

Abstract
Reactive oxygen and nitrogen species are overproduced in the cardiovascular system in response to the exposure to doxorubicin, a cardiotoxic anticancer compound. Oxidant-induced cell injury involves the activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) and pharmacological inhibition of PARP has recently been shown to improve myocardial contractility in doxorubicin-induced heart failure models. The current investigation, by utilizing an isolated perfused heart system capable of beat-to-beat intracellular calcium recording, addressed the following questions: (1) is intracellular calcium handling altered in hearts of rats after 6-week doxorubicin treatment, under baseline conditions, and in response to oxidative stress induced by hydrogen peroxide exposure in vitro; and (2) does pharmacological inhibition of PARP with the phenanthridinone-based PARP inhibitor PJ34 affect the changes in myocardial mechanical performance and calcium handling in doxorubicin-treated hearts under normal conditions and in response to oxidative stress. The results showed a marked elevation in intracellular calcium in the doxorubicin-treated hearts which was normalized by pharmacological inhibition of PARP. PARP inhibition also prevented the myocardial contractile disturbances and calcium overload that developed in response to hydrogen peroxide in the doxorubicin-treated hearts. We conclude that PARP activation contributes to the development of the disturbances in cellular calcium handling that develop in the myocardium in response to prolonged doxorubicin exposure.
Keywords: Peroxynitrite, Poly(ADP-ribose) polymerase (PARP), Adriamycin, Heart failure, Superoxide, Calcium
1. Introduction
Overactivation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) represents a novel mechanism of cell dysfunction in various pathophysiological conditions associated with the oxidative stress. Evidence is accumulating that in the myocardium, PARP activation is associated with ischemia-reperfusion injury, cardiopulmonary bypass, heart transplantation, circulatory shock, diabetes mellitus and side effects of a variety of drugs overviewed in [1,2].
Cardiotoxicity is a prominent and dose-limiting side effect of the anticancer drug doxorubicin. The cardiotoxic effects of doxorubicin have been extensively investigated. The effects on mechanical performance of the heart have been described; a variety of pathogenetic mechanisms have also been elucidated (including mitochondrial dysfunction, free radical production, changes in iron handling) and the cellular and molecular mechanisms of the myocyte apoptosis and necrosis have been characterized in much detail overviewed in [3-6]. Recent work demonstrated the close relationship between oxidative and nitrosative stress and PARP activation in doxorubicin-treated animals as well as in humans undergoing doxorubicin anticancer therapy [7-9]. Since proper calcium handling is of utmost importance for adequate cardiac mechanical performance, it remains to be explored whether alterations in cardiac myocyte calcium homeostasis are involved in doxorubicin-induced cardiomyopathy.
The current investigation, by utilizing an isolated, perfused heart system capable of beat-to-beat intracellular calcium recording, addresses the following questions: (1) is intracellular calcium handling altered in hearts of rats after 6-week doxorubicin treatment, under baseline conditions, and in response to oxidative stress induced by hydrogen peroxide exposure in vitro; and (2) does pharmacological inhibition of PARP mitigate or prevent the changes in myocardial mechanical performance and calcium handling? To inhibit the catalytic activity of PARP, the potent phenanthridinone-based PARP inhibitor compound, PJ34 was used [10].
2. Materials and methods
2.1. General
Rats were treated with a weekly subcutaneous injection of doxorubicin for six weeks. The PARP inhibitor PJ34 was administered in the drinking water of the animals throughout the experimental period. Six weeks after the onset of treatment, the hearts were excised and perfused according to Langendorff with Krebs–Henseleit solution. Measurement of left ventricular pressure was accomplished via introduction of a balloon into the left ventricle and coronary flow was obtained with an ultrasonic transducer. Indo-1 fluorescent dye was loaded into the heart for the determination of intracellular calcium concentration. Cyclic changes of intracellular free calcium levels (Cai2+ transient) were measured using surface fluorometry. The hearts were subjected to an oxidative stress with the application of H2O2 (600 μM). Hemodynamic performance and functional parameters of intracellular calcium cycling were assessed.
2.2. Animal used—induction of cardiomyopathy
Male Sprague-Dawley rats ranging in body weight from 100 to 120 g were maintained in an animal facility with a 12-h light:12-h dark cycle with free access to food and water. Experiments on animals followed the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and the study protocol was approved by the Laboratory Animals Committee of the Semmelweis University of Budapest.
Animals were allotted to four different experimental groups. Rats were treated with 2.5 mg/kg doxorubicin injection subcutaneously [11] once a week for six weeks (DOX Group: n = 5). Age-matched controls received saline only (Control Group: n = 6). In the remaining two experimental groups, doxorubicin-treated rats and their age-matched controls were given 20 mg/BW PJ34, dissolved in water to drink during the treatment period (DOX-PJ Group: n = 5, Control-PJ Group: n = 6, respectively). There were no differences in the amount of PJ34 intake between groups. The current treatment protocol has previously been shown to achieve maximal inhibition of myocardial PARP activity in vivo [12,13].
2.3. Langendorff heart preparation
The general procedure to measure hemodynamic parameters and fluorescence signals in a beating heart preparation was outlined previously in detail [14].
