

Abstract
Polychlorinated biphenyls (PCBs) are persistent environmental pollutants implicated in the development of pro-inflammatory events critical in the pathology of atherosclerosis and cardiovascular disease. PCB exposure of endothelial cells results in increased cellular oxidative stress, activation of stress and inflammatory pathways leading to increased expression of cytokines and adhesion molecules and ultimately cell death, all of which can lead to development of atherosclerosis. To date no studies have been performed to examine the direct effects of PCB exposure on the vasculature relaxant response which if impaired may predispose individuals to hypertension, an additional risk factor for atherosclerosis. Overactivation of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP) following oxidative/nitrosative stress in endothelial cells and subsequent depletion of NADPH has been identified as a central mediator of cellular dysfunction. The aim therefore was to investigate whether 2,2′,4,6,6′-pentachlorobiphenyl (PCB 104) directly causes endothelial cell dysfunction via increased oxidative stress and subsequent overactivation of PARP. Exposure of ex vivo rat aortic rings to PCB 104 impaired the acetylcholine-mediated relaxant response, an effect that was dependent on both concentration and exposure time. In vitro exposure of mouse endothelial cells to PCB 104 resulted in increased cellular oxidative stress through activation of the cytochrome p450 enzyme CYP1A1 with subsequent overactivation of PARP and NADPH depletion. Pharmacological inhibition of CYP1A1 or PARP protected against the PCB 104-mediated endothelial cell dysfunction. In conclusion, the environmental contaminants, PCBs, can activate PARP directly impairing endothelial cell function that may predispose exposed individuals to development of hypertension and cardiovascular disease.
Keywords: Polychlorinated biphenyl, Oxidative stress, PARP, Cardiovascular, Atherosclerosis
1. Introduction
Polychlorinated biphenyls (PCBs) belong to a class of widely dispensed and persistent environmental contaminants collectively referred to as halogenated aromatic hydrocarbons. The commercial production of these compounds was banned at the end of the 1970s but PCBs continue to be a health problem partly due to their formation as industrial by-products but mainly because of their persistence in the environment and our food chain [1,2]. The major source of exposure to PCBs in the general population is from dietary intake of contaminated foods that include fish, meat and dairy products. Epidemiology studies have demonstrated that cardiovascular diseases can be linked to environmental pollution. Human exposure to PCBs for example, among Swedish capacitor manufacturing workers and the Italian population of Serveso (exposed to PCBs as a result of an industrial accident in 1976), has resulted in a detectable increase in cardiovascular disease [3].
Exposure to PCBs can lead to cardiovascular toxicity and atherosclerosis in animal models [4] as well as having a pro-inflammatory effect on endothelial cells [5]. PCB-mediated dysfunction in the vascular endothelium has been linked to increased oxidative stress mediated through the activation of a cytochrome P450 oxidase, CYP1A1, following PCB activation of the Ah receptor [5–7] and mediated by caveolae signalling [8]. PCB-mediated cellular oxidative stress leads to subsequent activation of stress kinases [9], increased transcription of NF-κB [7], increased pro-inflammatory gene expression (cytokines, adhesion molecules) [10], disruption of endothelial cell barrier function [11], and endothelial cell apoptosis [12], all of which can lead to atherosclerosis [5,13].
In the early 1980s it was suggested that PCB exposure was linked to hypertension [14] and this has been confirmed with much larger population studies [15,16]. Hypertension is a contributing factor for development of atherosclerosis and though PCBs have already been shown to exert a pro-inflammatory effect on endothelial cells increasing the risk of developing atherosclerosis no studies on the direct effects of PCBs on endothelial cell function have been performed.
Endothelial dysfunction is commonly associated with pathology of the cardiovascular system where overproduction of reactive oxygen and nitrogen intermediates in endothelial cells leads to DNA damage and overactivation of a DNA repair enzyme, poly (ADP-ribose) polymerase (PARP), representing a final common pathway in the pathogenesis of many cardiovascular diseases. PARP activation has been implicated as a central mediator of endothelial cell dysfunction in a variety of disease states including diabetes [17,18], atherosclerosis [19], ageing [20], doxorubicin-induced dysfunction [21] and septic shock [22].
