

1. ABSTRACT
Cigarette smoking is the major cause of preventable morbidity and mortality in the United States and constitutes a major risk factor for atherosclerotic vascular disease, including coronary artery disease and stroke. Increasing evidence supports the hypothesis that oxidative stress and inflammation provide the pathophysiological link between cigarette smoking and CAD. Previous studies have shown that cigarette smoke activates leukocytes to release reactive oxygen and nitrogen species (ROS/RNS) and secrete pro-inflammatory cytokines, increases the adherence of monocytes to the endothelium and elicits airway inflammation. Here we present an overview of the direct effects of water-soluble cigarette smoke constituents on endothelial function, vascular ROS production and inflammatory gene expression. The potential pathogenetic role of peroxynitrite formation, and downstream mechanisms including poly (ADP-ribose) polymerase (PARP) activation in cardiovascular complications in smokers are also discussed.
Keywords: Endothelium, Cigarette Smoking, Smokers, Tobacco, Senescence, Inflammation, Gene Expression, Redox Status, Peroxynitrite, PARP-1, poly polymerase, ADP-ribose, PARP
2. INTRODUCTION
In the United States, an estimated 45.8 million adults (~22.5%) are current smokers and more than 400 000 people die from tobacco smoke–related illnesses each year. There is overwhelming evidence that tobacco smoking is an important factor contributing to premature vascular aging. The cardiovascular morbidity and mortality induced by cigarette smoking exceeds that attributable to lung cancer: 190 000 cardiovascular disease deaths per year are related to cigarette smoke, out of which 37 000 to 40 000 deaths are attributable to second hand smoke exposure (1). Although the precise molecular basis of smoking-induced vascular injury remains unclear, increasing evidence supports the hypothesis that oxidative-nitrosative stress and inflammation provide the pathophysiological link between cigarette smoking and coronary artery disease (CAD) (2-24).
Cigarette smoke contains reactive oxidants, which can get into the bloodstream and cause macromolecular damage in the endothelial cells. Cigarette smoking also elicits a marked activation of leukocytes and platelets, which can also contribute to the oxidative vascular damage in smokers. Circulating cigarette smoke constituents were also shown to induce and activate ROS producing enzyme systems within the vascular wall. The mechanisms by which endothelial oxidative stress leads to vascular inflammation and development of atherosclerosis have been the subject of intense studies in this field. This review focuses on the emerging evidence that reactive oxygen species (ROS) and activation of inflammatory pathways play a central role in accelerated cardiovascular aging in smokers, and discusses some of the signaling pathways involved in cigarette smoking-induced vascular inflammation.
3. CIGARETTE SMOKE-INDUCED VASCULAR OXIDATIVE-NITROSATIVE STRESS
The causes of cigarette smoking-induced vascular oxidative stress are likely multifaceted. Cigarette smoke can be divided into 2 phases: particulate matter and gas-phase smoke, which contain high concentrations of ROS, NO, peroxynitrite, and/or free radicals of organic compounds (25-39). In addition to these short-lived, highly reactive substances, inhaled particles encountered in cigarette smoke, especially in the presence of ROS (4) may evoke an inflammatory response in the lung activating immunocytes to produce ROS and promoting the production of proinflammatory cytokines. It has also been suggested that aqueous cigarette tar extracts contain pro-oxidant substances that have the potential to increase cellular production of ROS (11, 25, 26, 40-43). These include a semiquinones, hydroquinones, and quinones (25, 26), a series of α,ß-unsaturated aldehydes, such as acrolein and crotonaldehyde, α,ß-unsaturated ketones, and a number of saturated aldehydes (40-42). These water soluble components of cigarette smoke are likely to reach the systemic circulation and can directly promote vascular oxidative stress in systemic vascular beds. This hypothesis was supported by clinical and animal studies showing that cigarette smoke produces generalized endothelial dysfunction in virtually every vascular bed (2, 21, 44-47), which is usually an indicator of an increased oxidative stress.
Several lines of evidence suggest that cigarette smoke constituents can directly activate vascular ROS production. Clinical and animal studies have demonstrated that cigarette smoke (CS) produces generalized endothelial dysfunction by reducing NO bioavailability (2, 21, 44-48). It is generally accepted that tonic release of NO from the endothelium exerts vasculoprotective and cardioprotective effects, such as maintenance of normal coronary blood flow, inhibition of platelet aggregation and inflammatory cell adhesion to endothelial cells and disruption of pro-inflammatory cytokine-induced signaling pathways. Importantly, coronary vasodilatory capacity was shown to be impaired in smokers lowering the ischemic threshold and even short-term smoking increases the coronary vasomotor tone and markedly reduces the myocardial flow reserve (46, 47). Substantial evidence is accumulating suggesting that decreased NO bioavailability and increased generation of reactive oxygen species (ROS), including that of superoxide (O2.-; Figure 1), plays a critical role in cigarette smoke-related endothelial dysfunction (2, 7, 49, 50). Also, CSE is known to induce endothelial apoptosis by activating caspase-3 (51). In endothelial cells eNOS pre-activation by L-arginine substantially reduces CS-induced endothelial apoptosis; whereas eNOS inhibition accentuated CS-induced endothelial apoptosis (51). Thus, ROS-mediated reduction of endogenous NO production seems to be an important mechanism in smoking-induced endothelial damage (51). Because SOD enzymes catalyze the removal of O2.- with a rate constant of 2×109 mol-1 · L · s-1, it is likely that a significant portion of O2.- is dismutated, increasing also H2O2 levels. Indeed, in CSE-treated vessels there was a substantially increased H2O2 production (48).
Figure 1.
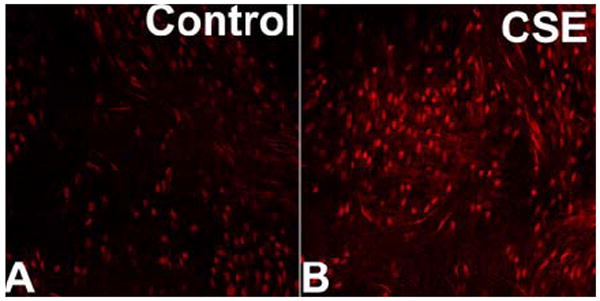
Representative confocal images of nuclear ethidium bromide (EB) staining of en face preparations of rat aortas incubated with cigarette smoke extract (CSE; 4 μg/mL, for 6 h; Panel B) and untreated controls (Panel A). Vessels were incubated with the dye dihydroethidium, which produces a red nuclear fluorescence when oxidized to EB by O2.-. On these images the EB-stained elongated nuclei of vascular smooth muscle cells and the round nuclei of endothelial cells are visualized. Identical results were observed in 4 separate experiments (original magnification: 20X).
Previous studies identified NAD (P)H oxidase (s) and xanthine oxidase as two potential intracellular sources of O2.- in various cell types that may be induced by components of cigarette smoke (2, 6, 49). We have recently shown that both in vivo chronic cigarette smoke exposure and in vitro treatment with aqueous cigarette smoke extract (CSE) elicit significant endothelial dysfunction in rat carotid arteries, which could be reversed by inhibition of the NAD (P)H oxidase (48). This finding accords with the increased NAD (P)H oxidase-dependent O2.- generation in these vessels upon CSE exposure (48). It is likely that water soluble components of cigarette smoke are directly responsible for the activation of the vascular NAD (P)H oxidase, because exposure of isolated arteries to CSE in vitro, in the absence of activated leukocytes, elicited significant O2.- production in a concentration-dependent manner (48). Further evidence for the central role for NAD (P)H oxidase activation in cigarette smoke-induced vascular oxidative stress came from the observations that CSE increased the expression of gp91phox subunit of NAD (P)H oxidase in rat arteries (48). Also, in human pulmonary artery endothelial cells gp91 docking sequence-tat peptide (similar to apocynin) was reported to inhibit CSE-induced O2.- generation (49). In this context it is important to note that NAD (P)H oxidase activation has been linked to the development of CAD in various pathophysiological conditions, including aging. We have previously shown that both endothelial cells and vascular smooth muscle cells exhibit an up-regulated O2.- generation in vessels of cigarette smoke-exposed animals (48). Similarly, CSE challenge elicited oxidative stress in both cell types (48), mimicking the effects of in vivo exposure to cigarette smoke. Recent studies support the idea that CSE in vitro may induces NAD (P)H oxidase (s) in other cell types as well (2, 6, 49). Cigarette smoke contains more than 4000 known components and at present it is unclear, which components activate NAD (P)H oxidase. Numerous experimental and epidemiological studies have demonstrated that polycyclic aromatic hydrocarbons (PAHs), which are major constituents of cigarette tobacco tar, are able to induce various cellular enzyme systems involved in ROS metabolism. Although nicotine may impair endothelium-mediated vasodilation in microvessels (52), in our recent studies it could not mimic the effect of serum from cigarette smoke-exposed rats or CSE on endothelial ROS production (48). Also, although nicotine at high concentrations may cause pro-inflammatory gene expression in cultured coronary endothelial cells (such as up-regulation of angiotensin-I converting enzyme, tissue-type plasminogen activator, plasminogen activator inhibitor-1 (53)), it does not mimic the effect of smoking on the endothelial expression of ICAM-1, matrix metalloproteinases and many other inflammatory mediators (53). A recent study suggested that acrolein, a thiol-reactive α,β-unsaturated aldehyde that is abundantly present in cigarette smoke, is a potent inducer of NAD (P)H oxidase-derived O2.- generation in pulmonary arterial endothelial cells (49). Because of their stability and water solubility, acrolein and other related compounds are likely to reach vascular beds remote from the primary site of exposure and, possibly, induce the production of ROS. It is now well established that NAD (P)H oxidase (s) are a major source of ROS in vascular cells and an increased NAD (P)H oxidase activity is responsible for enhanced endothelial O2.- production in aging and in pathophysiological conditions associated with accelerated vascular aging (54) such as, hyperhomocysteinemia (55) and hypertension (56, 57). Activity of arterial NAD (P)H oxidase is regulated by PKC-dependent signaling pathways (56, 57) and one could hypothesize that PKC activation by CS components (12, 58, 59) may contribute to the increased NAD (P)H oxidase activity observed in CS-exposed animals. Because NAD (P)H oxidase seems to represent a common pathway eliciting endothelial oxidative stress, further studies are needed to test the hypothesis that smoking will aggravate vascular injury in pathophysiological conditions associated with a high basal NAD (P)H oxidase activity (e.g. hypertension).