Six weeks after the initiation of doxorubicin and drug treatments, the animals were anesthetized by 40 mg/kg i.p. pentobarbital (Nembutal–Sanofi, Budapest, Hungary). Hearts were quickly removed and immediately rinsed in cold Krebs–Henseleit solution void of CaCl2 and subsequently mounted on a Langendorff perfusion apparatus (Experimetria Ltd., Budapest, Hungary). Retrograde aortic perfusion was initiated with a modified Krebs–Henseleit solution containing 118 mM NaCl, 4.3 mM KCl, 25 mM NaHCO3, 1.2 mM MgSO4, 1.2 mM KH2PO4, 0.5 mM NaEDTA, 2.0 mM CaCl2, 11 mM glucose, 5 mM pyruvate (all chemicals purchased from Sigma, Budapest, Hungary) which was equilibrated with 95% O2 and 5% CO2. The temperature of the buffer was maintained at 37 °C and pH was adjusted to 7.4. Perfusion pressure was set at 70 mmHg. Throughout the experiments, the hearts were immersed in a bath of Krebs–Henseleit solution kept at 37 °C.
Coronary flow was measured with an ultrasonic transducer (T-106, Transonic Systems Inc., Ithaca NY, USA) inserted into the perfusion line. Left ventricular pressure was measured using a latex balloon inserted through the mitral valve orifice into the left ventricle and connected to a pressure transducer (Electromedics Inc., Englewood, CO, USA). Left ventricular end-diastolic pressure (EDPlv) was set between 5 and 10 mmHg by adjusting the balloon volume. The left ventricular pressure (Plv) was assessed to determine the systolic, diastolic and developed (systolic minus diastolic) pressures and heart rate. The maximal positive and negative first derivative of pressure (+dPlv/dtmax, −dPlv/dtmax) were obtained as indices of inotropic and lusitropic state of the heart, respectively.
2.4. Indo-1 AM loading and fluorescence measurement
Hearts were allowed to equilibrate for 30 min before loading of the dye. Two hundred and fifty micrograms of Indo-1 AM was solubilized in a mixture of 0.25 ml dimethylsulfoxide and 0.08 g pluronic F-127 and added to 40 ml Krebs–Henseleit solution containing 118 mM NaCl, 5.5 mM KCl, 1.2 mM MgSO4, 25 mM HEPES, 11 mM glucose, 5 mM Na pyruvate, and 1.5 mM CaCl2. The final concentration of Indo-1 AM was 6.25 μM. The hearts were perfused in a recirculating mode with a peristaltic pump (Cole-Palmer Co., Chicago, IL, USA) connected to the aortic cannula. After 20 min of loading, fluorescence signals reached maximum levels and a washout period started in normal Langendorff mode for 15 min with the modified Krebs–Henseleit solution to eliminate fluorescence originating from Indo-1 AM present in the extracellular compartment.
Fluorescence excitation was provided by a 100 W d.c. mercury arc lamp (Oriel, Stratford, CT, USA). Illumination was filtered at 340 nm and directed through a randomized multifurcated fiber optic light guide onto a circular region of the epicardium of the left ventricular wall. The emitted light was detected by photomultiplier tubes (Electron Tubes Ltd., Ruislip, Middlesex, England). Fluorescence emission was recorded at 400 nm (Cai2+-bound dye) and 506 nm (Cai2+-free dye). Reflectance was also detected at 340 nm for the estimation of non-specific changes in tissue density (optical artifacts). To minimize UVexposure of the tissue and photo-bleaching of the intracellular dye, a shutter positioned in front of the excitation light was opened for approximately 1 min in every 5 min. Both the fluorescence and reflectance signals together with the hemodynamic parameters were recorded and stored with a multichannel AD converter (Hemosys, Experimetria, Budapest, Hungary) coupled to an IBM based computer [14].
2.5. Data analysis and interpretation
The ratio of the fluorescence signals at 400 and 506 nm is proportional to the cytoplasmic free calcium level. The individual fluorescence signals were corrected for closed shutter background and tissue autofluorescence of the unloaded tissue. Previously, we have shown that using this loading procedure Indo-1 is not sequestered into the mitochondria, hence, the apparent fluorescence signal is mainly of cytoplasmic origin [14]. Cai2+ concentration was calculated with the formula determined by Grynkiewicz et al. [15]. The Indo-1 dissociation constant (Kd) for calcium was determined previously at 844 nM [16]. The fluorescent ratios at zero Cai2+ (Rmin) and at saturating Cai2+ (Rmax) were determined in separate experiments. Two control hearts loaded with Indo-1 were treated with 20 μm; BAPTA-AM to define Rmin and two other control hearts were infused with 1 μM 4-bromo-calcium-ionophore (A23187) to determine Rmax. The calcium transient was assessed to determine systolic and diastolic Cai2+ and Cai2+ amplitude. Maximal rates of rise of Cai2+ (+dCai2+/dtmax) and decline of Cai2+ (−dCai2+/dtmax) were obtained as indices for the rate of calcium release from the sarcoplasmic reticulum (SR) and sequestration back into the SR, respectively.