Therefore the aim of this study was to examine the direct effects of 2,2′,4,6,6′-pentachlorobiphenyl (PCB 104, Fig. 1), an example of a highly ortho-chlorinated nonplanar PCB, on endothelial cell function and investigate whether increased cellular oxidative stress and activation of PARP is responsible for any deleterious effects.
Fig. 1.
Structure of 2,2′,4,6,6′-pentachlorobiphenyl (PCB 104), an example of a highly ortho-chlorinated nonplanar PCB.
2. Materials and methods
2.1. Reagents
Acetylcholine, α-naphthoflavone, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT), dicumarol, digitonin, dimethyl sulfoxide (DMSO), endothelial cell growth supplement, 7-ethoxyresorufin, heparin, 1-methoxy-5-methyl-phenazinium, nitro blue tetrazolium (NBT), phenylephrine, resorufin, sodium dodecyl sulfate (SDS), sodium nitroprusside and thiazolyl blue tetrazolium bromide (MTT) were obtained from Sigma/Aldrich (Poole, UK). 3H-NAD was obtained from PerkinElmer Ltd. (Windsor, UK). PJ-34 was purchased from Calbiochem (Nottingham, UK). Hams F12 culture media, penicillin, streptomycin and fetal bovine serum (FBS) were obtained from Lonza (Nottingham, UK). Sprague–Dawley rats used in the ex vivo studies were obtained from Charles River Laboratories (Kent, UK). PCB 104 (purity 99.1%) was obtained from QMX laboratories (Essex, UK). All other chemicals were of reagent grade and obtained from Fisher Scientific (Loughborough, UK).
2.2. Ex vivo
Thoracic aorta from male Sprague–Dawley rats (180–220 g) were dissected and cleared of periadventitial fat and connective tissue. Rings of 2–3 mm were cut and placed into Hams F12 medium supplemented with 3% FBS, endothelial cell growth supplement (0.03 mg/ml), heparin (50 U/ml) and 1% penicillin and streptomycin. PCB 104 was dissolved in DMSO to make a stock solution which was warmed to 37 °C before being added to pre-warmed media at 37 °C at the appropriate volumes to obtain concentrations of 1, 3, 10 and 20 μM. For the initial experiments rings were incubated with Hams F12 media alone (control) or with PCB 104 (1, 3, 10 or 20 μM), for 4 or 6 h at 37 °C with a 95% O2, 5% CO2 mix. Subsequent to these experiments rings were exposed to PCB 104 10 μM in combination with inhibitors of CYP1A1 (α-naphthoflavone 0.3 μM) and PARP (PJ-34 3 μM) for 4 h.
Subsequent to the relevant incubation the aortic rings were mounted in organ baths filled with warmed (37 °C) and gas-equilibrated (95% O2, 5% CO2) Krebs solution containing (in mmol/L) CaCl2 1.6, MgSO4 1.17, EDTA 0.026, NaCl 130, NaHCO3 14.9, KCl 4.7, KH2PO4 1.18, and glucose 5. Isometric tension of the rings was measured with isometric transducers (DMT), digitised using PowerLab and displayed on a Macintosh computer. A preload tension of 1.5 g was applied and the rings were equilibrated for 60 min, followed by measurements of the concentration-dependent contraction to phenylephrine (10−9 to 10−4 M) and in rings precontracted with phenylephrine (10−6 M), relaxation to acetylcholine (10−9 to 10−4 M) and sodium nitroprusside (10−12 to 10−5 M).
