight at 4°C in a humidity chamber. Primary antibody was omitted in negative controls. Secondary Alexa Fluor 488 goat anti-mouse antibody was diluted 1:200 in TBS and applied for 2 h at room temperature. Sections were mounted in Prolong Gold Antifade Reagent. At least, ten fields of each section were examined to select one representative image. Representative images were micro-photographed and PAR fluorescence was calculated for each microphotograph using the ImageJ 1.32 software, and was expressed as mean ± SEM for each experimental group. To evaluate neuronal PAR accumulation, a percentage of DRG neurons with weak, moderate, and intense PAR immunofluorescence was calculated for each experimental group.
iii) Intraepidermal nerve fiber density (INFD)
INFD was assessed as described (7–9,11,17). Three randomly chosen 5-μm sections from foodpad skin of each mouse were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol, and washed in water. Non-specific binding was blocked by 10% goat serum containing 1% BSA in TBS (Dako, Carpinteria, CA) for 2 h, and the Avidin/Biotin Blocking Kit, according to the manufacturer’s instructions. Then, rabbit polyclonal anti-protein gene product 9.5 (ubiquitin c-terminal hydrolase) antibody was applied in 1:2000 dilution. Secondary biotinylated goat anti-rabbit IgG (H+L) antibody was applied in 1:400 dilution, and the staining performed with the Vectastain Elite ABC Kit (Standard*). For visualization of specific binding sites, the DAB Substrate Kit containing 3,3-diaminobenzidine was used. Sections were counter-stained with Gill’s hematoxylin, dehydrated and mounted in Micromount mounting medium (Surgipath Medical Ind., Richmond, IL). Intraepidermal nerve fiber profiles were counted blindly by three independent investigators, under an Olympus BX-41 microscope, and the average values were used. Microphotographs of stained sections were taken on Axioscop 2 microscope (Zeiss) at magnification x4, and the length of epidermis was assessed with the ImagePro 3.0 program (Media Cybernetics). An average of 2.8±0.3 mm of the sample length was investigated to calculate a number of nerve fiber profiles per mm of epidermis.
Statistical analysis
The results are expressed as mean ± SEM. Data were subjected to equality of variance F test, and then to log transformation, if necessary, before one-way analysis of variance. Where overall significance (p<0.05) was attained, individual between group comparisons were made using the Student-Newman-Keuls multiple range test. Significance was defined at p≤0.05. When between-group variance differences could not be normalized by log transformation (datasets for body weights and plasma glucose), the data were analyzed by the non-parametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni/Dunn test for multiple comparisons.
Results
Weight gain during the 6-week study was 19% in non-diabetic wild-type and 11.5% in non-diabetic nNOS−/− mice (Table I). Both diabetic wild-type and diabetic nNOS−/− mice lost 7% of their initial body weights. Initial (after STZ injection) blood glucose concentrations were 108 and 146% higher in diabetic wild-type and diabetic nNOS−/− mice compared with the corresponding controls. Hyperglycemia progressed with the prolongation of diabetes. Final blood glucose concentrations were similar in diabetic wild-type and diabetic nNOS−/− mice.
Table I.
Initial and final body weights and blood glucose concentrations in experimental groups.
| Body weight (g) |
Blood glucose (mmol/l) |
|||
|---|---|---|---|---|
| Initial | Final | Initial | Final | |
| Non-diabetic wild-type | 25.8±1.0 | 30.7±0.6 | 7.1±0.4 | 6.6±0.4 |
| Non-diabetic iNOS−/− | 26.9±1.8 | 30.0±1.2 | 6.3±0.1 | 6.4±0.2 |
| Diabetic wild-type | 26.0±0.7 | 24.2±0.5a | 14.8±0.9a | 28.3±1.4a |
| Diabetic iNOS−/− | 26.6±0.8 | 24.8±0.5a | 15.5±1.2a | 29.9±1.6a |
Data are means ± SEM, n=8–11 per group.
Significantly different from the corresponding non-diabetic groups (p<0.01).
nNOS gene deficiency did not affect sciatic MNCV and resulted in 11% reduction in hind-limb SNCV in non-diabetic mice (Fig. 1). MNCV and SNCV were 19 and 20% lower in diabetic wild-type mice (p<0.01 for both comparisons), and 11 and 10% lower in diabetic nNOS−/− mice (p<0.05 and <0.01, respectively), compared with the corresponding non-diabetic groups.
Figure 1.