2.6. Perfusion protocol
After loading of the dye and 15 min washout, the fluorescence signals were recorded in the “basal” situation. Thereafter, the hearts were subjected to an oxidative stress by adding 600 μM H2O2 to the perfusion medium. To this end, the H2O2 solution was infused into the aortic line via a side port. Hemodynamic parameters and fluorescence signals were recorded about 2 min after the start of infusion. H2O2 is an oxidative agent and as such, it also may react directly or indirectly with the calcium tracer, Indo-1, causing deterioration of the fluorescence signal. As a consequence of this, a few hearts not suitable for analysis of the fluorescence signal due to low signal/noise ratios were omitted from the groups subjected to H2O2 challenge because in these hearts Indo-1 fluorescence quenched within the first 2–3 min of H2O2 infusion (Control, n = 4; DOX, n = 5; Control-PJ, n = 3; DOX-PJ, n = 4). The hearts were removed from the cannula at the end of the experiments, wiped off with a paper towel and weighed to determine heart mass.
2.7. Reagents
All reagents were obtained from Sigma–Aldrich (St. Louis, MO and Budapest, Hungary), unless indicated otherwise. The potent, novel, water-soluble phenanthridinone derivative PARP inhibitor, PJ34—the hydrochloride salt of N-(-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide—was synthesized as described [10].
2.8. Statistics
All data are expressed as mean value ± S.D. Statistical analysis was carried out using one-way analysis of variance followed by Fisher post hoc test. A statistically significant difference of p < 0.05 was accepted throughout. Effects of H2O2 infusion were analyzed by Student’s t-test and Fisher post hoc test as appropriate (p < 0.05).
3. Results
Six weeks of DOX treatment induced the expected changes in the animals [9,17]. As indicated in Table 1, the body and heart mass was markedly reduced as compared to their age-matched counterparts. Heart weight/body mass ratio of the animals was unaffected by DOX treatment.
Table 1.
General effects of DOX treatment
| Control (n = 6) | 6-week DOX (n = 5) | 6-week DOX-PJ (n = 5) | |
|---|---|---|---|
| Body mass (BM) (g) | 366 ± 30 | 285 ± 36* | 270 ± 18* |
| Heart mass (HM) (g) | 1.33 ± 0.16 | 1.05 ± 0.19* | 1.05 ± 0.16* |
| HM/BM (g/g) | 0.0036 ± 0.0005 | 0.0037 ± 0.0005 | 0.0039 ± 0.0004 |
Control vs. DOX or DOX-PJ. Data refer to mean and S.D., PJ34 treatment per se did not affect the above parameters (Control vs. Control-PJ, data not shown).
p < 0.05.
3.1. Cardiac mechanical performance
Treatment with DOX for six weeks did not induce significant changes in the hemodynamic parameters of the Langendorff-perfused hearts under basal conditions (Fig. 1, Table 2).
Fig. 1.
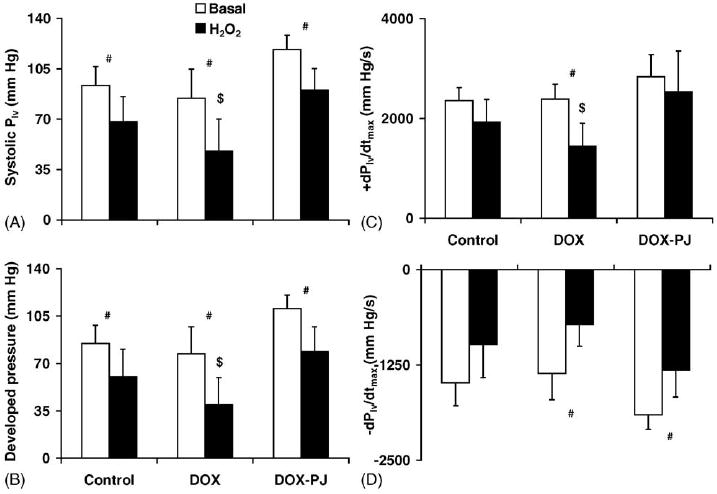
Hemodynamic changes in hearts of DOX-treated rats. The figure shows the contractile activity of the hearts in the different experimental groups: (A) peak systolic left ventricular pressure; (B) developed pressure; (C) +dPlv/dtmax; and (D) −dPlv/dtmax. #Indicates statistically significant (p < 0.05) difference between the basal state and the oxidative stress (H2O2). $Indicates statistically significant (p < 0.05) difference between DOX and DOX-PJ groups during oxidative stress. PJ34 treatment per se induced no change in the hemodynamic parameters of the perfused heart (Control vs. Control-PJ, data not shown).
Table 2.