2.3. In vitro
Mouse aortic endothelial cells [17] were plated at a density of 250,000 cells per well in a 12-well plate and grown in Ham’s F12 media supplemented with 10% fetal calf serum, heparin (50 U/ml) and 0.03 mg/ml endothelial cell growth supplement. Following 24 h the media was replaced and the cells treated with increasing concentrations of PCB 104 (1, 3, 10 or 20 μM) for 2, 4 or 6 h. In a second set of experiments mouse endothelial cells were treated with 10 μM PCB 104 in combination inhibitors of CYP1A1 (α-naphthoflavone 0.3 μM) and PARP (PJ-34 3 μM) for 4 h. The following measurements were then determined.
2.4. Oxidative stress
Cellular levels of oxidative stress were assessed by reduction of nitro blue tetrazolium (NBT) as previously described [23]. Briefly, mouse endothelial cells were grown in 12-well plates and incubated at 37 °C with PCB 104 ± pharmacological inhibitors for 2, 4 or 6 h in the presence of 25 μg/ml NBT. Following the incubation period the media was removed from the wells and replaced with 75% ethanol for 10 min then removed. The wells were then washed twice with 100% methanol followed by air-drying of the wells. The blue crystals were then solubilized in a mixture of potassium hydroxide and DMSO (5:6) before absorbance was measured at 700 nm and results expressed as a percentage of absorbance in untreated cells.
2.5. CYP1A1 activity
The activity of CYP1A1 was assayed by the o-dealkylation of ethoxyresorufin (EROD) [24]. Briefly, the media was removed and replaced with 250 μl fresh media containing 8 μM 7-ethoxyr-esorufin and 10 μM dicumarol for an additional 30 min incubation at 37 °C. Following the incubation period 100 μl was removed and mixed with 130 μl of absolute ethanol. Resorufin-associated fluorescence was measured in a multiwell fluorescence reader, with excitation/emission wavelengths of 530/590 nm and compared to a resorufin standard curve. Cellular protein was determined using the Bradford assay [25] and results expressed as pmol resorufin/mg protein/min.
2.6. PARP activity
PARP activity was measured as previously described [26]. Briefly, the media was removed and replaced with 0.5 ml HEPES (pH 7.5) containing 0.01% digitonin and 3H-NAD (0.5 μCi ml−1) and the cells incubated for 20 min at 37 °C. The cells were then scraped from the wells and placed in an Eppendorf tubes containing 200 μl of ice-cold 50% TCA (w/v), the tubes were then placed at 4 °C. After 4 h the tubes were centrifuged at 1800 × g for 10 min and the supernatant removed, the pellet was washed twice with 500 μl ice-cold 5% TCA. The pellet was solubilized in 250 μl NaOH (0.1 M) containing 2% SDS overnight at 37 °C, the PARP activity was then determined by measuring the radioactivity incorporated using a Wallac scintillation counter. The solubilized protein (250 μl) was mixed with 5 ml of scintillant (ScintiSafe Plus, Fisher) before being counted for 10 min. Results are expressed as a percentage of the PARP activity observed in untreated cells.
2.7. NADPH depletion
Cell NADPH levels were determined as previously described [27]. Briefly, the media was removed and the cells washed with once with PBS before 100 μl of fresh media containing 0.5 mM 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) and 20 nM 1-methoxy-5-methylphenazinium was added to each well. The plate was then incubated for 30 min and absorbences were read at 450 and 670 nm. Results were expressed as a percentage of the absorbance observed in untreated cells.
2.8. MTT assay
Cell viability was determined by the reduction of yellow MTT into a purple formazan product by mitochondrial dehydrogenases of metabolically active cells. Following the treatment period the experimental media was removed and 200 μl MTT (1 mg/ml) added. After 1 h the MTT solution was carefully removed and the purple crystals were solubilized in 100 μl of DMSO. The DMSO was transferred to an ELISA plate and absorbance measured at 550 nm with a 620 nm reference [26]. Results were expressed as a percentage of the absorbance observed in untreated cells.
2.9. Statistical analysis
Results are presented as mean ± standard error of the mean (SEM). Analysis of variance with Bonferroni’s correction or Student’s t-test was used to compare mean values as appropriate. Differences were considered significant when p < 0.05.