Mitochondria are also important sources of ROS in the cardiovascular system. There is growing evidence that cigarette smoke constituents impair mitochondrial function and elicit mitochondrial oxidative stress (Figure 2) in various cell types (60-65), including cardiovascular tissues (66). A recent study demonstrated that acrolein, a major toxicant in cigarette smoke, causes oxidative mitochondrial damage (61). In vitro treatment with CSE caused loss of cellular ATP and rapid depolarization of mitochondrial membrane potential, followed by apoptotic cell death (62). In smokers a higher level of oxidative mtDNA damage has been observed (66-68). These data support the hypothesis that cigarette smoke-induced mitochondrial damage and dysfunction may contribute an increased risk for cardiovascular disease in smokers. It should be noted that in addition of the NAD (P)H oxidase and mitochondria, other cellular sources (such as cytochrome P450, which may be induced by cigarette smoke constituents (69)) can also produce significant amounts of ROS, however, the role of these enzymes in CSE-induced vascular oxidative stress is not well understood. In parenchymal tissues a striking cigarette smoke-induced depletion of GSH content was shown (70-75). In addition, acrolein also readily inactivates thioredoxin and thioredoxin reductase (76), which effects have the potential to modulate redox signaling mechanisms.
Figure 2.
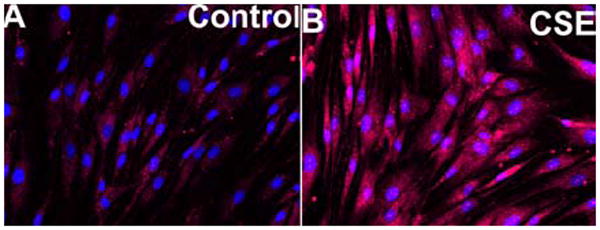
Incubation with cigarette smoke extract (CSE) elicits mitochondrial oxidative stress. Representative fluorescent images show intensive MitoSox staining (red fluorescence) in CSE-treated cells (CSE; 4 μg/mL, for 6 h; Panel B), whereas MitoSox fluorescence is significantly weaker in mitochondria of untreated control cells (Panel A). Cells were incubated with the mitochondrion-targeted O2.- sensitive dye MitoSox (Invitrogen), which produces a red fluorescence in the mitochondria when oxidized by O2.-. The DNA-binding dye Hoechst 33258 was used for nuclear counterstaining (original magnification: 20X).
In addition to the increased levels of O2.- and H2O2 (which have been implicated in pro-atherogenic vascular phenotypic alterations (77-79), including induction of pro-inflammatory gene expression (80-86)), a large body of experimental evidence accumulated over the past 15 years indicates that peroxynitrite (ONOO-) generation from NO and O2.- represents a major threat to the functional integrity of the vascular endothelium (55, 87-89). Cigarette smoke contains peroxynitrite that can penetrate into the blood stream and also contains agents that lead to increased ONOO- production within the cells (90-94). Importantly, CSE-induced increased ONOO- generation has been recently demonstrated in cultured endothelial cells (49). The damaging potential of peroxynitrite is explained by its peculiar chemistry involving direct oxidation as well as radical-mediated nitration reactions. These properties allow peroxynitrite to significantly alter the function of a considerable number of proteins (e.g. GSH peroxidase, myeloperoxidase), to degrade membrane structure by peroxidizing lipids, to turn off crucial metabolic functions within mitochondria, and to inflict serious damage to nucleic acids, activating a major pathway of cell injury and inflammation orchestrated by the nuclear enzyme PARP (87). Once severe enough to overwhelm repair mechanism, these various cytotoxic effects commit cells to death, either through the necrotic or apoptotic pathway. Peroxynitrite can also activate/modulate cell signaling pathways. The ability of peroxynitrite to nitrate tyrosine residues can interfere with signaling processes depending on tyrosine phosphorylation (87). Peroxynitrite was also shown to modulate MAP kinase pathways as well (87). There are also studies extant directly implicating peroxynitrite in activation of NF-κB -dependent signaling pathways (95). As a result, previous studies have shown that ONOO- can mediate a wide range of pro-inflammatory effects (95-102). We and others have shown that in coronary arteries expression of TNFα, which orchestrates pro-atherogenic vascular phenotypic changes (103), is frequently up-regulated in conditions associated with increased O2.- and ONOO- production, such as hyperhomocysteinemia (55), aging (104) and hypertension. Thus, future studies should elucidate the possible link between cigarette smoking, peroxynitrite generation and up-regulation of pro-inflammatory cytokine expression.
4. CIGARETTE SMOKE-INDUCED ENDOTHELIAL DNA DAMAGE
Cigarette smoking was reported to increase oxidative DNA modification in humans (105) and laboratory animals. These studies suggested that a link exists between oxidative DNA modification, accelerated aging and cancerogenesis. Earlier studies focused on cigarette smoking-induced DNA damage in the lung, however, it soon became obvious that systemic exposure to circulating cigarette smoke constituents results in an increased presence of elevated levels of DNA adducts in tissues not directly exposed to tobacco smoke. The literature is mainly focused on 8oxodG lesions caused by circulating cigarette smoke constituents (e.g. measurements of increased urinary excretion of 8-hydroxy-2’-deoxyguanosine in smokers (105)). It has been shown that cigarette smoke extracts increases similar DNA damage in cultured lung fibroblasts and endothelial cells (106) (Figure 3). Because endothelial cells in vivo represent the first line of defense against circulating toxic agents, it was expected that significant oxidative DNA damage can occur in the vasculature in vivo. Indeed, apart from 8oxodG, previous studies demonstrated the presence of cigarette smoking-related polycyclic aromatic hydrocarbon (PAH)-DNA adducts in human internal mammary artery specimens from smokers (107). The levels of these adducts are about 100 times less frequent than 8oxodG. However, the half-life of PAH-DNA adducts (at least in the lung) is much longer, about 1-2 years (108). Taken together, these findings provide strong evidence that cigarette smoke constituents can induce oxidative DNA damage in the cardiovascular system. Whereas most studies focus on oxidative DNA modifications, it has become clear that DNA repair processes have equal importance for accelerated aging. Repair of oxidative DNA damage is extensive and differences in DNA repair are proposed to be important for cancerogenesis and premature aging as well. There are a large number of enzyme systems that recognize oxidative DNA modifications and start a multistep process of repair. Previous research has suggested that there a positive correlation between maximum longevity and the rate or fidelity of DNA repair. Our studies comparing relative rate of DNA repair in long and short lived species of primates (human vs. marmoset; Podlutsky and Austad unpublished data 2007), rodents (white-footed mouse vs. house mouse (109)) and bat species indicate that long-lived species have superior DNA repair compared to related short-lived species. In humans many of the enzyme systems involved in DNA repair exhibit single nucleotide polymorphisms (SNPs), which are associated with an increased risk for cancer development. The relationship between cigarette smoke-induced oxidative DNA damage in the lung and parenchymal tissues and carcinogenesis are widely appreciated and there is good reason to believe that DNA damage also contribute to cardiovascular pathophysiological alterations. An important hypothesis put forward by Ames also suggest a direct link between oxidative DNA modification and the aging process (110-112). Thus, future studies should elucidate whether inter-individual differences in DNA repair capacity may determine susceptibility to CAD and accelerated cardiovascular aging in smokers.
Figure 3.
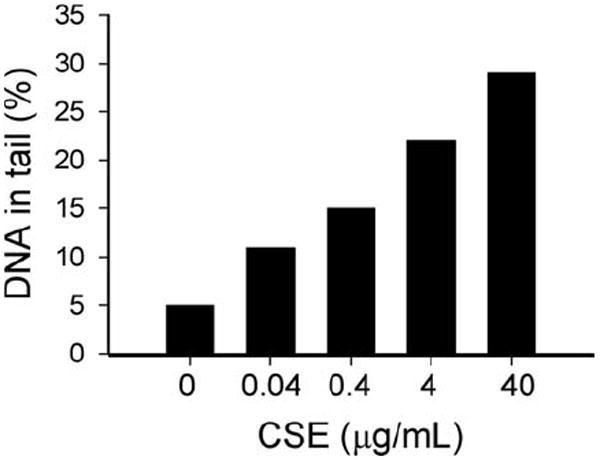
Water-soluble cigarette smoke constituents elicit endothelial DNA damage. Coronary arterial endothelial cells were treated with cigarette smoke extract (CSE for 6 h). Then, cells were harvested and the extent of DNA damage was examined by single-cell electrophoresis (“comet assay”) as we reported (109, 139). Damaged DNA migrates during electrophoresis from the nucleus towards the anode, forming a shape of a “comet” with a head (cell nucleus with intact DNA) and a tail (relaxed and broken DNA). Frequency distribution of tail DNA content in untreated control and CSE-exposed endothelial cells were obtained (median values for tail DNA content are shown in the graph).
5. CIGARETTE SMOKE-INDUCED VASCULAR INFLAMMATION
Several lines of evidence suggest that CS can induce inflammatory processes, in part, via induction of pro-inflammatory cytokines. CS exposure (especially the ultrafine particulate fraction) is known to activate circulating immunocytes in the lung, which then release pro-inflammatory cytokines. It appears that especially TNFα plays a crucial role in CS-induced pathophysiology (113). For example, mice lacking TNFα receptors (TNFR -/- mice) do not develop a pulmonary inflammatory infiltrate or matrix breakdown after CS exposure (114). Recent studies demonstrated that apart from being a target for circulating cytokines, vascular cells (endothelial and smooth muscle cells, fibroblasts) produce a wide range of cytokines (104, 115, 116), which orchestrate pro-atherogenic processes in an autocrine/paracrine manner. Recently we have shown that in vivo exposure of rats to cigarette smoke provokes an increase in the expression of pro-inflammatory cytokines (including IL-6, TNFα and IL-1β) and cytokine-sensitive inflammatory mediators (iNOS) in the vascular wall (48). Importantly, these pro-inflammatory phenotypic alterations could also be mimicked by in vitro CSE challenge (Figure 4) (48). Recent studies suggest that exposure of cultured human endothelial cells to CSE or serum from smokers also results in pro-inflammatory gene expression (12, 21, 24, 117, 118). Atherosclerosis is a chronic inflammatory disease and pathological and epidemiological evidence suggest that pro-inflammatory cytokines play a central role orchestrating the patholological processes underlying the development of the atherosclerotic plaque. The aforementioned findings clearly demonstrate that cigarette smoke components are able to elicit a pro-atherogenic microenvironment in the vascular wall in the absence of circulating factors and immunocytes. The findings that inhibition of NAD (P)H oxidase and administration of antioxidants can attenuate CSE-induced up-regulation of pro-inflammatory mediators, suggest a central role for NAD (P)H oxidase-derived ROS in CSE-induced vascular inflammation (48).