Hemodynamic parameters in isolated Control, DOX and DOX-PJ hearts under basal conditions
| Control (n = 6) | 6-week DOX (n = 5) | 6-week DOX-PJ (n = 5) | |
|---|---|---|---|
| Diastolic Plv (mmHg) | 8.4 ± 0.6 | 7.4 ± 0.2 | 7.9 ± 0.5 |
| Systolic Plv (mmHg) | 93 ± 13 | 84 ± 20 | 118 ± 10 |
| Developed pressure (mmHg) | 85 ± 13 | 77 ± 20 | 110 ± 10 |
| Heart rate (bpm) | 291 ± 21 | 297 ± 18 | 285 ± 12 |
| Coronary flow (ml/min) | 18 ± 3 | 13 ± 2* | 16 ± 2 |
| CF/HM (ml/min/g) | 13.4 ± 2.2 | 12.8 ± 1.6 | 15.2 ± 1.3 |
| +dPlv/dtmax (mmHg/s) | 2363 ± 255 | 2387 ± 301 | 2837 ± 442 |
| −dPlv/dtmax (mmHg/s) | −1483 ± 303 | −1363 ± 344 | −1900 ± 194 |
Control vs. DOX or DOX-PJ. Data refer to mean and S.D. CF refers to coronary flow. Plv and HM refers to left ventricular pressure and heart mass, respectively. PJ34 treatment per se did not affect the above hemodynamic parameters (Control vs. Control-PJ, data not shown).
p < 0.05.
Two minutes of oxidative stress (600 μM H2O2) impaired hemodynamic performance of the hearts. The effects of the oxidant were found to be more pronounced in the hearts of the animals that had previously received doxorubicin (Fig. 1). Pre-treatment of the animals with PJ34 provided protection against the deleterious effect of H2O2. The protective effects of PJ34 were apparent against all hemodynamic parameters measured in the current study; both inotropic and lusitropic states of the heart were maintained close to control levels after pharmacological inhibition of PARP (Fig. 1).
3.2. Intracellular calcium handling
Typical Cai2+ tracings are shown in Fig. 2. It can be observed that DOX treatment increased both the end-diastolic level and amplitude of the Cai2+ transient with a simultaneous reduction of peak systolic left ventricular pressure.
Fig. 2.
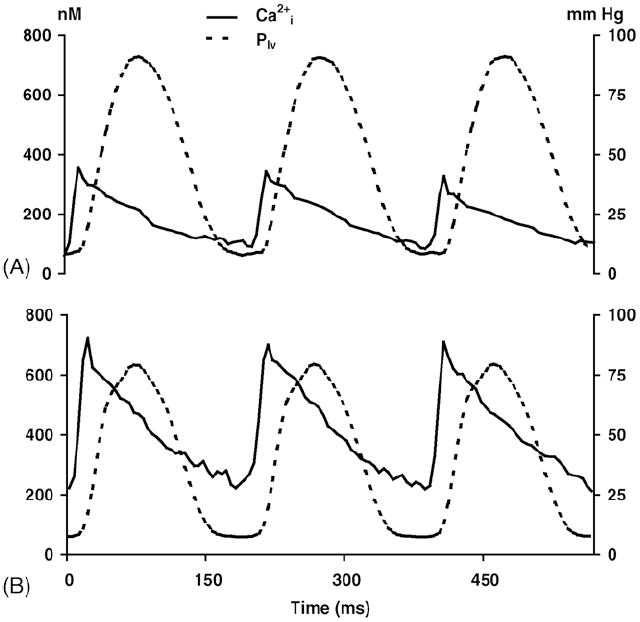
Cyclic changes of Cai2+ and left ventricular pressure. Representative tracings of Cai2+ transient and left ventricular pressure (Plv) of (A) an intact, perfused control rat heart and (B) DOX-treated heart, both under basal conditions. Note that the peak of the calcium transient precedes the peak of the left ventricular pressure by 30–40 ms.
One of the most prominent findings in the current study is the profound elevation of end-diastolic Cai2+ in the hearts of the DOX-treated animals. The end-diastolic calcium concentration doubled from the control 109 ± 22 to 222 ± 73 nM in the hearts of the animals subjected to DOX (Fig. 3). Pharmacological inhibition of PARP prevented this elevation of end-diastolic Cai2+ (DOX-PJ group). The oxidative challenge elicited by H2O2 triggered a marked additional elevation in the end-diastolic Cai2+ in the hearts of the DOX-treated animals. This increase was significantly mitigated by pre-treatment of the animals with PJ34 (DOX-PJ versus DOX group, end-diastolic Cai2+ level, 226 ± 75 nM versus 548 ± 298 nM, respectively).
Fig. 3.
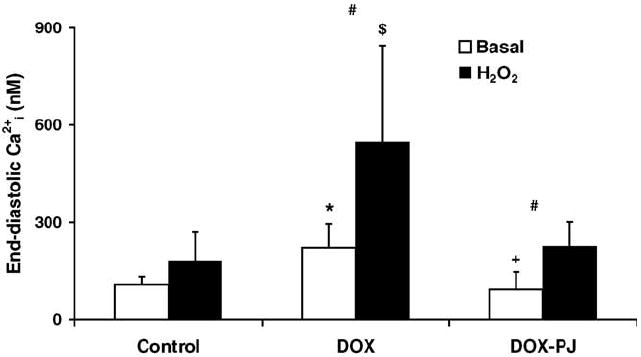
Changes in end-diastolic Cai2+ in DOX-induced cardiomyopathy. The figure shows changes in end-diastolic Cai2+ levels, prior to (basal) and during oxidative stress (H2O2) in the different experimental groups. *Indicates statistically significant (p < 0.05) difference between DOX and Control groups under basal condition. +Indicates statistically significant (p < 0.05) difference between DOX and DOX-PJ groups under basal condition. #Indicates statistically significant (p < 0.05) difference between basal state and oxidative stress (H2O2). $Indicates statistically significant (p < 0.05) difference between DOX and Control, or DOX-PJ groups during oxidative stress. PJ34 treatment per se did not alter the end-diastolic level of Cai2+ in the perfused heart (Control vs. Control-PJ, data not shown).