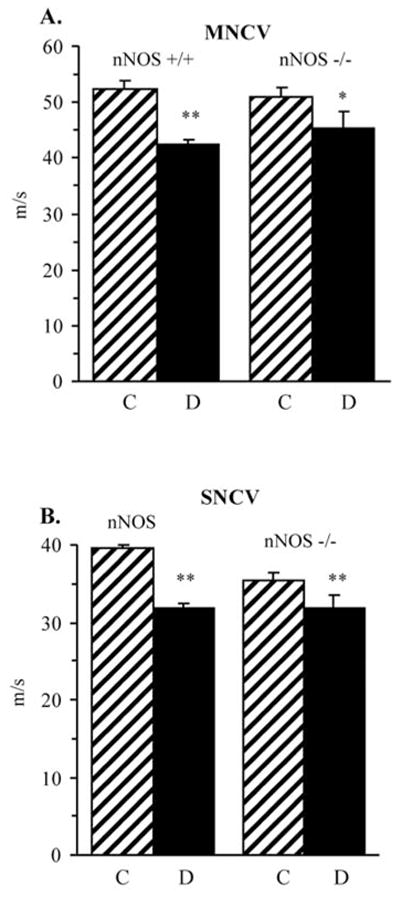
Sciatic motor nerve conduction velocities (A) and hind-limb digital sensory nerve conduction velocities (B) in control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=8–10 per group. C, control mice; D, diabetic mice. *p<0.05 and **p<0.01 vs corresponding non-diabetic groups.
The latency of hind paw withdrawal in response to radiant heat was increased by 100% in diabetic wild-type mice compared with the control group (p<0.01), consistent with clearly manifest thermal hypoalgesia (Fig. 2A). This is in agreement with the results of tail-flick test which also revealed increased thermal response latencies in the diabetic wild-type mice (Fig. 2B). The severity of hypoalgesia by paw withdrawal test was lower in diabetic nNOS−/− mice, compared with diabetic wild-type mice.
Figure 2.
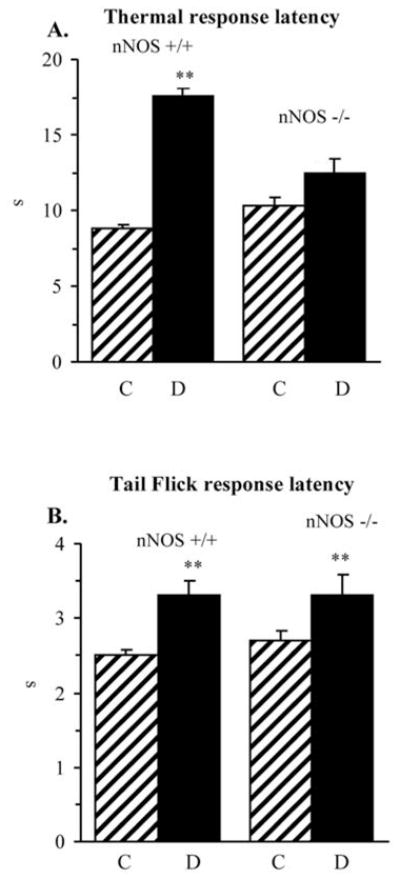
Paw withdrawal latencies in response to radiant heat (A) and tail-flick test response latencies (B) in control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=8–11 per group. C, control mice; D, diabetic mice. **p<0.01 vs corresponding non-diabetic groups; ##p<0.01 vs diabetic wild-type mice.
Non-diabetic nNOS−/− mice displayed 47% reduction in tactile response thresholds, compared with non-diabetic wild-type mice (Fig. 3). Diabetic wild-type mice developed clearly manifest tactile allodynia, whereas tactile response threshold were similarly reduced in non-diabetic and diabetic nNOS−/− mice, compared with wild-type controls.
Figure 3.
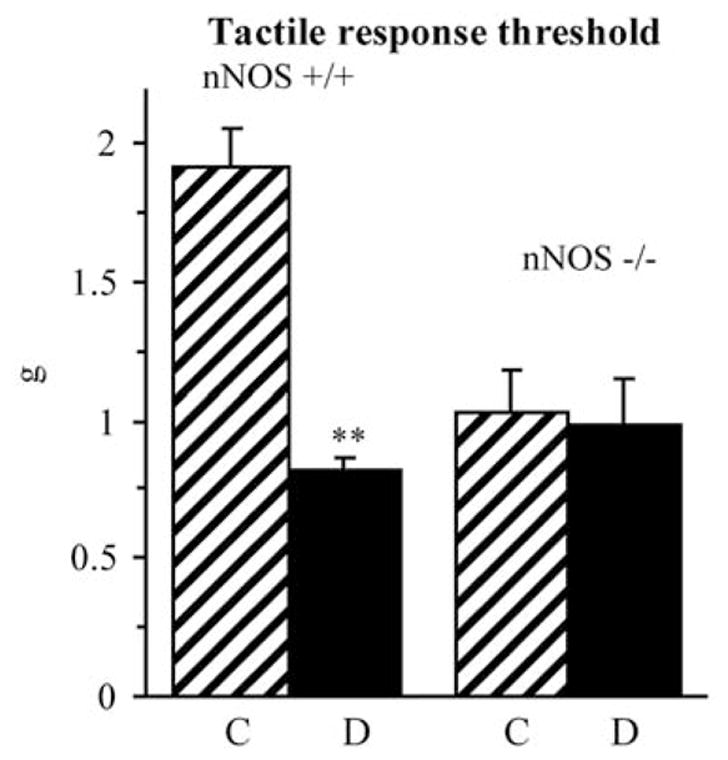
Tactile response thresholds in response to stimulation with flexible von Frey filaments in control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=8–11 per group. C, control mice; D, diabetic mice. **p<0.01 vs non-diabetic control mice.