3. Results
3.1. PCB 104 exposure results in dose- and time-dependent endothelial cell dysfunction
The effect of short-term exposure to PCB 104 on rat endothelial cell function was investigated by exposing aortic rings to PCB 104 1, 3, 10 or 20 μM for 2, 4 or 6 h. Endothelial cell function was assessed by the ability of the endothelial cell to produce nitric oxide in response to acetylcholine and cause subsequent vascular smooth muscle cell relaxation.
Acetylcholine-mediated relaxation of rat aortic rings is impaired following exposure to PCB 104 at 3, 10 or 20 μM (Fig. 2a and b) for 4 or 6 h. There was a significant increase in the EC50 concentration of acetylcholine following PCB exposure as compared to untreated rings (Table 1), however though the mean EC50 increases both with higher doses and longer exposure time this did not reach statistical significance (Table 1). There was no effect on endothelial cell function following 2 h exposure to PCB 104 at any of the three concentrations tested (data not shown). In addition, exposure to 1 μM PCB 104 had no effect on endothelial cell function at any exposure time tested (data not shown). This impairment of endothelial cell function was both dose-dependent and time-dependent particularly at the higher, 10 and 20 μM, concentrations of PCB 104. PCB 104 exposure had no effect on vascular smooth muscle contraction to phenylephrine or on relaxation in response to sodium nitroprusside (data not shown).
Fig. 2.
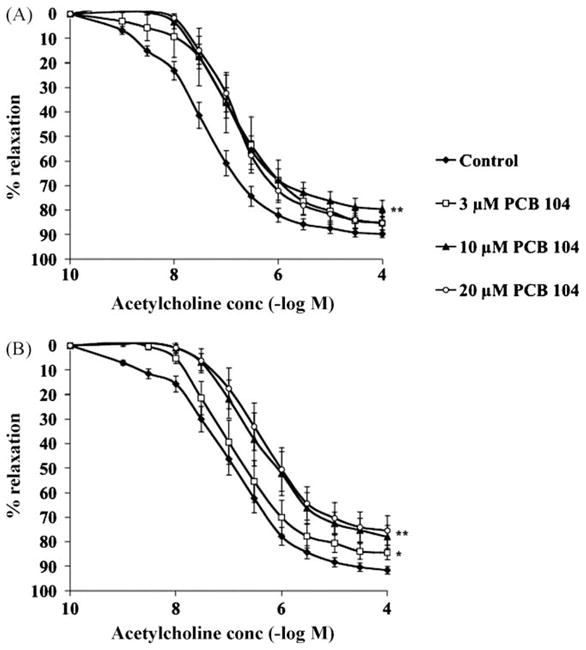
Exposure of ex vivo aortic rings to PCB 104 for 4 h (A) or 6 h (B) dose-dependently impairs the acetylcholine-relaxant response. Thoracic aortic rings obtained from male Sprague–Dawley rats were exposed to 1, 3, 10 or 20 μM PCB 104 for 2, 4 or 6 h. Following PCB exposure acetylcholine-induced endothelium-dependent relaxation was determined. Data is expressed as mean ± SEM from 6 animals; *p < 0.05 and **p < 0.01 vs. untreated rings.
Table 1.
Exposure of ex vivo aortic rings to PCB 104 for 4 or 6 h increases the EC50 of acetylcholine. Thoracic aortic rings obtained from male Sprague–Dawley rats were exposed to 3, 10 or 20 μM PCB 104 for 4 or 6 h. Following PCB exposure acetylcholine-induced endothelium-dependent relaxation was determined and EC 50 measured. Data is expressed as mean ± SEM from 6 animals.
| Exposure time | 4 h | 6 h |
|---|---|---|
| Control | 1.1 × 10−7 ± 0.2 × 10−7 M | 0.8 × 10−7 ± 0.2 × 10−7 M |
| PCB 104 3 μM | 2.0 × 10−7 ± 0.6 × 10−7 M* | 5.6 × 10−7 ± 3.3 × 10−7 M* |
| PCB 104 10 μM | 4.1 × 10−7 ± 1.2 × 10−7 M* | 8.3 × 10−7 ± 3.7 × 10−7 M* |
| PCB 104 20 μM | 9.4 × 10−7 ± 1 × 10−7 M* | 15.4 × 10−7 ± 7.7 × 10−7 M* |
p < 0.05 vs. untreated rings.