The analysis of the Cai2+ transients revealed that the systolic levels and, hence, the amplitude increased in the hearts of DOX-treated rats (Fig. 4A,B). Also, the release of calcium from SR was accelerated in the DOX group, as indicated by the enhanced +dCai2+/dtmax (Fig. 4C). Oxidative stress by H2O2 did not cause any significant alterations in the dynamic parameters of the Cai2+ transient in any of the experimental groups.
Fig. 4.
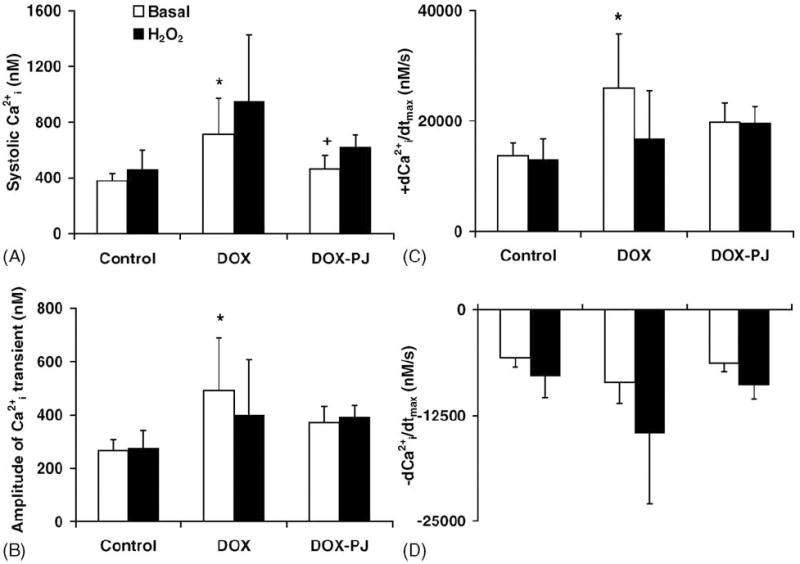
Changes in the dynamic parameters of Cai2+ transient in DOX-induced cardiomyopathy. The figure shows parameters calculated from the Cai2+ transients of the hearts in the different experimental groups: (A) peak systolic Cai2+; (B) amplitude of Cai2+; (C) +dCai2+/dtmax; and (D) −dCai2+/dtmax. *Indicates statistically significant (p < 0.05) difference between DOX and Control groups under basal condition. +Indicates statistically significant (p < 0.05) difference between DOX and DOX-PJ groups under basal condition. PJ34 treatment per se induced no change in the dynamic parameters of the Cai2+ transient in the perfused heart (Control vs. Control-PJ, data not shown).
When sensitivity of the contractile machinery was estimated from the ratio of peak systolic pressure (Plv) and peak systolic Cai2+ there was a statistically significant (p < 0.05) difference suppressive effect of doxorubicin which was prevented by the PARP inhibitor (Fig. 5).
Fig. 5.
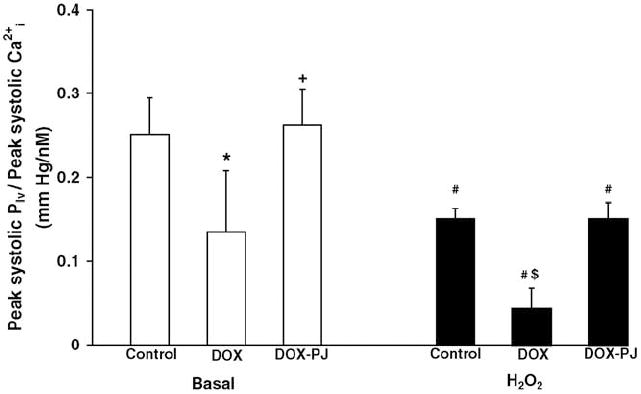
Calcium sensitivity of the myocardium during systole. Sensitivity of the contractile machinery was calculated as the ratio of peak systolic pressure (Plv) and peak systolic Cai2+. *Indicates statistically significant (p < 0.05) difference between DOX and Control groups under basal condition. +Indicates statistically significant (p < 0.05) difference between DOX and DOX-PJ groups under basal condition. #Indicates statistically significant (p < 0.05) difference between basal state and oxidative stress (H2O2). $Indicates statistically significant (p < 0.05) difference between DOX on the one hand and Control and DOX-PJ groups on the other during oxidative stress. PJ34 treatment per se induced no change in the Cai2+ sensitivity of the myocardium (Control vs. Control-PJ, data not shown).