INFD was reduced by 38% in diabetic wild-type mice and by 27% in diabetic nNOS−/− mice compared with the corresponding controls (p<0.05 for both comparisons, Fig. 4). Note, however, that nNOS gene deficiency was associated with a 32% decrease in INFD in non-diabetic mice.
Figure 4.
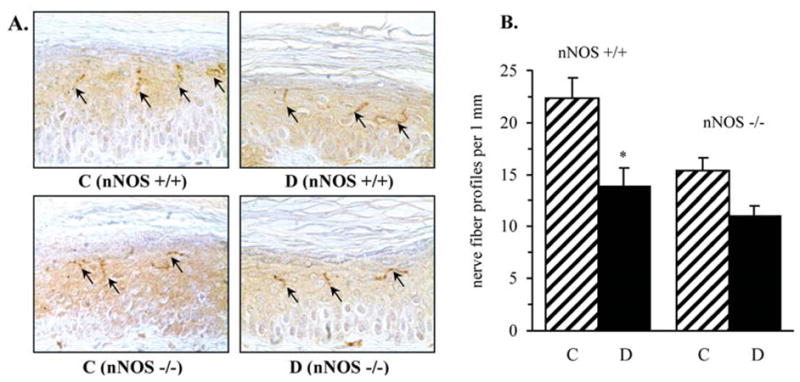
Intraepidermal nerve fiber profiles in control and diabetic wild-type and nNOS−/− mice. (A) Representative images of intraepidermal nerve fiber profiles, magnification x200; (B) skin fiber density. Mean ± SEM, n=8–11 per group. C, control mice; D, diabetic mice. *p<0.05 vs control mice.
NT immunofluorescence was 31% higher in non-diabetic nNOS−/− mice compared with non-diabetic wild-type mice, but the difference between two non-diabetic groups did not achieve statistical significance. NT immunofluorescence increased by 69 and 37% in the sciatic nerves of diabetic wild-type and nNOS−/− mice, respectively, compared with the corresponding non-diabetic controls (p<0.01, Fig. 5). DRG NT immunofluorescence was comparable in non-diabetic wild-type and nNOS−/− mice (Fig. 6). It was increased by 56% in diabetic wild-type mice compared with non-diabetic controls (p<0.01). In contrast, diabetic nNOS−/− mice were protected against peroxynitrite injury in DRG and maintained nitrotyrosine immunoreactivity at the level detected in non-diabetic mice.
Figure 5.
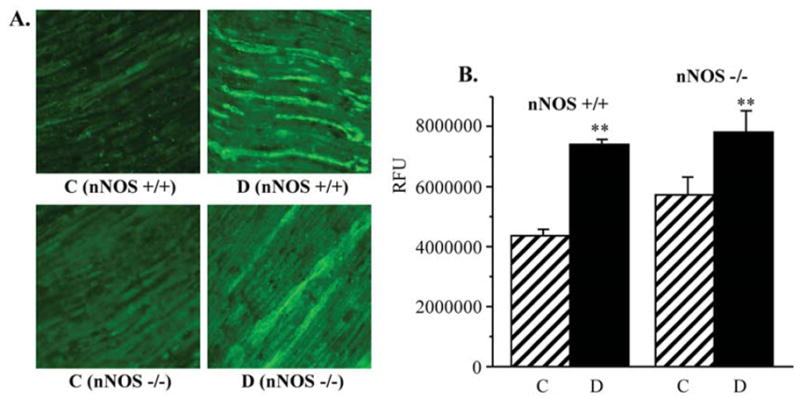
(A) Representative microphotographs of immunofluorescent staining of nitrotyrosine in sciatic nerves of control and diabetic wild-type and nNOS−/− mice. Magnification x40. (B) Nitrotyrosine fluorescence counts in sciatic nerves of control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=7–11 per group. C, control mice; D, diabetic mice. **p<0.01 vs non-diabetic control mice.
Figure 6.
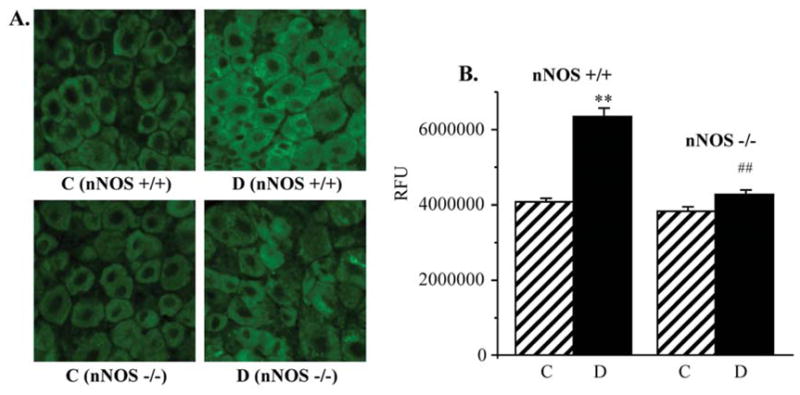
(A) Representative microphotographs of immunofluorescent staining of nitrotyrosine in dorsal root ganglia of control and diabetic wild-type and nNOS−/− mice. Magnification x40. (B) Nitrotyrosine fluorescence counts in dorsal root ganglia of control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=7–11 per group. C, control mice; D, diabetic mice. **p<0.01 vs non-diabetic control mice. ##p<0.01 vs diabetic wild-type mice.