Figure 4.
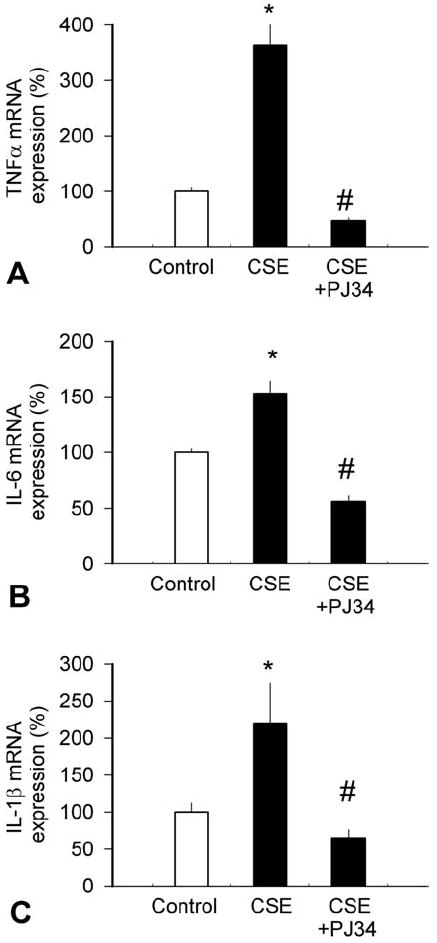
Water-soluble cigarette smoke constituents up-regulate vascular inflammatory cytokine expression. Isolated rat carotid arteries were maintained in organoid culture in the presence and absence of cigarette smoke extract (CSE) and mRNA expression of TNFα (A), IL-6 (B) and IL-1β (C) were assessed by real-time QRT-PCR, as reported (48). β-actin was used for internal normalizations. Some vessels were pre-incubated with the potent PARP inhibitor PJ34 (142-144, 146). Data are mean ± S.E.M. (n=4-6 for each group). *P<0.05 vs. untreated, # P<0.05 vs. CSE only.
NF-κB is thought to induce the transcription of a large range of genes implicated in inflammation, including cytokines, chemokines and adhesion molecules (119-121). All of these factors are known to predispose arteries to atherosclerosis (122). NF-κB has been shown to be activated by increased levels of ROS in many cell types (123-129), providing an important link between oxidative stress and pro-inflammatory cytokine expression in blood vessels. In line with the evidence that cigarette smoke induces significant endothelial oxidative stress (Figure 1), CSE was shown to induce NF-κB activation in human coronary arterial endothelial cells (Figure 5A) (48). In addition, there are studies extant showing that CSE elicits NF-κB activation in human histiocytic lymphoma cells and other cell lines (130). A recent study also showed that NF-κB activity in peripheral blood mononuclear cells of smokers compared to non-smokers is significantly higher (131). CS was also shown to elicit rapid increases in whole-lung NF-κB activation (132) and gene expression of proinflammatory cytokines after cigarette smoke exposure (113), an effect that could be attenuated by pretreatment with the free radical scavenger SOD (132). The findings that apocynin and catalase were able to reduce CSE-induced activation of NF-κB (24) in endothelial cells (48) (Figure 5A) provide strong evidence that NAD (P)H oxidase-derived H2O2 promotes vascular inflammation via NF-κB activation. A central role for H2O2 in endothelial NFκB activation is suggested by the findings that administration of exogenous H2O2 can mimic the effect of CSE on NF-κB activity in endothelial cells (48, 133). NF-κB response elements are present in the promoter region of many inflammatory genes (such as iNOS and adhesion molecules) as well as many cytokines (e.g. the TNFα gene (134, 135) and the IL-6 gene (136)), which seem to be up-regulated by cigarette smoke. Interestingly, in a monocyte-macrophage cell line cigarette smoke-induced proinflammatory cytokine release seems to be regulated also by SIRT1 (a member of the sirtuin family of class III histone/protein deacetylases) by its interaction with NF-κB (137). In these cells CSE exposure resulted in time-dependent decreases in SIRT1 activity and levels, which was concomitant to increased NF-κB -dependent cytokine production (137). Importantly, resveratrol, an activator of SIRT1, inhibited CSE-mediated proinflammatory cytokine release in cultured monocytic cells (137). It is possible that SIRT1 also plays an important role in controlling vascular inflammatory processes (138), because in CSE-treated cultured coronary arterial endothelial cells resveratrol also attenuates the expression of inflammatory cytokines (Ungvari and Csiszar, unpublished data, 2006). SIRT1 also seems to be important for protecting endothelial cells from oxidative stress-induced apoptosis (139).
Figure 5.
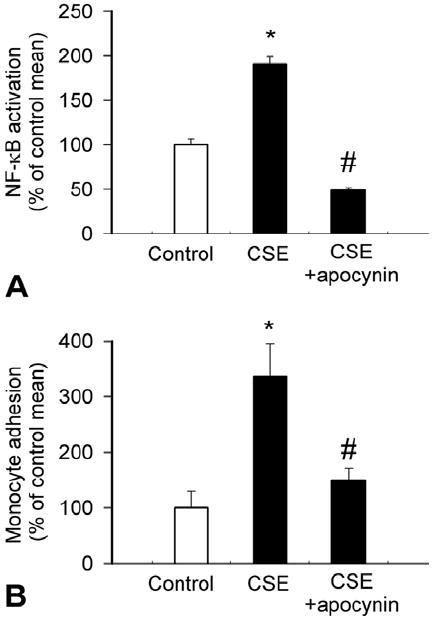
Water-soluble cigarette smoke constituents elicit NF-κB activation and enhance monocyte adhesiveness via increasing NAD (P)H oxidase-derived ROS generation in primary coronary arterial endothelial cells (CAECs). Panel A: Reporter gene assay showing the effect of cigarette smoke extract (CSE) on NF-κB reporter activity in CAECs. The effects of pre-treatment with NAD (P)H oxidase inhibitor apocynin (3×10-4 mol/L) on CSE-induced NF-κB reporter activity in CAECs is also shown. Endothelial cells were transiently co-transfected with NF-κB-driven firefly luciferase and CMV-driven ranilla luciferase constructs followed by CSE stimulation. Cells were then lysed and subjected to luciferase activity assay. After normalization relative luciferase activity was obtained from four to seven independent transfections. *p<0.05. (Data are mean ± S.E.M. *p<0.05. vs. untreated; #P<0.05 vs. CSE only). B: Treatment of carotid arteries and aortas with cigarette smoke extract (CSE) significantly increased adhesion of fluorescently labeled PMA-stimulated monocyte enriched peripheral blood mononuclear cells (PBMC). The effects of CSE were also assessed after pretreatment with the NAD (P)H oxidase inhibitor apocynin (3×10-4 mol/L) and DPI (10-5 mol/L) or catalase (200 U/mL). Data are mean ± S.E.M. *p<0.05. vs. control. C: Treatment of CAECs with CSE significantly increased the adhesion of fluorescently labeled PMA-stimulated monocytes. Pre-treatment with the NAD (P)H oxidase inhibitor apocynin significantly attenuated CSE-induced monocyte adhesion. Data are mean ± S.E.M. *p<0.05. vs. control. #p<0.05 vs. CSE alone. Figure is redrawn based on data from reference (48).
6. CIGARETTE SMOKE-INDUCED PARP ACTIVATION
In recent years, evidence has accumulated that another important mechanism mediating the deleterious effects of endothelial oxidative stress is the activation of poly (ADP-ribose) polymerase (PARP), an enzyme present in the nucleus of eukaryotic cells (140, 141). PARP activation has been demonstrated in vitro in various cells exposed to ROS (140, 141), as well as to ONOO-. In vivo, endothelial and cardiac PARP activation has been shown to act as a common oxidant-induced effector in various pathophysiological conditions associated with accelerated cardiovascular aging (142-150). Importantly, CSE induces PARP activity in coronary arterial endothelial cells (Figure 6). CSE was also reported to induce PARP activity in alveolar epithelial cells (151). PARP-1 is known to participate in the regulation of gene transcription in many cell types (152). In particular, PARP-1 has been shown to play a key role in NF-κB-driven expression of pro-inflammatory cytokines (153). Recently, PAR-binding domains have been detected in NF-κB subunits (154) and a co-activator role of PARP-1 for NF-κB -dependent pro-inflammatory gene expression has been proposed (145, 155, 156). Consistent with the stimulatory effect of CSE on NF-κB activation, the potent PARP inhibitor PJ34 (157) provided significant anti-inflammatory effects in CSE-treated coronary arterial endothelial cells (Figure 4). Drugs inhibiting PARP-1 activity also reduce NF-κB-dependent transcription of TNFα in glial cells (153) and prevent pro-inflammatory gene expression in cultured endothelial cells (158). In addition, both NF-κB activity and NF-κB -driven transcription of pro-inflammatory cytokines is markedly impaired in PARP-1 knockout animals (159, 160). Despite the relevance of PARP-1 to transcriptional regulation of pro-inflammatory cytokines in cultured endothelial cells, the importance of PARP-1 activity in pro-inflammatory phenotypic alterations in coronary arteries induced by cigarette smoking remains to be established. In addition to the aforementioned pathways cigarette smoke exposure has been shown to activate a number of other redox-sensitive signaling pathways as well. For example, CSE was shown to activate MAP kinase pathways and stress activated protein kinase/c-Jun N-terminal protein kinases in endothelial cells (51).
Figure 6.