4. Discussion
There are multiple mechanisms of doxorubicin-induced myocardial functional disturbances leading to cardiomyopathy overviewed in [7,8]. However, it appears that oxidative and nitrosative stress plays a clear role in this process. Doxorubicin undergoes redox cycling to generate free radicals that are responsible for mediating the various cytopathologies associated. Pharmacological neutralization of oxidant and free radical actions (via iron chelators, antioxidant vitamins, peroxynitrite decomposition catalyst and inducible nitric oxide synthase inhibitors) have been shown to improve cardiac function and protect against doxorubicin-induced cardiomyopathy [7,8,17]. The downstream intracellular mechanisms of doxorubicin-induced cell death and myocardial contractile dysfunction are less understood. Matrix metalloproteinase activation, due to reactive oxidants such as peroxynitrite, may be involved [18]. There is also evidence that doxorubicin-triggered oxidants promote mitochondrial dysfunction and activate a variety of cell death pathways including cytochrome c release, caspase-3 activation [19,20], DNA strand breakage [9,19] as well as up-regulation of various pro-inflammatory signal transduction pathways and transcriptional regulation of multiple regulatory and inflammatory proteins [21].
One of the more recently identified pathways of doxorubicin cardiotoxicity is related to PARP activation [9]. PARP is an abundant nuclear enzyme of eukaryotic cells with multiple regulatory functions [1]. When activated by DNA single-strand breaks, PARP initiates an energy consuming cycle by transferring ADP ribose units from NAD+ to nuclear proteins resulting in rapid depletion of the intracellular NAD+ and ATP pools, slowing the rate of glycolysis and mitochondrial respiration eventually leading to cellular dysfunction and death [1,2]. Overactivation of PARP represents an important mechanism of tissue damage in various pathophysiological conditions associated with increased oxidative stress including myocardial reperfusion injury [1,2] as well as drug-induced cardiomyopathy [9,22].
It is well established that doxorubicin induces profound disturbances in myocardial calcium handling [8,21,23]. The focus of the current study was to investigate whether the PARP pathway regulates the alterations in calcium handling provoked by doxorubicin treatment. We have utilized a 6-week doxorubicin treatment protocol which represents a relatively early stage in the sequel of doxorubicin-mediated pathophysiological events. At this point, contractile function has not yet significantly declined in the isolated perfused hearts ex vivo under basal conditions but calcium handling was already impaired (as evidenced by increased intracellular end-diastolic calcium concentration), indicative of early-stage myocardial calcium overload. While this early-stage increase in myocardial intracellular calcium does not necessarily or immediately impair contractile function, it has been shown to contribute to the late-stage morphological and functional alterations associated with doxorubicin cardiomyopathy [21,23]. Furthermore, the estimated Cai2+ sensitivity of the contractile machinery is significantly decreased in the doxorubicin-treated hearts. In Fig. 5, the steady-state relationship between Cai2+ and force (peak systolic pressure) was characterized by calculating the ratio of peak systolic Plv and peak systolic Cai2+. Even without manifest signs of cardiac failure, doxorubicin treatment reduced the calcium sensitivity of the myocardium by about 50% which was almost completely reversed by PJ34 treatment. The exact pathological mechanism of the doxorubicin-induced reduction in Cai2+ sensitivity has not yet been elucidated.
Our results show that in the early stage of doxorubicin-induced heart failure investigated in the current study, “basal” contractile function of the isolated Langendorff-perfused heart was not overtly impaired. These findings do not corroborate the in vivo observations showing significant depression of cardiac function (see also [17,24,25]). The lack of decline in contractile performance under basal conditions in our study may be explained by the fact that these Langendorff-perfused heart preparations are less challenged than the in vivo hearts (no external work performed), hampering a proper comparison between the in vitro and in vivo setting. However, the doxorubicin-treated hearts in our study became markedly sensitive to the deleterious effect of hydrogen peroxide. A possible explanation for this finding is that the prolonged production of reactive oxidants and free radicals in the doxorubicin-treated heart diminishes the oxygen free radical scavenging capacity, i.e., superoxide dismutase (SOD), glutathione peroxidase (GSHpx) and catalase activity, and hence suppresses endogenous antioxidant levels (such as glutathione) [26,27]. Therefore, it is conceivable that in the heart, the vital antioxidant system is depressed and, therefore, the hearts became more sensitive to an acute oxidative insult. Similar mechanisms may also contribute to the increased susceptibility of patients with doxorubicin therapy to cardiovascular events.
The current results demonstrate that the PARP system plays a regulatory role in the alterations in doxorubicin-induced cellular calcium handling. Both the “basal” intracellular calcium elevation as well as the hydrogen peroxide induced acute enhancement of the intracellular calcium levels were mitigated in the hearts of animals that have received the potent, competitive inhibitor of PARP activity (PJ34) during the in vivo doxorubicin treatment. PARP has two fundamental mechanisms of actions influencing both cellular function and pathophysiological events as overviewed in [1]. The first set of mechanism interferes with the increased catalytic activation due to DNA strand breaks followed by enhanced consumption of cellular NAD+ and ATP. By doing so, it unfavorably competes with other cellular ATP-requiring processes. The second mechanism relates to the regulatory action of PARP on the expression of a variety of genes and gene products. With respect to the current results (ability of PARP to promote the disturbances in cellular calcium handling), either of the above two sets of mechanisms or the combination of the two may be involved. It is well established that doxorubicin impairs SERCA2a-mediated calcium re-sequestration into the SR which is also an energy-requiring process. It is also known that doxorubicin leads to depletion in cellular NAD+ and ATP levels [28]. Thus, it is conceivable that diminished myocardial energetic status induces enhanced calcium influx into the intracellular space through “leaky” cell membrane and/or impairs the ability of the cardiac muscle cells to re-sequester calcium. This way, PARP inhibition may exert its beneficial effects through restoration of calcium re-sequestration via normalization of cellular energetic pools and restoration of the function of calcium pumps. It is also conceivable that PARP, on a transcriptional level, is involved in the regulation of proteins essential for myocardial calcium handling in the doxorubicin-treated hearts. For instance, there is evidence that the transcriptional regulation and the expression of multiple proteins involved in myocardial calcium handling is altered in response to doxorubicin, including down-regulation of the SR Ca2+-ATPase (SERCA2a), and the ryanodine receptor (RYR2), and up-regulation of the sarcolemmal calcium channel (dihydropyridine receptor, DHPR) [29-31].