PAR fluorescence was similar in DRG of non-diabetic wild-type and nNOS−/− mice (Fig. 7). It was 23% higher in DRG of diabetic wild-type mice compared with non-diabetic controls (p<0.01). In contrast, diabetic nNOS−/− mice displayed normal DRG PAR immunofluorescence (Fig. 8). The percentage of DRG neurons with weak PAR immunofluorescence was lower, and of those with moderate and intense immunofluorescence higher in diabetic wild-type mice compared with the corresponding control group. nNOS gene deficiency reduced the percentage of neurons with moderate and intense PAR fluorescence and increased the percentage of neurons with weak PAR fluorescence in diabetic mice.
Figure 7.
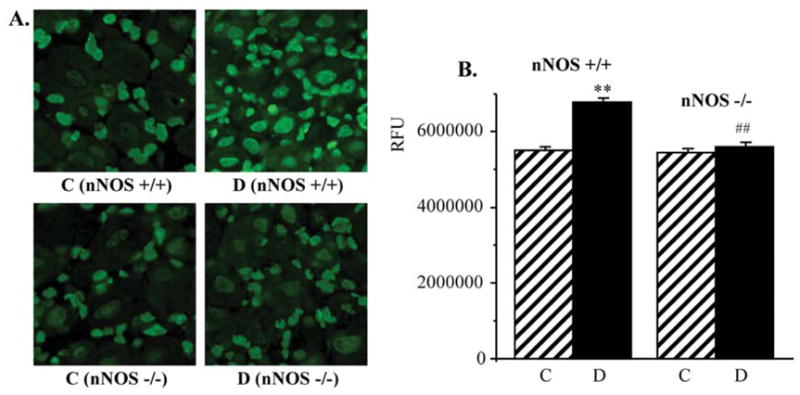
(A) Representative microphotographs of immunofluorescent staining of poly(ADP-ribose) in dorsal root gangia of control and diabetic wild-type and nNOS−/− mice. Magnification x40. (B) Poly(ADP-ribose) fluorescence counts in dorsal root ganglia of control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=7–11 per group. C, control mice; D, diabetic mice. **p<0.01 vs non-diabetic control mice. ##p<0.01 vs diabetic wild-type mice.
Figure 8.
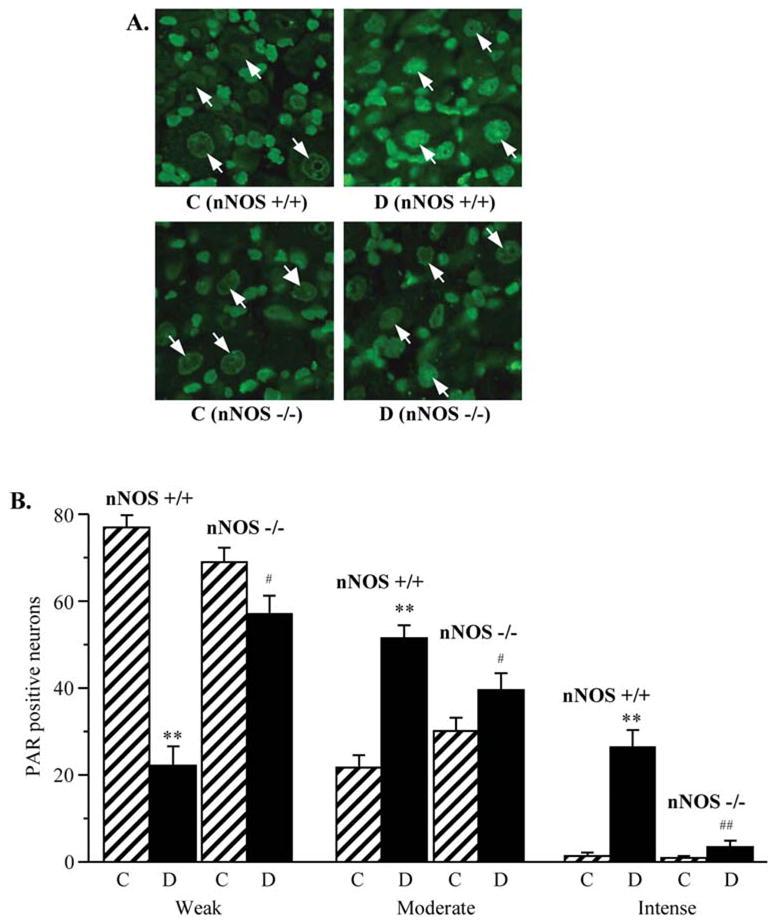
(A) Representative microphotographs of immunofluorescent staining of poly(ADP-ribose) in dorsal root gangion neurons of control and diabetic wild-type and nNOS−/− mice. Magnification x100. (B) Percentage of dorsal root ganglion neurons with weak, moderate, and intense poly(ADP-ribose) immunofluorescence in experimental groups. The number of dorsal root ganglion neurons with weak, moderate and intense poly(ADP-ribose) immunofluorescence was expressed as a percentage of neurons with identifiable poly(ADP-ribose) immunofluorescence in the dorsal root ganglia of control and diabetic wild-type and nNOS−/− mice. Mean ± SEM, n=8–11 per group. C, control mice; D, diabetic mice. **p<0.01 vs non-diabetic control mice; #,##p<0.01 vs diabetic wild-type mice.