3.2. PCB 104 exposure increases endothelial cell oxidative stress, CYP1A1 activity, and PARP activity while depleting NADPH levels
To investigate the mechanism by which PCB 104 may be causing cell dysfunction mouse endothelial cells were exposed to PCB 104 in vitro and cellular levels of oxidative stress, CYP1A1 activity, PARP activity and NADPH were measured.
PCB 104 between 1 and 10 μM dose- and time-dependently increased endothelial cell oxidative stress (Fig. 3a) as measured by NBT reduction. This increased oxidative stress following PCB 104 exposure is likely mediated by increased CYP1A1 activity. Exposure of endothelial cells to PCB 104 dose- and time-dependently increased CYP1A1 activity as measured by EROD activity (Fig. 3b). Although 2 h exposure to 20 μM PCB 104 increases both oxidative stress and EROD activity (Fig. 3a and b), there is, however, a significant decrease in the measured oxidative stress and EROD activity after 4 or 6 h exposure at this PCB 104 concentration that may indicate that at this very high concentration of PCB 104 either CYP1A1 becomes inhibited or the endothelial cells themselves are becoming metabolically dysfunctional.
Fig. 3.

In vitro exposure of endothelial cells to PCB 104 dose- and time-dependently increased cellular levels of oxidative stress (A), CYP1A1 activity (B), and PARP activity (C) while depleting NADPH levels (D). Mouse endothelial cells were exposed to PCB 104 (1, 3, 10 or 20 μM) for 2, 4 or 6 h prior to the measurements being taken. Data is expressed as mean ± SEM from 4 separate experiments with 3–6 replicates per experiment; *p < 0.01 vs. untreated cells.
PCB 104 exposure dose- and time-dependently increased PARP activity as measured by NAD incorporation (Fig. 3c). As NAD is the substrate for PARP, depletion of NAD related high-energy phosphate levels following PARP overactivation has been identified as a mechanism of cellular dysfunction, particularly in endothelial cells where NADPH is an essential cofactor for nitric oxide synthase and subsequent nitric oxide production. PCB 104 exposure significantly depleted endothelial cell NADPH levels (Fig. 3d) an effect that appears to be linked to PARP activity as the higher the PARP activity the more cellular NADPH is depleted (Fig. 3c and d).
3.3. Inhibition of PARP and CYP1A1 protects against PCB 104-mediated endothelial cell dysfunction
To determine whether PCB 104-mediated activation of CYP1A1 and PARP mediates the observed endothelial cell dysfunction we pharmacologically inhibited CYP1A1 with α-naphthoflavone [28] and PARP with PJ-34 [29].
Exposure to 10 μM PCB 104 for 4 h significantly impaired endothelial cell function as assessed by acetylcholine-mediated vascular ring relaxation (Fig. 4) and increased the EC50 for acetylcholine from 1.2 × 10−7 ± 0.2 × 10−7 to 5.0 × 10−7 ± 1 × 10−7 M (p < 0.05). Inhibition of PARP with PJ-34 (3 μM) or CYP1A1 activity with α-naphthoflavone (0.3 μM) completely protected against the PCB 104-mediated endothelial cell dysfunction (Fig. 4) as well as returning the EC50 for acetylcholine to 0.6 × 10−7 ± 0.2 × 10−7 and 0.8 × 10−7 ± 0.3 × 10−7 M respectively (p < 0.05 vs. PCB 104 10 μM). Neither PJ-34 nor α-naphthoflavone alone had any effect on vascular ring contraction induced by phenylephrine or relaxation by acetylcholine (data not shown).