It is important to stress that the extent to which administration of a PARP inhibitor over a longer time period prevents the development of doxorubicin cardiotoxicity is currently unknown. The longest in vivo studies so far have been several weeks up to a month of DOX in the presence of PARP inhibitors [9,17]. These studies primarily investigated functional alterations such as myocardial contractility (as opposed to morphological alterations and pathological signs of cardiomyopathy which tend to develop over longer periods of time). Clearly, longer term studies are required. Nevertheless, a number of studies indicate that PARP inhibitors beneficially affect remodeling in chronic models of heart failure (even though these were induced by various other interventions such as LAD ligation or ascending aortic banding) [12,32]. Overall, it is conceivable that PARP inhibition would also beneficially affect chronic morphological and functional alterations in DOX hearts, but this issue remains to be directly tested in future studies.
The most important novel findings of the present study include: (1) the demonstration of an increase in end-diastolic calcium levels in doxorubicin-treated hearts; (2) development of a decline in calcium sensitivity of the contractile machinery in the diseased hearts; (3) the enhanced susceptibility of the doxorubicin-treated hearts towards a subsequent acute oxidative stress; and (4) the protective effects of PJ34, strongly suggesting that the activation of nuclear enzyme PARP is involved in the functional alterations of the doxorubicin-treated heart. Overall, the results of the current study are consistent with the notion that reactive oxidant species and the PARP pathway plays a pathogenetic role in the development of doxorubicin-induced cardiac dysfunction. Normalization of cellular calcium handling appears to be an additional mode of the cardioprotective effects of PARP inhibitors.
Acknowledgments
This work was supported by grants from OTKA (Hungarian Research Fund, T038121, T047095 and T042969) and the National Institutes of Health (R01 HL 59266) and by the Socrates program of the European Community and the Department of Biomedical Engineering, Eindhoven University of Technology, Eindhoven, The Netherlands.
References
- 1.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 2.Szabo G, Liaudet L, Hagl S, Szabo C. Poly(ADP-ribose) polymerase activation in the reperfused myocardium. Cardiovasc Res. 2004;61:471–80. doi: 10.1016/j.cardiores.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 3.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 4.Minotti G, Recalcati S, Menna P, Salvatorelli E, Corna G, Cairo G. Links doxorubicin cardiotoxicity and the control of iron metabolism: quinone-dependent and independent mechanisms. Methods Enzymol. 2004;378:340–61. doi: 10.1016/S0076-6879(04)78025-8. [DOI] [PubMed] [Google Scholar]
- 5.Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002;234–235:119–24. [PubMed] [Google Scholar]
- 6.De Beer EL, Bottone AE, Voest EE. Doxorubicin and mechanical performance of cardiac trabeculae after acute and chronic treatment: a review. Eur J Pharmacol. 2001;415:1–11. doi: 10.1016/s0014-2999(01)00765-8. [DOI] [PubMed] [Google Scholar]
- 7.Fogli S, Nieri P, Breschi MC. The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. Fed Am Soc Exp Biol J. 2004;18:664–75. doi: 10.1096/fj.03-0724rev. [DOI] [PubMed] [Google Scholar]
- 8.Singal PK, Iliskovic N, Li T, Kumar D. Adriamycin cardiomyopathy: pathophysiology and prevention. Fed Am Soc Exp Biol J. 1997;11:931–6. doi: 10.1096/fasebj.11.12.9337145. [DOI] [PubMed] [Google Scholar]
- 9.Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300:862–7. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 10.Jagtap P, Soriano FG, Virag L, Liaudet L, Mabley J, Szabo E, et al. Novel phenanthridinone inhibitors of poly (adenosine 5’-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit Care Med. 2002;30:1071–82. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 11.Solem LE, Heller LJ, Wallace KB. Dose-dependent increase in sensitivity to calcium-induced mitochondrial dysfunction and cardiomyocyte cell injury by doxorubicin. J Mol Cell Cardiol. 1996;28:1023–32. doi: 10.1006/jmcc.1996.0095. [DOI] [PubMed] [Google Scholar]
- 12.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–16. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 13.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–21. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 14.Ivanics T, Miklos Z, Dezsi L, Ikrenyi K, Toth A, Roemen TH, et al. Concomitant accumulation of intracellular free calcium and arachidonic acid in the ischemic-reperfused rat heart. Mol Cell Biochem. 2001;226:119–28. doi: 10.1023/a:1012739722150. [DOI] [PubMed] [Google Scholar]