Discussion
nNOS is expressed in many cell types in peripheral nervous system including Schwann cells of peripheral nerve (36), DRG neurons (37,38), and motoneurons, oligodendrocytes, and astrocytes of spinal cord (39,40). One can not exclude that it is also expressed in vasa nervorum considering recent report of the presence of nNOS in vascular endothelium (41). Note, that nNOS is constitutively expressed in some types of neurons, and can be induced in others (42). Sciatic nerve (43) or pelvic nerve (44) transection induced nitric oxide synthase expression in small to medium sized DRG neurons suggesting that this change may be limited to, or most prominent in, C-fiber afferents.
The findings of the present study suggest that nNOS is required for maintaining the normal peripheral nerve function and small sensory nerve fibre innervation. The latter is not surprising because nNOS is the main producer of nitric oxide which serves as a neurotransmitter in PNS (45). nNOS gene deficiency in non-diabetic mice affected both large and small sensory nerve fibers and was associated with reduction in sensory nerve conduction velocity, tactile response thresholds, and loss of intraepidermal innervation. Other report suggests that nNOS plays a critical role in Schwann cells proliferation during peripheral nerve regeneration (46). On the other hand, nNOS overexpression was implicated in neuropathic pain resulting from nerve injury or peripheral inflammation (47,48).
nNOS expression was reduced in sciatic nerve of C57BLKS/J-m+/+Leprdb homozygous (db/db) mice compared with non-diabetic controls, and in high glucose-exposed cultured mouse Schwann cells compared with the cells cultured at normal glucose concentration (36). The 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor rosuvastatin reversed reduction of nNOS expression in sciatic nerves of diabetic mice, and prevented high glucose-induced nNOS down-regulation in mouse Schwann cells, by mechanisms that involved phosphoinositide 3-kinase and phosphorylation of Akt (36). In the same study, co-treatment of diabetic mice with rosuvastatin and the selective nNOS inhibitor 1,2-trifluoromethylphenyl imidazole, blunted beneficial effects of rosuvastatin on MNCV and SNCV deficits, thermal sensitivity, and nerve vascularity which suggests an important role for nNOS-derived NO in peripheral nerve function. Of interest, NOS enzymatic activity in DRG was increased rather than reduced in rats with STZ-diabetes of 12-month duration (37). This activation, however, was not due to an increase in nNOS mRNA expression or immunoreactivity.
In the present study, diabetic wild-type mice displayed clearly manifest nitrosative stress in both peripheral nerve and DRG. nNOS gene deficiency prevented diabetes-induced peroxynitrite injury manifest by nitrotyrosine accumulation, in DRG neurons, but not in peripheral nerve which is indicative of different roles of nNOS-derived NO in peroxynitrite formation in these two tissue-sites of PDN. Despite the lack of neuronal nitrosative stress, diabetic nNOS−/− mice displayed reduction in MNCV and SNCV, sensory disorders, and intraepidermal nerve fiber loss, although nerve conduction deficits and thermal hypoalgesia were less severe in diabetic nNOS−/− mice, than in diabetic wild-type mice. Note, that in our previous study (11), diabetic iNOS−/− mice which were protected from nitrosative stress in peripheral nerve, but not DRG neurons, preserved normal nerve conduction velocities and thermal sensitivity. These findings suggest that nitrosative stress in peripheral nerve, rather than DRG neurons, contributes to the development of early PDN. The roles iNOS and nNOS in advanced PDN still remain to be established.