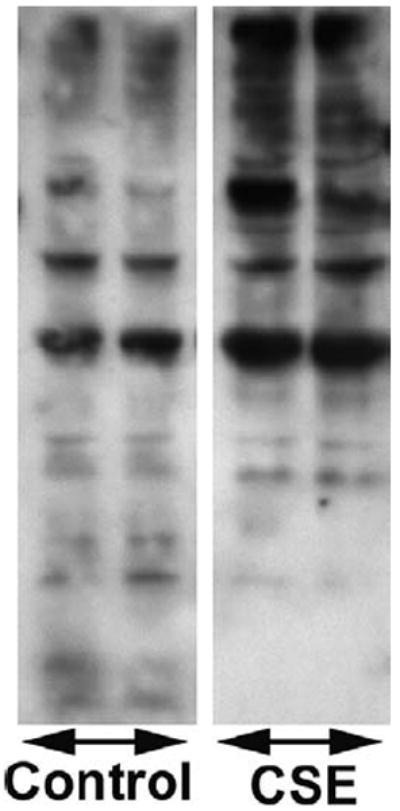
Water-soluble cigarette smoke constituents elicit PARP activation in human coronary arterial endothelial cells. Endothelial cells were treated with cigarette smoke extract (CSE). Then, cells were harvested and the poly (ADP-ribose) polymer content was assessed by Western blotting.
7. CIGARETTE SMOKE-INDUCED ENDOTHELIAL ACTIVATION
Previous studies suggested that even moderate cigarette smoking leads to an activation of the circulating monocytes and their increased adhesion to the endothelium (161). Both in vivo exposure of rats to cigarette smoke and in vitro incubation of vessels with CSE enhance adhesion of activated monocytes to the endothelial surface (162-164) (Figure 5B). The role of water soluble components of cigarette smoke is supported by the findings that serum collected from smokers increases endothelial expression of adhesion molecules, including ICAM-1 (21, 24, 48, 164). Our recent findings support a role for NAD (P)H oxidase-derived H2O2 p in endothelial activation by cigarette smoke constituents (48). Also, increasing plasma vitamin C concentrations in smokers by oral supplementation decreased monocyte adhesion to values found in nonsmokers (165).
8. ACCELERATED VASCULAR AGING IN CIGARETTE SMOKERS
Epidemiological studies suggest that advanced age itself promotes the development of atherosclerosis and it seems that the cumulative effects of advanced age and cigarette smoking have deleterious consequences on cardiovascular morbidity and mortality in the elderly. Since Harmon proposed the original free radical theory of aging (166), considerable evidence has been published that cardiovascular aging in various tissues is associated with an increased oxidative-nitrosative stress and impaired bioavailability of vasoprotective NO (88, 89, 104, 109, 116, 167-175). Importantly, vascular NAD (P)H oxidase activity/expression is up-regulated in aging (89, 169, 176), accompanied by a down-regulation of antioxidant mechanisms, such as ecSOD (167). On the basis of the aforementioned findings, we posit that aged arteries are more susceptible to cigarette smoke-induced oxidative stress. Our recent data that CSE elicits an exaggerated ROS production in aged vessels seems to support this premise (Csiszar and Ungvari, unpublished data 2006). Previously we have demonstrated that a pro-inflammatory shift develops in the vascular cytokine expression profile in aged coronary arteries (89, 104, 116). Importantly, expression of the pro-inflammatory cytokines TNFα, TNFβ (which acts on the same receptor as TNFα) and that of IL-6 increase in aged endothelial and/or smooth cells (89, 104, 177, 178) and recent studies suggest that NF-κB binding increases in aging (175, 179). Thus, we hypothesize that age-related oxidative/nitrosative stress induces chronic activation of NF-κB and/or PARP-1 (even in the absence of exogenous pro-oxidant factors). Because multiple intracellular antioxidant and anti-inflammatory mechanisms (e.g. NO signaling) are likely to be impaired in aging (167, 180-182), we hypothesize that aged vessels are especially vulnerable to the pro-inflammatory effects of cigarette smoke. We propose that because the NF-κB/PARP system is already “primed” in aged vessels, cigarette smoke-induced oxidative stress will result in an enhanced vascular inflammatory response. More research on cigarette smoke-induced oxidative/nitrosative stress, pro-inflammatory vascular cytokine expression and endothelial activation in aged vessels is evidently needed. Future studies should determine whether advanced age renders endothelial and smooth muscle cells in coronary arteries more vulnerable toward the pro-oxidant, pro-inflammatory effects of cigarette smoke.
9. PERSPECTIVE
In conclusion, water soluble components of cigarette smoke increase NAD (P)H-oxidase derived ROS generation in endothelial and smooth muscle cell, which induce a pro-inflammatory vascular phenotype likely via mechanisms that involve NF-κB activation (Figure 7). It is likely that cigarette smoking-induced oxidative-nitrosative stress, PARP-1 activation and vascular inflammation will support atherosclerotic plaque formation in the coronary and carotid arteries increasing the morbidity of myocardial infarction and stroke. The available data support a role for novel anti-inflammatory treatments, including PARP-1 inhibitors, in pharmacological vasculoprotection in accelerated vascular aging (144, 149, 183).
Figure 7.
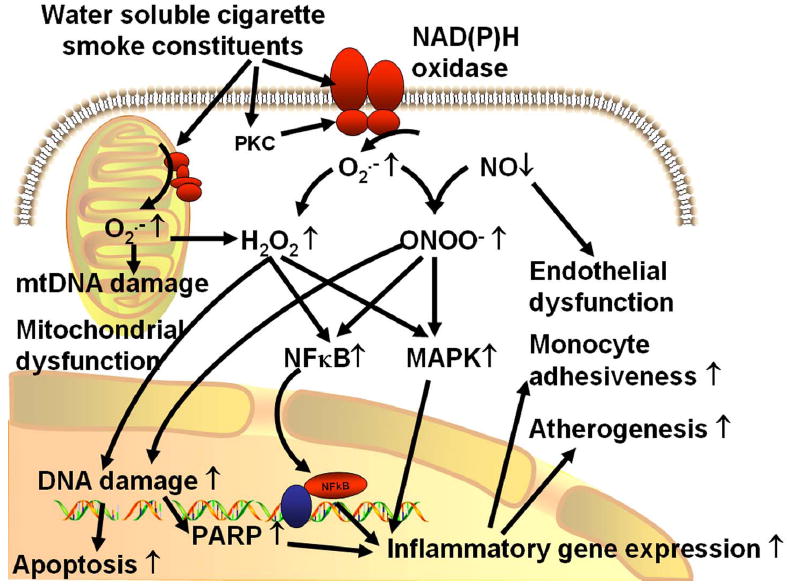
Proposed scheme for the mechanisms by which water soluble components of cigarette smoke promote pro-inflammatory phenotypic alterations in the blood vessels. The model predicts that cigarette smoke induces an increased generation O2.- by NAD (P)H oxidase, which scavenges vasodilator NO resulting in an enhanced ONOO- formation and endothelial dysfunction. Cigarette smoke also impairs mitochondrial function eliciting mitochondrial oxidative stress. Increased cellular levels of NAD (P)H oxidase- and mitochondria-derived O2.-, H2O2 and/or ONOO- activate redox-sensitive signaling pathways, including MAP kinases and the transcription factor NF-κB, up-regulating inflammatory gene expression. Oxidative-nitrosative stress (in particular, the enhanced ONOO- levels and increased production of hydroxyl radicals) also elicits nuclear DNA damage promoting endothelial apoptosis and activating the nuclear enzyme PARP-1, which importantly contributes to NF-κB -dependent regulation of gene transcription. The resulting pro-inflammatory vascular phenotype will likely increase monocyte recruitment to the vascular wall and promote the development of atherosclerosis, especially if other risk factors (e.g. hypertension, hypercholesterolemia, hyperhomocysteinemia) are also present.
Acknowledgments
This work was supported by grants from the American Heart Association (0430108N and 0435140N, to A.C. and Z.U.), the NIH (HL077256 and HL43023, to A.C. M.W. and Z.U.), Philip Morris U.S.A. Inc. and Philip Morris International (to Z.U.), the San Antonio Area Foundation (to A.P.) and in part funded by the Intramural Research Program of the National Institutes of Health/NIAAA (P.P. and P.M.).
Apologies are extended to all those whose findings or opinions pertinent to this topic were not referenced or discussed due to limitations of space or inadvertent omissions.