- 15.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 16.Bassani JW, Bassani RA, Bers DM. Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophys J. 1995;68:1453–60. doi: 10.1016/S0006-3495(95)80318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virag L, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 18.Bai P, Mabley JG, Liaudet L, Virag L, Szabo C, Pacher P. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncol Rep. 2004;11:505–8. [PubMed] [Google Scholar]
- 19.Green PS, Leeuwenburgh C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochim Biophys Acta. 2002;1588:94–101. doi: 10.1016/s0925-4439(02)00144-8. [DOI] [PubMed] [Google Scholar]
- 20.Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C. Doxorubicin treatment in vivo causes cytochrome c release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 2002;62:4592–8. [PubMed] [Google Scholar]
- 21.Takahashi S, Denvir MA, Harder L, Miller DJ, Cobbe SM, Kawakami M, et al. Effects of in vitro and in vivo exposure to doxorubicin (adriamycin) on caffeine-induced Ca2+ release from sarcoplasmic reticulum and contractile protein function in ‘chemically-skinned’ rabbit ventricular trabeculae. Jpn J Pharmacol. 1998;76:405–13. doi: 10.1254/jjp.76.405. [DOI] [PubMed] [Google Scholar]
- 22.Szabados E, Fischer GM, Toth K, Csete B, Nemeti B, Trombitas K, et al. Role of reactive oxygen species and poly-ADP-ribose polymerase in the development of AZT-induced cardiomyopathy in rat. Free Radic Biol Med. 1999;26:309–17. doi: 10.1016/s0891-5849(98)00199-3. [DOI] [PubMed] [Google Scholar]
- 23.Rossi F, Filippelli W, Russo S, Filippelli A, Berrino L. Cardiotoxicity of doxorubicin: effects of drugs inhibiting the release of vasoactive substances. Pharmacol Toxicol. 1994;75:99–107. doi: 10.1111/j.1600-0773.1994.tb00330.x. [DOI] [PubMed] [Google Scholar]
- 24.Sacco G, Bigioni M, Evangelista S, Goso C, Manzini S, Maggi CA. Cardioprotective effects of zofenopril, a new angiotensin-converting enzyme inhibitor, on doxorubicin induced cardiotoxocity in the rat. Eur J Pharmacol. 2001;414:71–8. doi: 10.1016/s0014-2999(01)00782-8. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz ER, Pollick C, Dow J, Patterson M, Birnbaum Y, Kloner RA. A small animal model of non-ischemic cardiomyopathy and its evaluation by transthoracic echocardiography. Cardiovasc Res. 1998;39:216–23. doi: 10.1016/s0008-6363(98)00009-1. [DOI] [PubMed] [Google Scholar]
- 26.Luo X, Evrovsky Y, Cole D, Trines J, Benson LN, Lehotay DC. Doxorubicin-induced acute changes in cytotoxic aldehydes, antioxidant status and cardiac function in the rat. Biochim Biophys Acta. 1997;1360:45–52. doi: 10.1016/s0925-4439(96)00068-3. [DOI] [PubMed] [Google Scholar]
- 27.Mohamed HE, El-Swefy SE, Hagar HH. The protective effect of glutathione administration on adriamycin-induced acute cardiac toxicity in rats. Pharmacol Res. 2000;42:115–21. doi: 10.1006/phrs.1999.0630. [DOI] [PubMed] [Google Scholar]
- 28.Vidal RF, Eksborg S, Sundberg M, Carlberg M, Elfsson B, Andersson BS. Doxorubicin- and daunorubicin-induced energy deprivation and nucleotide degradation in isolated cardiomyocytes. Toxicology. 1996;114:1–10. doi: 10.1016/s0300-483x(96)03410-5. [DOI] [PubMed] [Google Scholar]
- 29.Huang XM, Zhu WH, Kang ML. Study on the effect of doxorubicin on expressions of genes encoding myocardial sarcoplasmic reticulum Ca2+ transport proteins and the effect of taurine on myocardial protection in rabbits. J Zhejiang Univ Sci. 2003;4:114–20. doi: 10.1631/jzus.2003.0114. [DOI] [PubMed] [Google Scholar]
- 30.Gambliel HA, Burke BE, Cusack BJ, Walsh GM, Zhang YL, Mushlin PS, et al. Doxorubicin and C-13 deoxydoxorubicin effects on ryanodine receptor gene expression. Biochem Biophys Res Commun. 2002;291:433–8. doi: 10.1006/bbrc.2002.6380. [DOI] [PubMed] [Google Scholar]
- 31.Boucek RJ, Jr, Miracle A, Anderson M, Engelman R, Atkinson J, Dodd DA. Persistent effects of doxorubicin on cardiac gene expression. J Mol Cell Cardiol. 1999;31:1435–46. doi: 10.1006/jmcc.1999.0972. [DOI] [PubMed] [Google Scholar]
- 32.Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, et al. Poly(ADP-ribose) polymerase promotes cardiac remodeling, contractile failure and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther Nov. 2004 doi: 10.1124/jpet.104.077164. Epub ahead of print. [DOI] [PubMed] [Google Scholar]