The importance of oxidative-nitrosative stress in peripheral nerve, but not DRG, in early PDN is further supported by the pattern of changes in PARP activity. Evidence for the important role for PARP activation in diabetic complications including endothelial dysfunction (49), cardiomyopathy (50), peripheral (14–17) and autonomic (51) neuropathy, nephropathy (52), and retinopathy (53,54) is emerging. Accumulation of poly(ADP-ribosyl)ated proteins, a sign of PARP activation, has been documented in peripheral nerve and DRG neurons of animal models of Type 1 and Type 2 diabetes and high glucose-exposed cultured human Schwann cells (6–11,14–17,22,35,55) PARP activation has been implicated in neurovascular dysfunction, motor and sensory nerve conduction velocity deficits, peripheral nerve energy failure, thermal hyper- and hypoalgesia, mechanical hypoalgesia, tactile allodynia, and intraepidermal nerve fiber loss characteristic for PDN (14–17,56). In the present study, PARP activation was clearly manifest in both sciatic nerve and DRG of diabetic wild-type mice consistent with our previous studies (6,8,9,11). Diabetic nNOS mice displayed near normal poly(ADP-ribosyl)ated fluorescence in DRG neurons, but, nevertheless, developed PDN. In contrast, iNOS gene deficiency did not protect against diabetes-associated accumulation of poly(ADP-ribosyl)ated proteins in DRG neurons (11). However, diabetic iNOS−/− mice displayed near normal poly(ADP-ribosyl)ation in peripheral nerve, and had normal nerve conduction and milder sensory neuropathy than wild-type mice with diabetes of similar severity and duration (11). These findings suggest that PARP activation in peripheral nerve rather than DRG neurons plays a pathogenetic role in early diabetic neuropathy. This conclusion is consistent with our findings in the diabetic rat model (10,16). Rats with 4–6-weeks STZ-diabetes displayed PARP activation manifest by increased poly(ADP-ribosyl)ated protein immunoreactivity (14,15), immunofluorescence (16) or expression by Western blot analysis (57) in the peripheral nerve, but not DRG [(10), Obrosova et al, unpublished data). Nevertheless, PARP inhibitor treatment of rats with short-term (4–6 weeks) STZ-diabetes counteracted multiple functional manifestations of PDN (14–16) which, again, suggests that PARP activation in peripheral nerve [i.e., endothelial and Schwann cells (14)], but not DRG neurons, contributes to the development of early PDN.
In conclusion, nNOS is required for maintaining the normal peripheral nerve function and small sensory nerve fibre innervation and appears to be the main source of nitric oxide for diabetes-associated peroxynitrite formation in DRG neurons. In contrast, this isoform does not seem to contribute to peroxynitrite formation in diabetic peripheral nerve. nNOS gene deficiency does not protect from development of nerve conduction deficit, sensory neuropathy, and intraepidermal nerve fiber loss. These findings, together with our previous studies, suggest that oxidative-nitrosative stress in axons and Schwann cells, rather than DRG neurons, plays a major role in, at least, early PDN.
Acknowledgments
The study was supported by the American Diabetes Association Research Grant 7-05-RA-102, the Juvenile Diabetes Research Foundation International Grant 1-2005-223, the National Institutes of Health Grant DK 071566-01 (all to I.G.O.), and the Intramural Research Program of the National Institutes of Health/National Institute of Alcohol Abuse and Alcoholism (to P.P.).
References
- 1.Virág L, Szabó E, Gergely P, Szabó C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett. 2003;140–141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- 2.Pacher P, Obrosova IG, Mabley JG, Szabó C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12:267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 5.Szabó C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, Soriano FG, van Duzer JH, Williams W, Salzman AL, Groves JT. Part I. Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med. 2002;8:571–580. [PMC free article] [PubMed] [Google Scholar]
- 6.Obrosova IG, Mabley JG, Zsengellér Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 7.Vareniuk I, Pavlov IA, Drel VR, Lyzogubov VV, Ilnytska O, Bell SR, Tibrewala J, Groves JT, Obrosova IG. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Exp Neurol. 2007;205:425–436. doi: 10.1016/j.expneurol.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 8.Drel VR, Pacher P, Vareniuk I, Pavlov I, Ilnytska O, Lyzogubov VV, Tibrewala J, Groves JT, Obrosova IG. A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin-diabetic mice. Eur J Pharmacol. 2007;569:48–58. doi: 10.1016/j.ejphar.2007.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drel VR, Pacher P, Vareniuk I, Pavlov IA, Ilnytska O, Lyzogubov VV, Bell SR, Groves JT, Obrosova IG. Evaluation of the peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate on peripheral neuropathy in a mouse model of type 1 diabetes. Int J Mol Med. 2007;20:783–792. [PMC free article] [PubMed] [Google Scholar]