Abbreviations
- CAD
coronary artery disease
- TNFα
tumor necrosis factor α
- CS
cigarette smoke
- CSE
cigarette smoke extract
- PARP
poly (ADP-ribose) polymerase
- NF-κB
nuclear factor κB
- ROS
reactive oxygen species
- PAH
polycyclic aromatic hydrocarbons
Footnotes
11. REFERENCES
- 1.Law MR, Wald NJ. Environmental tobacco smoke and ischemic heart disease. Prog Cardiovasc Dis. 2003;46:31–38. doi: 10.1016/s0033-0620(03)00078-1. [DOI] [PubMed] [Google Scholar]
- 2.Raij L, DeMaster EG, Jaimes EA. Cigarette smoke-induced endothelium dysfunction: role of superoxide anion. J Hypertens. 2001;19:891–897. doi: 10.1097/00004872-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Burke A, Fitzgerald GA. Oxidative stress and smoking-induced vascular injury. Prog Cardiovasc Dis. 2003;46:79–90. doi: 10.1016/s0033-0620(03)00076-8. [DOI] [PubMed] [Google Scholar]
- 4.Churg A. Interactions of exogenous or evoked agents and particles: the role of reactive oxygen species. Free Radic Biol Med. 2003;34:1230–1235. doi: 10.1016/s0891-5849(03)00175-8. [DOI] [PubMed] [Google Scholar]
- 5.Ijzerman RG, Serne EH, van Weissenbruch MM, de Jongh RT, Stehouwer CD. Cigarette smoking is associated with an acute impairment of microvascular function in humans. Clin Sci (Lond) 2003;104:247–252. doi: 10.1042/CS20020318. [DOI] [PubMed] [Google Scholar]
- 6.Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003;107:416–421. doi: 10.1161/01.cir.0000046448.26751.58. [DOI] [PubMed] [Google Scholar]
- 7.Fennessy FM, Moneley DS, Wang JH, Kelly CJ, Bouchier-Hayes DJ. Taurine and vitamin C modify monocyte and endothelial dysfunction in young smokers. Circulation. 2003;107:410–415. doi: 10.1161/01.cir.0000046447.72402.47. [DOI] [PubMed] [Google Scholar]
- 8.Iwado Y, Yoshinaga K, Furuyama H, Ito Y, Noriyasu K, Katoh C, Kuge Y, Tsukamoto E, Tamaki N. Decreased endothelium-dependent coronary vasomotion in healthy young smokers. Eur J Nucl Med Mol Imaging. 2002;29:984–990. doi: 10.1007/s00259-002-0818-1. [DOI] [PubMed] [Google Scholar]
- 9.Tsuchiya M, Asada A, Kasahara E, Sato EF, Shindo M, Inoue M. Smoking a single cigarette rapidly reduces combined concentrations of nitrate and nitrite and concentrations of antioxidants in plasma. Circulation. 2002;105:1155–1157. doi: 10.1161/hc1002.105935. [DOI] [PubMed] [Google Scholar]
- 10.Barua RS, Ambrose JA, Srivastava S, DeVoe MC, Eales-Reynolds LJ. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: an in vitro demonstration in human coronary artery endothelial cells. Circulation. 2003;107:2342–2347. doi: 10.1161/01.CIR.0000066691.52789.BE. [DOI] [PubMed] [Google Scholar]
- 11.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Ye Y, Zhu M, Cho C. Increased interleukin-8 expression by cigarette smoke extract in endothelial cells. Environ Toxicol Pharmacol. 2000;9:19–23. doi: 10.1016/s1382-6689(00)00056-9. [DOI] [PubMed] [Google Scholar]
- 13.Neunteufl T, Priglinger U, Heher S, Zehetgruber M, Soregi G, Lehr S, Huber K, Maurer G, Weidinger F, Kostner K. Effects of vitamin E on chronic and acute endothelial dysfunction in smokers. J Am Coll Cardiol. 2000;35:277–283. doi: 10.1016/s0735-1097(99)00542-2. [DOI] [PubMed] [Google Scholar]
- 14.Heitzer T, Just H, Munzel T. Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation. 1996;94:6–9. doi: 10.1161/01.cir.94.1.6. [DOI] [PubMed] [Google Scholar]
- 15.Heitzer T, Yla-Herttuala S, Luoma J, Kurz S, Munzel T, Just H, Olschewski M, Drexler H. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia. Role of oxidized LDL. Circulation. 1996;93:1346–1353. doi: 10.1161/01.cir.93.7.1346. [DOI] [PubMed] [Google Scholar]
- 16.Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, Munzel T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86:E36–41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 17.Newby DE, Wright RA, Labinjoh C, Ludlam CA, Fox KA, Boon NA, Webb DJ. Endothelial dysfunction, impaired endogenous fibrinolysis, and cigarette smoking: a mechanism for arterial thrombosis and myocardial infarction. Circulation. 1999;99:1411–1415. doi: 10.1161/01.cir.99.11.1411. [DOI] [PubMed] [Google Scholar]
- 18.Blann AD, Kirkpatrick U, Devine C, Naser S, McCollum CN. The influence of acute smoking on leucocytes, platelets and the endothelium. Atherosclerosis. 1998;141:133–139. [PubMed] [Google Scholar]
- 19.Sugiyama S, Kugiyama K, Ohgushi M, Matsumura T, Ota Y, Doi H, Ogata N, Oka H, Yasue H. Supersensitivity of atherosclerotic artery to constrictor effect of cigarette smoke extract. Cardiovasc Res. 1998;38:508–515. doi: 10.1016/s0008-6363(98)00027-3. [DOI] [PubMed] [Google Scholar]
- 20.Ota Y, Kugiyama K, Sugiyama S, Ohgushi M, Matsumura T, Doi H, Ogata N, Oka H, Yasue H. Impairment of endothelium-dependent relaxation of rabbit aortas by cigarette smoke extract--role of free radicals and attenuation by captopril. Atherosclerosis. 1997;131:195–202. doi: 10.1016/s0021-9150(97)06106-6. [DOI] [PubMed] [Google Scholar]
- 21.Adams MR, Jessup W, Celermajer DS. Cigarette smoking is associated with increased human monocyte adhesion to endothelial cells: reversibility with oral L-arginine but not vitamin C. J Am Coll Cardiol. 1997;29:491–497. doi: 10.1016/s0735-1097(96)00537-2. [DOI] [PubMed] [Google Scholar]
- 22.Meeder JG, Blanksma PK, van der Wall EE, Anthonio RL, Willemsen AT, Pruim J, Vaalburg W, Lie KI. Long-term cigarette smoking is associated with increased myocardial perfusion heterogeneity assessed by positron emission tomography. Eur J Nucl Med. 1996;23:1442–1447. doi: 10.1007/BF01254465. [DOI] [PubMed] [Google Scholar]
- 23.Kugiyama K, Yasue H, Ohgushi M, Motoyama T, Kawano H, Inobe Y, Hirashima O, Sugiyama S. Deficiency in nitric oxide bioactivity in epicardial coronary arteries of cigarette smokers. J Am Coll Cardiol. 1996;28:1161–1167. doi: 10.1016/S0735-1097(96)00325-7. [DOI] [PubMed] [Google Scholar]
- 24.Shen Y, Rattan V, Sultana C, Kalra VK. Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytes. Am J Physiol. 1996;270:H1624–1633. doi: 10.1152/ajpheart.1996.270.5.H1624. [DOI] [PubMed] [Google Scholar]
- 25.Pryor WA, Stone K, Zang LY, Bermudez E. Fractionation of aqueous cigarette tar extracts: fractions that contain the tar radical cause DNA damage. Chem Res Toxicol. 1998;11:441–448. doi: 10.1021/tx970159y. [DOI] [PubMed] [Google Scholar]
- 26.Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med. 1995;19:161–167. doi: 10.1016/0891-5849(94)00236-d. [DOI] [PubMed] [Google Scholar]
- 27.Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 1983;47:345–355. doi: 10.1289/ehp.8347345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakayama T, Church DF, Pryor WA. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic Biol Med. 1989;7:9–15. doi: 10.1016/0891-5849(89)90094-4. [DOI] [PubMed] [Google Scholar]
- 29.Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med. 1995;19:161–167. doi: 10.1016/0891-5849(94)00236-d. [DOI] [PubMed] [Google Scholar]
- 30.Stone KK, Bermudez E, Pryor WA. Aqueous extracts of cigarette tar containing the tar free radical cause DNA nicks in mammalian cells. Environ Health Perspect. 1994;102:173–178. doi: 10.1289/ehp.94102s10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pryor WA. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environmental health perspectives. 1997;105(Suppl 4):875–882. doi: 10.1289/ehp.97105s4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pryor WA, Stone K, Zang LY, Bermudez E. Fractionation of aqueous cigarette tar extracts: fractions that contain the tar radical cause DNA damage. Chem Res Toxicol. 1998;11:441–448. doi: 10.1021/tx970159y. [DOI] [PubMed] [Google Scholar]
- 33.Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Annals of the New York Academy of Sciences. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. discussion 27-18. [DOI] [PubMed] [Google Scholar]
- 34.Pryor WA, Arbour NC, Upham B, Church DF. The inhibitory effect of extracts of cigarette tar on electron transport of mitochondria and submitochondrial particles. Free Radic Biol Med. 1992;12:365–372. doi: 10.1016/0891-5849(92)90085-u. [DOI] [PubMed] [Google Scholar]
- 35.Borish ET, Pryor WA, Venugopal S, Deutsch WA. DNA synthesis is blocked by cigarette tar-induced DNA single-strand breaks. Carcinogenesis. 1987;8:1517–1520. doi: 10.1093/carcin/8.10.1517. [DOI] [PubMed] [Google Scholar]
- 36.Borish ET, Cosgrove JP, Church DF, Deutsch WA, Pryor WA. Cigarette tar causes single-strand breaks in DNA. Biochem Biophysi Res Commun. 1985;133:780–786. doi: 10.1016/0006-291x(85)90972-6. [DOI] [PubMed] [Google Scholar]
- 37.Pryor WA, Terauchi K, Davis WH., Jr Electron spin resonance (ESR) study of cigarette smoke by use of spin trapping techniques. Environ Health Perspects. 1976;16:161–176. doi: 10.1289/ehp.7616161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Perspect. 1983;47:345–355. doi: 10.1289/ehp.8347345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pryor WA, Hales BJ, Premovic PI, Church DF. The radicals in cigarette tar: their nature and suggested physiological implications. Science (New York, N Y. 1983;220:425–427. doi: 10.1126/science.6301009. [DOI] [PubMed] [Google Scholar]
- 40.Bock FG, Swain AP, Stedman RL. Carcinogenesis assay of subfractions of cigarette smoke condensate prepared by solvent-solvent separation of the neutral fraction. J Natl Cancer Inst. 1972;49:477–483. [PubMed] [Google Scholar]
- 41.Schmeltz I, Stills CD, Chamberlain WJ, Stedman RL. Analysis of cigarette smoke fraction by combined gas chromatography--infrared spectrophotometry. Anal Chem. 1965;37:1614–1616. doi: 10.1021/ac60231a603. [DOI] [PubMed] [Google Scholar]
- 42.Swain AP, Cooper JE, Stedman RL. Large-scale fractionation of cigarette smoke condensate for chemical and biologic investigations. Cancer Res. 1969;29:579–583. [PubMed] [Google Scholar]