Fig. 4.
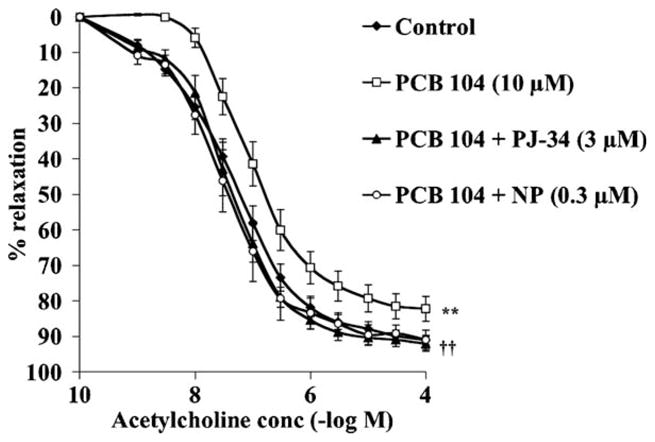
PCB 104-mediated impairment of endothelium-dependent aortic ring relaxation is prevented by pharmacological inhibition of CYP1A1 or PARP. Exposure of thoracic aortic rings to 10 μM PCB 104 for 4 h significantly impaired acetylcholine-induced endothelium-dependent relaxation, an effect prevented by simultaneous incubation of the rings with the CYP 1A1 inhibitor α-naphthoflavone (NP, 0.3 μM) or the PARP inhibitor PJ-34 (3 μM). Data is expressed as mean ± SEM from 6 animals; **p < 0.01 vs. untreated rings and ††p < 0.01 vs. PCB 104 treated rings.
To confirm that both these pharmacological inhibitors were affecting the cellular activities of PARP and CYP1A1 we carried out in vitro experiments exposing endothelial cells to 10 μM PCB 104 for 4 h and measuring oxidative stress, CYP1A1 activity, PARP activity and NADPH depletion.
PCB 104 exposure significantly increased endothelial cell oxidative stress (Fig. 5a), CYP1A1 activity (Fig. 5b), PARP activity (Fig. 5c) and NADPH depletion (Fig. 5d). The CYP1A1 inhibitor α-naphthoflavone reduced PCB 104-mediated increased EROD activity to levels observed in control cells (Fig. 5b) indicating that this concentration of α-naphthoflavone, 0.3 μM, was an effective inhibitory concentration. Inhibiting PCB 104-induced CYP1A1 activity also prevented the increased cellular oxidative stress (Fig. 5a), PARP activity (Fig. 5c) and NADPH depletion (Fig. 5d).
Fig. 5.

Effect of CYP1A1 and PARP inhibition on the PCB 104-mediated increase in endothelial cell oxidative stress (A), CYP1A1 activity (B), PARP activity (C) and NADPH depletion (D). Inhibition of CYP1A1 with α-naphthoflavone (0.3 μM) prevents the increase in oxidative stress, CYP1A1 and PARP activity, plus the NADPH depletion observed following 4 h exposure to PCB 104 (10 μM). Inhibition of PARP either pharmacologically with PJ-34 (3 μM) or via gene knockout had no effect on PCB 104-mediated increase in oxidative stress or CYP1A1 activity but did inhibit the increase in PARP activity and NADPH depletion. Data is expressed as mean ± SEM from 4 separate experiments with 3–6 replicates per experiment; *p < 0.05, **p < 0.01 vs. untreated cells and †p < 0.01 vs. PCB 104 treated cells.