- 10.Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am J Physiol Endocrinol Metab. 2007;293:E1645–E1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- 11.Vareniuk I, Pavlov IA, Obrosova IG. Inducible nitric oxide synthase gene deficiency counteracts multiple manifestations of peripheral neuropathy in a streptozotocin-induced mouse model of diabetes. Diabetologia. 2008;51:2126–2133. doi: 10.1007/s00125-008-1136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nangle MR, Cotter MA, Cameron NE. Effects of the peroxynitrite decomposition catalyst, FeTMPyP, on function of corpus cavernosum from diabetic mice. Eur J Pharmacol. 2004;502:143–148. doi: 10.1016/j.ejphar.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 13.Zheng L, Du Y, Miller C, Gubitosi-Klug RA, Ball S, Berkowitz BA, Kern TS. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia. 2007;50:1987–1996. doi: 10.1007/s00125-007-0734-9. [DOI] [PubMed] [Google Scholar]
- 14.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 15.Li F, Szabó C, Pacher P, Southan GJ, Abatan OI, Charniauskaya T, Stevens MJ, Obrosova IG. Evaluation of orally active poly(ADP-ribose) polymerase inhibitor in streptozotocin-diabetic rat model of early peripheral neuropathy. Diabetologia. 2004;47:710–717. doi: 10.1007/s00125-004-1356-0. [DOI] [PubMed] [Google Scholar]
- 16.Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N, Pacher P, Yorek MA, Obrosova IG. Poly(ADP-Ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55:1686–1694. doi: 10.2337/db06-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obrosova IG, Xu W, Lyzogubov VV, Ilnytska O, Mashtalir N, Vareniuk I, Pavlov IA, Zhang J, Slusher B, Drel VR. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic Biol Med. 2008;44:972–981. doi: 10.1016/j.freeradbiomed.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cotter MA, Cameron NE. Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life Sci. 2003;73:1813–1824. doi: 10.1016/s0024-3205(03)00508-3. [DOI] [PubMed] [Google Scholar]
- 19.Inkster ME, Cotter MA, Cameron NE. Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur J Pharmacol. 2007;561:63–71. doi: 10.1016/j.ejphar.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 20.Davidson EP, Kleinschmidt TL, Oltman CL, Lund DD, Yorek MA. Treatment of streptozotocin-induced diabetic rats with AVE7688, a vasopeptidase inhibitor: effect on vascular and neural disease. Diabetes. 2007;56:355–362. doi: 10.2337/db06-1180. [DOI] [PubMed] [Google Scholar]
- 21.Oltman CL, Davidson EP, Coppey LJ, Kleinschmidt TL, Lund DD, Adebara ET, Yorek MA. Vascular and neural dysfunction in Zucker diabetic fatty rats: a difficult condition to reverse. Diabetes Obes Metab. 2008;10:64–74. doi: 10.1111/j.1463-1326.2007.00814.x. [DOI] [PubMed] [Google Scholar]
- 22.Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Low PA, Nickander KK. Oxygen free radical effects in sciatic nerve in experimental diabetes. Diabetes. 1991;40:873–877. doi: 10.2337/diab.40.7.873. [DOI] [PubMed] [Google Scholar]
- 24.Stevens MJ, Obrosova I, Cao X, van Huysen C, Greene DA. Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes. 2000;49:1006–1015. doi: 10.2337/diabetes.49.6.1006. [DOI] [PubMed] [Google Scholar]
- 25.Cameron NE, Eaton SE, Cotter MA, Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001;44:1973–1988. doi: 10.1007/s001250100001. [DOI] [PubMed] [Google Scholar]
- 26.Cotter MA, Ekberg K, Wahren J, Cameron NE. Effects of proinsulin C-peptide in experimental diabetic neuropathy: vascular actions and modulation by nitric oxide synthase inhibition. Diabetes. 2003;52:1812–1817. doi: 10.2337/diabetes.52.7.1812. [DOI] [PubMed] [Google Scholar]
- 27.Yagihashi S, Yamagishi SI, Wada Ri R, Baba M, Hohman TC, Yabe-Nishimura C, Kokai Y. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain. 2001;124:2448–2458. doi: 10.1093/brain/124.12.2448. [DOI] [PubMed] [Google Scholar]
- 28.Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, Yagihashi S, Oates PJ, Ellery CA, Chung SS, Chung SK. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes. 2006;55:1946–1953. doi: 10.2337/db05-1497. [DOI] [PubMed] [Google Scholar]
- 29.Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience. 2007;145:303–313. doi: 10.1016/j.neuroscience.2006.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller KA, Ryals JM, Feldman EL, Wright DE. Abnormal muscle spindle innervation and large-fiber neuropathy in diabetic mice. Diabetes. 2008;57:1693–1701. doi: 10.2337/db08-0022. [DOI] [PubMed] [Google Scholar]
- 31.Beiswenger KK, Calcutt NA, Mizisin AP. Dissociation of thermal hypoalgesia and epidermal denervation in streptozotocin-diabetic mice. Neurosci Lett. 2008;442:267–272. doi: 10.1016/j.neulet.2008.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, Brussee V, Liu W, Durand J, Nguyen MD, Schmidt AM, Zochodne DW. Receptor for advanced glycation and products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 33.Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intra-epidermal nerve fiber loss in experimental diabetes. Diabetes. 2007;56:2997–3005. doi: 10.2337/db07-0740. [DOI] [PubMed] [Google Scholar]