- 43.Squadrito GL, Cueto R, Dellinger B, Pryor WA. Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radic Biol Med. 2001;31:1132–1138. doi: 10.1016/s0891-5849(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 44.Celermajer DS, Sorensen KE, Bull C, Robinson J, Deanfield JE. Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J Am Coll Cardiol. 1994;24:1468–1474. doi: 10.1016/0735-1097(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 45.Celermajer DS, Sorensen KE, Georgakopoulos D, Bull C, Thomas O, Robinson J, Deanfield JE. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation. 1993;88:2149–2155. doi: 10.1161/01.cir.88.5.2149. [DOI] [PubMed] [Google Scholar]
- 46.Czernin J, Waldherr C. Cigarette smoking and coronary blood flow. Prog Cardiovasc Dis. 2003;45:395–404. doi: 10.1053/pcad.2003.00104. [DOI] [PubMed] [Google Scholar]
- 47.Czernin J, Sun K, Brunken R, Bottcher M, Phelps M, Schelbert H. Effect of acute and long-term smoking on myocardial blood flow and flow reserve. Circulation. 1995;91:2891–2897. doi: 10.1161/01.cir.91.12.2891. [DOI] [PubMed] [Google Scholar]
- 48.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD (P)H oxidase activation. Am J Physiol. 2007;292:H130–139. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 49.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. 2004;24:1031–1036. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 50.Nagy J, Demaster EG, Wittmann I, Shultz P, Raij L. Induction of endothelial cell injury by cigarette smoke. Endothelium. 1997;5:251–263. doi: 10.3109/10623329709052590. [DOI] [PubMed] [Google Scholar]
- 51.Raveendran M, Wang J, Senthil D, Utama B, Shen Y, Dudley D, Zhang Y, Wang XL. Endogenous nitric oxide activation protects against cigarette smoking induced apoptosis in endothelial cells. FEBS letters. 2005;579:733–740. doi: 10.1016/j.febslet.2004.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayhan WG, Sharpe GM. Superoxide dismutase restores endothelium-dependent arteriolar dilatation during acute infusion of nicotine. J Appl Physiol. 1998;85:1292–1298. doi: 10.1152/jappl.1998.85.4.1292. [DOI] [PubMed] [Google Scholar]
- 53.Zhang S, Day I, Ye S. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis. 2001;154:277–283. doi: 10.1016/s0021-9150(00)00475-5. [DOI] [PubMed] [Google Scholar]
- 54.Griendling KK, Sorescu D, Ushio-Fukai M. NAD (P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 55.Ungvari Z, Csiszar A, Edwards JG, Kaminski PM, Wolin MS, Kaley G, Koller A. Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD (P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2003;23:418–424. doi: 10.1161/01.ATV.0000061735.85377.40. [DOI] [PubMed] [Google Scholar]
- 56.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD (P)H oxidase. Circulation. 2003;108:1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 57.Ungvari Z, Csiszar A, Kaminski PM, Wolin MS, Koller A. Chronic high pressure-induced arterial oxidative stress: Involvement of protein kinase C-dependent NAD (P)H oxidase and local renin-angiotensin system. Am J Pathol. 2004;165:219–226. doi: 10.1016/S0002-9440(10)63290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kashyap R, Floreani AA, Heires AJ, Sanderson SD, Wyatt TA. Protein kinase C-alpha mediates cigarette smoke extract- and complement factor 5a-stimulated interleukin-8 release in human bronchial epithelial cells. J Investig Med. 2002;50:46–53. doi: 10.2310/6650.2002.33517. [DOI] [PubMed] [Google Scholar]
- 59.Mur C, Claria J, Rodela S, Lario S, Campistol JM, Titos E, Inigo P, Cases A, Abian J, Esmatjes E. Cigarette smoke concentrate increases 8-epi-PGF2alpha and TGFbeta1 secretion in rat mesangial cells. Life Sci. 2004;75:611–621. doi: 10.1016/j.lfs.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 60.Anbarasi K, Vani G, Devi CS. Protective effect of bacoside A on cigarette smoking-induced brain mitochondrial dysfunction in rats. J Environ Pathol Toxicol Oncol. 2005;24:225–234. doi: 10.1615/jenvpathtoxoncol.v24.i3.80. [DOI] [PubMed] [Google Scholar]
- 61.Jia L, Liu Z, Sun L, Miller SS, Ames BN, Cotman CW, Liu J. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci. 2007;48:339–348. doi: 10.1167/iovs.06-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, Lee JS, Postma DS, Kauffman HF, Choi AM. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol. 2007;36:409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Gairola C, Aleem HM. Cigarette smoke in vitro effects of condensate fractions on mitochondrial respiration. Life Sci. 1974;14:2199–2207. doi: 10.1016/0024-3205(74)90102-7. [DOI] [PubMed] [Google Scholar]
- 64.Gairola C, Aleem MI. Cigarette smoke: effect of aqueous and nonaqueous fractions on mitochondrial function. Nature. 1973;241:287–288. doi: 10.1038/241287a0. [DOI] [PubMed] [Google Scholar]
- 65.Gvozdjakova A, Bada V, Sany L, Kucharska J, Kruty F, Bozek P, Trstansky L, Gvozdjak J. Smoke cardiomyopathy: disturbance of oxidative processes in myocardial mitochondria. Cardiovasc Res. 1984;18:229–232. doi: 10.1093/cvr/18.4.229. [DOI] [PubMed] [Google Scholar]
- 66.Knight-Lozano CA, Young CG, Burow DL, Hu ZY, Uyeminami D, Pinkerton KE, Ischiropoulos H, Ballinger SW. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation. 2002;105:849–854. doi: 10.1161/hc0702.103977. [DOI] [PubMed] [Google Scholar]
- 67.Lewis PD, Fradley SR, Griffiths AP, Baxter PW, Parry JM. Mitochondrial DNA mutations in the parotid gland of cigarette smokers and non-smokers. Mutat Res. 2002;518:47–54. doi: 10.1016/s1383-5718(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 68.Ballinger SW, Bouder TG, Davis GS, Judice SA, Nicklas JA, Albertini RJ. Mitochondrial genome damage associated with cigarette smoking. Cancer Res. 1996;56:5692–5697. [PubMed] [Google Scholar]
- 69.Thirman MJ, Albrecht JH, Krueger MA, Erickson RR, Cherwitz DL, Park SS, Gelboin HV, Holtzman JL. Induction of cytochrome CYPIA1 and formation of toxic metabolites of benzo (a)pyrene by rat aorta: a possible role in atherogenesis. Proc Natl Acad Sci U S A. 1994;91:5397–5401. doi: 10.1073/pnas.91.12.5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leuchtenberger C, Leuchtenberger R, Zbinden I. Gas vapour phase constituents and SH reactivity of cigarette smoke influence lung cultures. Nature. 1974;247:565–567. doi: 10.1038/247565a0. [DOI] [PubMed] [Google Scholar]
- 71.Reddy S, Finkelstein EI, Wong PS, Phung A, Cross CE, van der Vliet A. Identification of glutathione modifications by cigarette smoke. Free Rad Biol Med. 2002;33:1490–1498. doi: 10.1016/s0891-5849(02)01079-1. [DOI] [PubMed] [Google Scholar]
- 72.Nardini M, Finkelstein EI, Reddy S, Valacchi G, Traber M, Cross CE, van der Vliet A. Acrolein-induced cytotoxicity in cultured human bronchial epithelial cells. Modulation by alpha-tocopherol and ascorbic acid. Toxicology. 2002;170:173–185. doi: 10.1016/s0300-483x(01)00540-6. [DOI] [PubMed] [Google Scholar]
- 73.Nguyen H, Finkelstein E, Reznick A, Cross C, van der Vliet A. Cigarette smoke impairs neutrophil respiratory burst activation by aldehyde-induced thiol modifications. Toxicology. 2001;160:207–217. doi: 10.1016/s0300-483x(00)00450-9. [DOI] [PubMed] [Google Scholar]
- 74.Muller T, Gebel S. The cellular stress response induced by aqueous extracts of cigarette smoke is critically dependent on the intracellular glutathione concentration. Carcinogenesis. 1998;19:797–801. doi: 10.1093/carcin/19.5.797. [DOI] [PubMed] [Google Scholar]
- 75.Muller T, Haussmann HJ, Schepers G. Evidence for peroxynitrite as an oxidative stress-inducing compound of aqueous cigarette smoke fractions. Carcinogenesis. 1997;18:295–301. doi: 10.1093/carcin/18.2.295. [DOI] [PubMed] [Google Scholar]
- 76.Park YS, Misonou Y, Fujiwara N, Takahashi M, Miyamoto Y, Koh YH, Suzuki K, Taniguchi N. Induction of thioredoxin reductase as an adaptive response to acrolein in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2005;327:1058–1065. doi: 10.1016/j.bbrc.2004.12.104. [DOI] [PubMed] [Google Scholar]
- 77.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 78.Patel RP, Moellering D, Murphy-Ullrich J, Jo H, Beckman JS, Darley-Usmar VM. Cell signaling by reactive nitrogen and oxygen species in atherosclerosis. Free Radic Biol Med. 2000;28:1780–1794. doi: 10.1016/s0891-5849(00)00235-5. [DOI] [PubMed] [Google Scholar]
- 79.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 80.Soriano FG, Virag L, Szabo C. Diabetic endothelial dysfunction: role of reactive oxygen and nitrogen species production and poly (ADP-ribose) polymerase activation. J Mol Med. 2001;79:437–448. doi: 10.1007/s001090100236. [DOI] [PubMed] [Google Scholar]
- 81.De Nigris F, Lerman LO, Condorelli M, Lerman A, Napoli C. Oxidation-sensitive transcription factors and molecular mechanisms in the arterial wall. Antioxid Redox Signal. 2001;3:1119–1130. doi: 10.1089/152308601317203620. [DOI] [PubMed] [Google Scholar]
- 82.Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, Schieffer B. Mechanical Stretch Enhances mRNA Expression and Proenzyme Release of Matrix Metalloproteinase-2 (MMP-2) via NAD (P)H Oxidase-Derived Reactive Oxygen Species. Circ Res. 2003;92:E80–E86. doi: 10.1161/01.RES.0000077044.60138.7C. [DOI] [PubMed] [Google Scholar]
- 83.Satriano JA, Shuldiner M, Hora K, Xing Y, Shan Z, Schlondorff D. Oxygen radicals as second messengers for expression of the monocyte chemoattractant protein, JE/MCP-1, and the monocyte colony-stimulating factor, CSF-1, in response to TNF-alpha and immunoglobulin G. Evidence for involvement of NADPH-dependent oxidase. J Clin Invest. 1993;92:1564–1571. doi: 10.1172/JCI116737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kranzhofer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kubler W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1623–1629. doi: 10.1161/01.atv.19.7.1623. [DOI] [PubMed] [Google Scholar]
- 85.Schieffer B, Luchtefeld M, Braun S, Hilfiker A, Hilfiker-Kleiner D, Drexler H. Role of NAD (P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circ Res. 2000;87:1195–1201. doi: 10.1161/01.res.87.12.1195. [DOI] [PubMed] [Google Scholar]