The PARP inhibitor PJ-34 not only reduced the PCB 104-mediated increase in PARP activity but also reduced the significantly reduced basal activity observed in control cells (Fig. 5c). Inhibition of PARP also significantly attenuated the NADPH depletion (Fig. 5d) following PCB 104 exposure and in control cells even increased NADPH levels above that seen in untreated cells (Fig. 5d). However, inhibition of PARP had no effect on either cellular oxidative stress or CYP1A1 activity (Fig. 5a and b). Similar results were seen with endothelial cells isolated from the PARP-1 deficient mouse where PCB 104 exposure increased cellular oxidative stress and CYP1A1 activity levels (Fig. 5a and b) but failed to dramatically affect PARP activity or NADPH depletion (Fig. 5c and d). However, in PARP-1−/− cells there was a minor increase in PARP activity and NADPH depletion that may reflect activation of PARP-2 or other PARP isoforms, which are also present in mammalian nuclei though at much lower levels than PARP-1. This is not observed in PJ-34 treated endothelial cells as PJ-34 is a global PARP inhibitor and inhibits not only PARP-1 but also PARP-2.
3.4. PCB 104-mediated loss of cell viability is prevented by inhibition of CYP1A1 and PARP
Exposure to PCBs has been shown to cause loss of endothelial cell viability and increase apoptosis. Endothelial cell viability following 48 h exposure to PCB 104 was measured using the MTT assay and found to be dose-dependently reduced (Fig. 6). Exposure to PCB 104 for 24 h also causes loss of endothelial cell viability but only at 10 and 20 μM (data not shown). This loss of cell viability following PCB 104 exposure was prevented by inhibition of CYP1A1, with α-naphthoflavone, or PARP, either pharmacologically with PJ-34 or in PARP-1 gene deficient cells (Fig. 6).
Fig. 6.
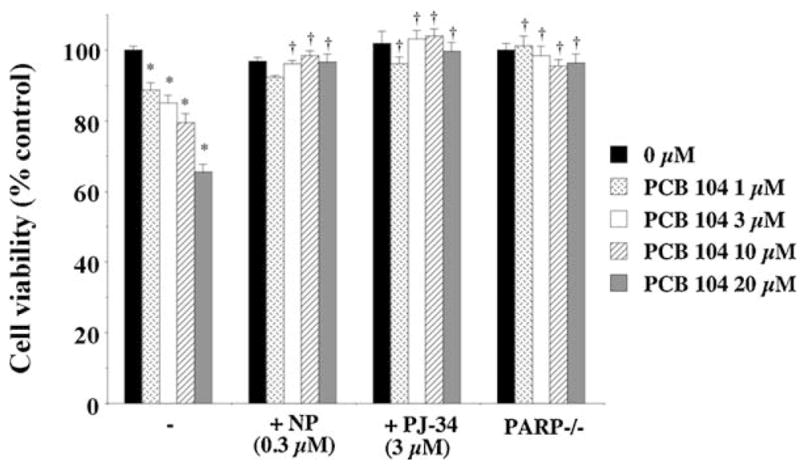
Exposure of endothelial cells to PCB 104 dose-dependently reduced cell viability, an effect attenuated by inhibition of CYP1A1 and PARP either pharmacologically or through gene deletion. Data is expressed as mean ± SEM from 4 separate experiments with 3–6 replicates per experiment; *p < 0.05, **p < 0.01 vs. untreated cells and †p < 0.01 vs. PCB 104 treated cells.
4. Discussion
The data presented here demonstrates for the first time that exposure of endothelial cells to the environmental contaminant PCB 104, for a relatively short exposure period, causes direct cellular dysfunction. Serum concentrations of PCBs can reach approximately 3 μM in people exposed to these toxicants [30,31] and possibly 10–20 μM in the extracellular space [32], it is exposure to these concentrations of PCB 104 which caused direct endothelial cell dysfunction in our study. PCB-mediated activation of the cytochrome P450 enzyme CYP1A1 and subsequently increased cellular oxidative stress appears to be the mediator of PCB-induced endothelial cell dysfunction with overactivation of the DNA repair enzyme PARP and subsequent depletion of cellular NADPH being the mechanism of dysfunction. Pharmacological inhibition of CYP1A1 or PARP protected endothelial cell function from PCB 104-mediated impairment. Previous work has established long-term exposure to PCBs as having pro-inflammatory effects on endothelial cells increasing the risk of developing atherosclerosis, our data indicates that PCBs cause direct endothelial cell dysfunction in a much shorter time period increasing the risk of developing hypertension and cardiovascular disease.