- 34.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 35.Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N, Nadler JL, Drel VR. High-fat diet induced neuropathy of pre-diabetes and obesity: effects of ‘healthy’ diet and aldose reductase inhibition. Diabetes. 2007;56:2598–2608. doi: 10.2337/db06-1176. [DOI] [PubMed] [Google Scholar]
- 36.Ii M, Nishimura H, Kusano KF, Qin G, Yoon YS, Wecker A, Asahara T, Losordo DW. Neuronal nitric oxide synthase mediates statin-induced restoration of vasa nervorum and reversal of diabetic neuropathy. Circulation. 2005;112:93–102. doi: 10.1161/CIRCULATIONAHA.104.511964. [DOI] [PubMed] [Google Scholar]
- 37.Zochodne DW, Verge VM, Cheng C, Höke A, Jolley C, Thomsen K, Rubin I, Lauritzen M. Nitric oxide synthase activity and expression in experimental diabetic neuropathy. J Neuropathol Exp Neurol. 2000;59:798–807. doi: 10.1093/jnen/59.9.798. [DOI] [PubMed] [Google Scholar]
- 38.Burnand RC, Price SA, McElhaney M, Barker D, Tomlinson DR. Expression of axotomy-inducible and apoptosis-related genes in sensory nerves of rats with experimental diabetes. Brain Res Mol Brain Res. 2004;132:235–240. doi: 10.1016/j.molbrainres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Wu W, Liuzzi FJ, Schinco FP, Depto AS, Li Y, Mong JA, Dawson TM, Snyder SH. Neuronal nitric oxide synthase is induced in spinal neurons by traumatic injury. Neuroscience. 1994;61:719–726. doi: 10.1016/0306-4522(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 40.Cheng C, Li X, Gao S, Niu S, Chen M, Qin J, Guo Z, Zhao J, Shen A. Expression of CAPON after spinal cord injury in rats. J Mol Neurosci. 2008;34:109–119. doi: 10.1007/s12031-007-9019-5. [DOI] [PubMed] [Google Scholar]
- 41.Capettini L, Cortes SF, Gomes MA, Silva G, Pesquero JL, Lopes MJ, Teixeira MM, Lemos VS. Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am J Physiol Heart Circ Physiol. 2008;295:H2503–H2511. doi: 10.1152/ajpheart.00731.2008. [DOI] [PubMed] [Google Scholar]
- 42.Vizzard MA, Erdman SL, De Groat WC. Increased expression of neuronal nitric oxide synthase (NOS) in visceral neurons after nerve injury. J Neurosci. 1995;15:4033–4045. doi: 10.1523/JNEUROSCI.15-05-04033.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vizzard MA, Erdman SL, De Groat WC. Increased expression of neuronal nitric oxide synthase in dorsal root ganglion neurons after systemic capsaicin administration. Neuroscience. 1995;67:1–5. doi: 10.1016/0306-4522(95)00137-8. [DOI] [PubMed] [Google Scholar]
- 44.Vizzard MA, Erdman SL, De Groat WC. The effect of rhizotomy on NADPH diaphorase staining in the lumbar spinal cord of the rat. Brain Res. 1993;607:349–353. doi: 10.1016/0006-8993(93)91530-6. [DOI] [PubMed] [Google Scholar]
- 45.Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen A, Gao S, Ben Z, Wang H, Jia J, Tao T, Niu S, Li X, Cheng C. Identification and potential role of PSD-95 in Schwann cells. Neurol Sci. 2008;29:321–330. doi: 10.1007/s10072-008-0989-z. [DOI] [PubMed] [Google Scholar]
- 47.Jin XG, He SQ, Yan XT, Zhang G, Wan L, Wang J, Li Y, Tian X, Tian Y, Luo A. Variants of neural nitric oxide synthase in the spinal cord of neuropathic rats and their effects on nuclear factor-kappaB (NF-kappaB) activity in PC12 Cells. J Pain. 2009;10:80–89. doi: 10.1016/j.jpain.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 48.Lam HH, Hanley DF, Trapp BD, Saito S, Raja S, Dawson TM, Yamaguchi H. Induction of spinal cord neuronal nitric oxide synthase (NOS) after formalin injection in the rat hind paw. Neurosci Lett. 1996;210:201–214. doi: 10.1016/0304-3940(96)12702-6. [DOI] [PubMed] [Google Scholar]
- 49.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 50.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó E, Szabó C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 51.Gibson TM, Cotter MA, Cameron NE. Effects of poly(ADP-ribose) polymerase inhibition on dysfunction of non-adrenergic non-cholinergic neurotransmission in gastric fundus in diabetic rats. Nitric Oxide. 2006;15:344–350. doi: 10.1016/j.niox.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Szabó C, Biser A, Benko R, Böttinger E, Suszták K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006;55:3004–3012. doi: 10.2337/db06-0147. [DOI] [PubMed] [Google Scholar]
- 53.Obrosova IG, Minchenko AG, Frank RN, Seigel GM, Zsengeller Z, Pacher P, Stevens MJ, Szabó C. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int J Mol Med. 2004;14:55–64. [PubMed] [Google Scholar]
- 54.Zheng L, Szabó C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 55.Stevens MJ, Li F, Drel VR, Abatan OI, Kim H, Burnett D, Larkin D, Obrosova IG. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J Pharmacol Exp Ther. 2007;320:458–464. doi: 10.1124/jpet.106.109702. [DOI] [PubMed] [Google Scholar]
- 56.Obrosova IG, Julius UA. Role for poly(ADP-ribose) polymerase activation in diabetic nephropathy, neuropathy and retinopathy. Curr Vasc Pharmacol. 2005;3:267–283. doi: 10.2174/1570161054368634. [DOI] [PubMed] [Google Scholar]
- 57.Li F, Drel VR, Szabó C, Stevens MJ, Obrosova IG. Low-dose poly(ADP-ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54:1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]