- 86.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 87.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Csiszar A, Pacher P, Kaley G, Ungvari Z. Role of oxidative and nitrosative stress, longevity genes and poly (ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr Vasc Pharmacol. 2005;3:285–291. doi: 10.2174/1570161054368616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 90.Yamaguchi Y, Nasu F, Harada A, Kunitomo M. Oxidants in the gas phase of cigarette smoke pass through the lung alveolar wall and raise systemic oxidative stress. J Pharmacol Sci. 2007;103:275–282. doi: 10.1254/jphs.fp0061055. [DOI] [PubMed] [Google Scholar]
- 91.Yamaguchi Y, Haginaka J, Morimoto S, Fujioka Y, Kunitomo M. Facilitated nitration and oxidation of LDL in cigarette smokers. Eur J Clin Invest. 2005;35:186–193. doi: 10.1111/j.1365-2362.2005.01472.x. [DOI] [PubMed] [Google Scholar]
- 92.Sawa T, Tatemichi M, Akaike T, Barbin A, Ohshima H. Analysis of urinary 8-nitroguanine, a marker of nitrative nucleic acid damage, by high-performance liquid chromatography-electrochemical detection coupled with immunoaffinity purification: association with cigarette smoking. Free Rad Biol Med. 2006;40:711–720. doi: 10.1016/j.freeradbiomed.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 93.Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Peroxynitrite-mediated oxidative modification of low-density lipoprotein by aqueous extracts of cigarette smoke and the preventive effect of fluvastatin. Atherosclerosis. 2004;172:259–265. doi: 10.1016/j.atherosclerosis.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 94.Petruzzelli S, Puntoni R, Mimotti P, Pulera N, Baliva F, Fornai E, Giuntini C. Plasma 3-nitrotyrosine in cigarette smokers. Am J Respir Crit Care Med. 1997;156:1902–1907. doi: 10.1164/ajrccm.156.6.9702075. [DOI] [PubMed] [Google Scholar]
- 95.Jozsef L, Khreiss T, El Kebir D, Filep JG. Activation of TLR-9 induces IL-8 secretion through peroxynitrite signaling in human neutrophils. J Immunol. 2006;176:1195–1202. doi: 10.4049/jimmunol.176.2.1195. [DOI] [PubMed] [Google Scholar]
- 96.Szabo C, Ferrer-Sueta G, Zingarelli B, Southan GJ, Salzman AL, Radi R. Mercaptoethylguanidine and guanidine inhibitors of nitric-oxide synthase react with peroxynitrite and protect against peroxynitrite- induced oxidative damage. J Biol Chem. 1997;272:9030–9036. doi: 10.1074/jbc.272.14.9030. [DOI] [PubMed] [Google Scholar]
- 97.Lancel S, Tissier S, Mordon S, Marechal X, Depontieu F, Scherpereel A, Chopin C, Neviere R. Peroxynitrite decomposition catalysts prevent myocardial dysfunction and inflammation in endotoxemic rats. J Am Coll Cardiol. 2004;43:2348–2358. doi: 10.1016/j.jacc.2004.01.047. [DOI] [PubMed] [Google Scholar]
- 98.Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H (2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 99.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, Deb A, Szabo E, Ungvari Z, Wolin MS, Szabo C. A potent peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;18:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 100.Zouki C, Jozsef L, Ouellet S, Paquette Y, Filep JG. Peroxynitrite mediates cytokine-induced IL-8 gene expression and production by human leukocytes. J Leukoc Biol. 2001;69:815–824. [PubMed] [Google Scholar]
- 101.Filep JG, Beauchamp M, Baron C, Paquette Y. Peroxynitrite mediates IL-8 gene expression and production in lipopolysaccharide-stimulated human whole blood. J Immunol. 1998;161:5656–5662. [PubMed] [Google Scholar]
- 102.Gagnon C, Leblond FA, Filep JG. Peroxynitrite production by human neutrophils, monocytes and lymphocytes challenged with lipopolysaccharide. FEBS Lett. 1998;431:107–110. doi: 10.1016/s0014-5793(98)00741-8. [DOI] [PubMed] [Google Scholar]
- 103.Young JL, Libby P, Schonbeck U. Cytokines in the pathogenesis of atherosclerosis. Thromb Haemost. 2002;88:554–567. [PubMed] [Google Scholar]
- 104.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. Faseb J. 2003;17:1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 105.Loft S, Vistisen K, Ewertz M, Tjonneland A, Overvad K, Poulsen HE. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: influence of smoking, gender and body mass index. Carcinogenesis. 1992;13:2241–2247. doi: 10.1093/carcin/13.12.2241. [DOI] [PubMed] [Google Scholar]
- 106.Chen HW, Chien ML, Chaung YH, Lii CK, Wang TS. Extracts from cigarette smoke induce DNA damage and cell adhesion molecule expression through different pathways. Chem Biol Interact. 2004;150:233–241. doi: 10.1016/j.cbi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 107.Zhang YJ, Weksler BB, Wang L, Schwartz J, Santella RM. Immunohistochemical detection of polycyclic aromatic hydrocarbon-DNA damage in human blood vessels of smokers and non-smokers. Atherosclerosis. 1998;140:325–331. doi: 10.1016/s0021-9150(98)00136-1. [DOI] [PubMed] [Google Scholar]
- 108.Phillips DH. Smoking-related DNA and protein adducts in human tissues. Carcinogenesis. 2002;23:1979–2004. doi: 10.1093/carcin/23.12.1979. [DOI] [PubMed] [Google Scholar]
- 109.Csiszar A, Labinskyy N, Xiangmin Z, F H, Serpillon S, Huang Z, Ballabh P, Levy R, Hintze TH, Wolin MS, Austad SN, Podlutsky A, Ungvari Z. Vascular O2.- and H2O2 production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007 doi: 10.1111/j.1474-9726.2007.00339.x. in press. [DOI] [PubMed] [Google Scholar]
- 110.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ames BN. Endogenous oxidative DNA damage, aging, and cancer. Free Radic Res Commun. 1989;7:121–128. doi: 10.3109/10715768909087933. [DOI] [PubMed] [Google Scholar]
- 112.Ames BN. Endogenous DNA damage as related to cancer and aging. Mut Res. 1989;214:41–46. doi: 10.1016/0027-5107(89)90196-6. [DOI] [PubMed] [Google Scholar]
- 113.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 114.Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor Necrosis Factor-{alpha} Drives 70% of Cigarette Smoke-Induced Emphysema in the Mouse. Am J Respir Crit Care Med. 2004 doi: 10.1164/rccm.200404-511OC. [DOI] [PubMed] [Google Scholar]
- 115.Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging. 2001;22:837–842. doi: 10.1016/s0197-4580(01)00276-7. [DOI] [PubMed] [Google Scholar]
- 116.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17:21–30. doi: 10.1152/physiolgenomics.00136.2003. [DOI] [PubMed] [Google Scholar]
- 117.Nordskog BK, Blixt AD, Zieske AW, Hellmann GM. MMP-1 polymorphic expression in aortic endothelial cells: possible role in lesion development in smokers and nonsmokers. Cardiovasc Toxicol. 2004;4:75–83. doi: 10.1385/ct:4:1:75. [DOI] [PubMed] [Google Scholar]
- 118.Nordskog BK, Blixt AD, Morgan WT, Fields WR, Hellmann GM. Matrix-degrading and pro-inflammatory changes in human vascular endothelial cells exposed to cigarette smoke condensate. Cardiovasc Toxicol. 2003;3:101–117. doi: 10.1385/ct:3:2:101. [DOI] [PubMed] [Google Scholar]
- 119.Tedgui A, Mallat Z. Anti-inflammatory mechanisms in the vascular wall. Circ Res. 2001;88:877–887. doi: 10.1161/hh0901.090440. [DOI] [PubMed] [Google Scholar]
- 120.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang YH, Lin JX, Vilcek J. Interleukin-6 induction by tumor necrosis factor and interleukin-1 in human fibroblasts involves activation of a nuclear factor binding to a kappa B-like sequence. Mol Cell Biol. 1990;10:3818–3823. doi: 10.1128/mcb.10.7.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD, Kraemer RT, Mirza UA, Chait BT, Burk SC, Quilliam LA. Redox regulation of cell signalling. Nature. 1996;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 124.Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- 125.Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 126.Piette J, Piret B, Bonizzi G, Schoonbroodt S, Merville MP, Legrand-Poels S, Bours V. Multiple redox regulation in NF-kappaB transcription factor activation. Biol Chem. 1997;378:1237–1245. [PubMed] [Google Scholar]
- 127.Hishikawa K, Oemar BS, Yang Z, Luscher TF. Pulsatile stretch stimulates superoxide production and activates nuclear factor-kappa B in human coronary smooth muscle. Circ Res. 1997;81:797–803. doi: 10.1161/01.res.81.5.797. [DOI] [PubMed] [Google Scholar]
- 128.Bowie A, O’Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 129.Schmidt KN, Amstad P, Cerutti P, Baeuerle PA. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-kappa B. Chem Biol. 1995;2:13–22. doi: 10.1016/1074-5521(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 130.Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB (alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis. 2002;23:1511–1518. doi: 10.1093/carcin/23.9.1511. [DOI] [PubMed] [Google Scholar]
- 131.van den Berg R, Haenen GR, van den Berg H, Bast A. Nuclear factor-kappaB activation is higher in peripheral blood mononuclear cells of male smokers. Environ Toxicol Pharmacol. 2001;9:147–151. doi: 10.1016/s1382-6689(00)00070-3. [DOI] [PubMed] [Google Scholar]
- 132.Nishikawa M, Kakemizu N, Ito T, Kudo M, Kaneko T, Suzuki M, Udaka N, Ikeda H, Okubo T. Superoxide mediates cigarette smoke-induced infiltration of neutrophils into the airways through nuclear factor-kappaB activation and IL-8 mRNA expression in guinea pigs in vivo. Am J Respir Cell Mol Biol. 1999;20:189–198. doi: 10.1165/ajrcmb.20.2.3305. [DOI] [PubMed] [Google Scholar]
- 133.Csiszar A, Smith KE, Koller A, Kaley G, Edwards JG, Ungvari Z. Regulation of bone morphogenetic protein-2 expression in endothelial cells: role of nuclear factor-kappaB activation by tumor necrosis factor-alpha, H2O2, and high intravascular pressure. Circulation. 2005;111:2364–2372. doi: 10.1161/01.CIR.0000164201.40634.1D. [DOI] [PubMed] [Google Scholar]
- 134.Pelletier C, Varin-Blank N, Rivera J, Iannascoli B, Marchand F, David B, Weyer A, Blank U. Fc epsilonRI-mediated induction of TNF-alpha gene expression in the RBL- 2H3 mast cell line: regulation by a novel NF-kappaB-like nuclear binding complex. J Immunol. 1998;161:4768–4776. [PubMed] [Google Scholar]
- 135.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tanabe O, Akira S, Kamiya T, Wong GG, Hirano T, Kishimoto T. Genomic structure of the murine IL-6 gene. High degree conservation of potential regulatory sequences between mouse and human. J Immunol. 1988;141:3875–3881. [PubMed] [Google Scholar]
- 137.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol. 2007;292:L567–576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 138.Csiszar A, Smith K, Labinskyy N, Orosz Z, Rivera A, Ungvari Z. Resveratrol attenuates TNF-{alpha}-induced activation of coronary arterial endothelial cells: role of NF-{kappa}B inhibition. Am J Physiol. 2006;291:H1694–1699. doi: 10.1152/ajpheart.00340.2006. [DOI] [PubMed] [Google Scholar]
- 139.Ungvari Z, Orosz Z, Rivera A, Labinskyy N, Xiangmin Z, Olson S, Podlutsky A, Csiszar A. Resveratrol increases vascular oxidative stress resistance. Am J Physiol. 2007;292:H2417–2424. doi: 10.1152/ajpheart.01258.2006. [DOI] [PubMed] [Google Scholar]
- 140.Szabo C, Dawson VL. Role of poly (ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 141.Liaudet L, Soriano FG, Szabo E, Virag L, Mabley JG, Salzman AL, Szabo C. Protection against hemorrhagic shock in mice genetically deficient in poly (ADP-ribose)polymerase. Proc Natl Acad Sci U S A. 2000;97:10203–10208. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly (adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 143.Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, Szabo C. Activation of poly (ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300:862–867. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 144.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly (ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002;9:659–664. [PubMed] [Google Scholar]
- 145.Hasko G, Mabley JG, Nemeth ZH, Pacher P, Deitch EA, Szabo C. Poly (ADP-ribose) polymerase is a regulator of chemokine production: relevance for the pathogenesis of shock and inflammation. Mol Med. 2002;8:283–289. [PMC free article] [PubMed] [Google Scholar]
- 146.Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: the role of poly (ADP-ribose) polymerase activation. Br J Pharmacol. 2002;135:1347–1350. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly (ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 148.Ungvari Z, Gupte SA, Recchia FA, Batkai S, Pacher P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol. 2005;3:221–229. doi: 10.2174/1570161054368607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pacher P, Vaslin A, Benko R, Mabley JG, Liaudet L, Hasko G, Marton A, Batkai S, Kollai M, Szabo C. A new, potent poly (ADP-ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther. 2004;311:485–491. doi: 10.1124/jpet.104.069658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Batkai S, Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Cravatt BF, Csiszar A, Ungvari ZI, Pacher P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression and apoptosis in mice lacking fatty acid amide hydrolase. Am J Physiol Heart Circ Physiol. 2007;293:H909–918. doi: 10.1152/ajpheart.00373.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kamp DW, Srinivasan M, Weitzman SA. Cigarette smoke and asbestos activate poly-ADP-ribose polymerase in alveolar epithelial cells. J Investig Med. 2001;49:68–76. doi: 10.2310/6650.2001.34092. [DOI] [PubMed] [Google Scholar]
- 152.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly (ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 153.Chiarugi A, Moskowitz MA. Poly (ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem. 2003;85:306–317. doi: 10.1046/j.1471-4159.2003.01684.x. [DOI] [PubMed] [Google Scholar]
- 154.Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly (ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275:40974–40980. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 155.Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem. 2001;276:45588–45597. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- 156.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 157.Mabley JG, Jagtap P, Perretti M, Getting SJ, Salzman AL, Virag L, Szabo E, Soriano FG, Liaudet L, Abdelkarim GE, Hasko G, Marton A, Southan GJ, Szabo C. Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm Res. 2001;50:561–569. doi: 10.1007/PL00000234. [DOI] [PubMed] [Google Scholar]
- 158.Sharp C, Warren A, Oshima T, Williams L, Li JH, Alexander JS. Poly ADP ribose-polymerase inhibitors prevent the upregulation of ICAM-1 and E-selectin in response to Th1 cytokine stimulation. Inflammation. 2001;25:157–163. doi: 10.1023/a:1011032313445. [DOI] [PubMed] [Google Scholar]
- 159.Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. Embo J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ha HC, Hester LD, Snyder SH. Poly (ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci U S A. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bergmann S, Siekmeier R, Mix C, Jaross W. Even moderate cigarette smoking influences the pattern of circulating monocytes and the concentration of sICAM-1. Respir Physiol. 1998;114:269–275. doi: 10.1016/s0034-5687(98)00098-x. [DOI] [PubMed] [Google Scholar]
- 162.Dovgan PS, Edwards JD, Zhan X, Wilde M, Agrawal DK. Cigarette smoking increases monocyte adherence to cultured endothelial cell monolayer. Biochem Biophys Res Commun. 1994;203:929–934. doi: 10.1006/bbrc.1994.2271. [DOI] [PubMed] [Google Scholar]
- 163.Lehr HA, Kress E, Menger MD, Friedl HP, Hubner C, Arfors KE, Messmer K. Cigarette smoke elicits leukocyte adhesion to endothelium in hamsters: inhibition by CuZn-SOD. Free Radic Biol Med. 1993;14:573–581. doi: 10.1016/0891-5849(93)90138-k. [DOI] [PubMed] [Google Scholar]
- 164.Kalra VK, Ying Y, Deemer K, Natarajan R, Nadler JL, Coates TD. Mechanism of cigarette smoke condensate induced adhesion of human monocytes to cultured endothelial cells. J Cell Physiol. 1994;160:154–162. doi: 10.1002/jcp.1041600118. [DOI] [PubMed] [Google Scholar]
- 165.Weber C, Erl W, Weber K, Weber PC. Increased adhesiveness of isolated monocytes to endothelium is prevented by vitamin C intake in smokers. Circulation. 1996;93:1488–1492. doi: 10.1161/01.cir.93.8.1488. [DOI] [PubMed] [Google Scholar]
- 166.Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 167.Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, Oury TD, Wolin MS, Kaley G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol. 2004;286:H2249–2256. doi: 10.1152/ajpheart.00854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hamilton CA, Brosnan MJ, McIntyre M, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37:529–534. doi: 10.1161/01.hyp.37.2.529. [DOI] [PubMed] [Google Scholar]
- 169.van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ungvari Z, Csiszar A, Kaley G. Vascular Inflammation in Aging. Herz. 2004;29:733–740. doi: 10.1007/s00059-004-2625-x. [DOI] [PubMed] [Google Scholar]
- 171.Labinskyy N, Csiszar A, Veress G, Stef G, Pacher P, Oroszi G, Wu J, Ungvari Z. Vascular dysfunction in aging: potential effects of resveratrol, an anti-inflammatory phytoestrogen. Curr Med Chem. 2006;13:989–996. doi: 10.2174/092986706776360987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Csiszar A, Labinskyy N, Orosz Z, Ungvari Z. Altered mitochondrial energy metabolism may play a role in vascular aging. Med Hypotheses. 2006;67:904–908. doi: 10.1016/j.mehy.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 173.Labinskyy N, Csiszar A, Orosz Z, Smith K, Rivera A, Buffenstein R, Ungvari Z. Comparison of endothelial function, O2.- and H2O2 production, and vascular oxidative stress resistance between the longest-living rodent, the naked mole rat, and mice. Am J Physiol. 2006;291:H2698–2704. doi: 10.1152/ajpheart.00534.2006. [DOI] [PubMed] [Google Scholar]
- 174.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective Effects of Anti-Tumor Necrosis Factor-{alpha} Treatment in Aging. Am J Pathol. 2007;170:388–698. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ungvari ZI, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith KE, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-kB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 176.Adler A, Messina E, Sherman B, Wang Z, Huang H, Linke A, Hintze TH. NAD (P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am J Physiol Heart Circ Physiol. 2003;285:H1015–1022. doi: 10.1152/ajpheart.01047.2002. [DOI] [PubMed] [Google Scholar]
- 177.Belmin J, Bernard C, Corman B, Merval R, Esposito B, Tedgui A. Increased production of tumor necrosis factor and interleukin-6 by arterial wall of aged rats. Am J Physiol. 1995;268:H2288–2293. doi: 10.1152/ajpheart.1995.268.6.H2288. [DOI] [PubMed] [Google Scholar]
- 178.Spaulding CC, Walford RL, Effros RB. Calorie restriction inhibits the age-related dysregulation of the cytokines TNF-alpha and IL-6 in C3B10RF1 mice. Mech Ageing Dev. 1997;93:87–94. doi: 10.1016/s0047-6374(96)01824-6. [DOI] [PubMed] [Google Scholar]
- 179.Helenius M, Hanninen M, Lehtinen SK, Salminen A. Aging-induced up-regulation of nuclear binding activities of oxidative stress responsive NF-kB transcription factor in mouse cardiac muscle. J Mol Cell Cardiol. 1996;28:487–498. doi: 10.1006/jmcc.1996.0045. [DOI] [PubMed] [Google Scholar]
- 180.Haendeler J, Hoffmann J, Diehl JF, Vasa M, Spyridopoulos I, Zeiher AM, Dimmeler S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ Res. 2004;94:768–775. doi: 10.1161/01.RES.0000121104.05977.F3. [DOI] [PubMed] [Google Scholar]
- 181.Godin DV, Nichols CR, Hoekstra KA, Garnett ME, Cheng KM. Alterations in aortic antioxidant components in an experimental model of atherosclerosis: a time-course study. Mol Cell Biochem. 2003;252:193–203. doi: 10.1023/a:1025548111491. [DOI] [PubMed] [Google Scholar]
- 182.Moon SK, Thompson LJ, Madamanchi N, Ballinger S, Papaconstantinou J, Horaist C, Runge MS, Patterson C. Aging, oxidative responses, and proliferative capacity in cultured mouse aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2001;280:H2779–2788. doi: 10.1152/ajpheart.2001.280.6.H2779. [DOI] [PubMed] [Google Scholar]
- 183.Mukhopadhyay P, Pacher P, Hu F, de Cabo R, Ballabh P, Ungvari ZI. Vasoprotective Effects of Resveratrol and SIRT1: Attenuation of Cigarette Smoke-induced Oxidative Stress and Pro-inflammatory Phenotypic Alterations. Am J Physiol Heart Circ Physiol. 2008 doi: 10.1152/ajpheart.00235.2008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]