PCBs have previously been shown to increase reactive oxygen species in human breast cancer cells and cause PARP activation [33]. When PARP becomes activated, using NAD+ as a substrate, it catalyzes the building of homopolymers of adenosine diphosphate ribose units. NAD+ levels regulate an array of vital cellular processes, NAD+ serves as a cofactor for glycolysis and the tricarboxylic acid cycle, thus providing ATP for most cellular processes, NAD+ also serves as the precursor for NADP, which acts as a cofactor for the pentose shunt, for bioreductive synthetic pathways, and is involved in the maintenance of glutathione (GSH) pools [34]. PCB 104 increased PARP activity in endothelial cells and the resultant effects both on the metabolic pathways of the cell and anti-oxidant defence systems are likely to be the major mechanism by which these environmental contaminants cause endothelial cell dysfunction. Indeed, the cellular glutathione status has been previously shown to modulate PCB-mediated endothelial cell apoptosis [9] with PCB-exposure reducing endothelial cell glutathione levels and increasing cell death, the increased PARP activation by PCBs may account for the effect on cellular glutathione levels.
Activation of PARP in endothelial cells has been shown to impair relaxation of the blood vessel in response to vasodilators such as acetylcholine [17], an effect which appears to be mediated by a decrease in the levels of cellular high-energy phosphates and suppression of NAD and NADPH levels [18]. Endothelial cell-mediated control of blood pressure is via production of nitric oxide from nitric oxide synthase, an enzyme dependent on NADPH [35]. Depletion of cellular NADPH levels in endothelial cells exposed to PCB 104 therefore is likely to result in a decreased production of nitric oxide from endothelial cells thus reducing vasodilatation and potentially increasing the risk of developing hypertension. This disruption of endothelial cell regulation of blood pressure by PCB 104 may account for the results observed in a recent study where high serum PCB levels in humans were positively associated with hypertension [16]. Impaired endothelial nitric oxide bioavailability and subsequent hypertension development is a significant risk factor in developing atherosclerosis [36]. Indeed endothelial cell regulation of blood pressure in the apolipoprotein E deficient mouse model of atherosclerosis, as assessed by endothelial cell-mediated relaxation of thoracic aorta, has been shown to be significantly impaired [37,38], with function being restored by inhibition of PARP [39].
Previous studies have shown PCBs to have a pro-inflammatory and hence pro-atherosclerotic effect on endothelial cells [5,13] coupled with our data indicating PCB-mediated PARP activation in endothelial cells leading to cell dysfunction and predisposing individuals to hypertension suggests PCB exposure poses a significant cardiovascular health problem. The pro-inflammatory effects of PCBs on endothelial cells include activation of stress kinases [9], increased transcription of NF-κB [7], increased pro-inflammatory cytokine production, increased cellular expression of adhesion molecules [10], disruption of cell barrier function [11], and increased apoptosis [12], are all effects which may ultimately be mediated by PCB-induced PARP activation. PARP has been identified as having a role in atherosclerosis development [40,41]. In animal models of atherosclerosis PARP inhibitors have been shown to be beneficial in reducing plaque size, increasing their stability and promoting their regression [42,43].
The activation of transcription factors NF-κB and AP-1, both upregulated in endothelial cells by PCBs [7] and saturated fatty acids [44], has been shown to be regulated by PARP [45,46]. Inflammatory cytokine production, again observed in endothelial cells following PCB and fatty acid exposure [13,44], can also be blocked by PARP inhibitors [29,47]. In diabetes [48] and atherosclerosis [49] PARP activation has been shown to mediate endothelial cell expression of adhesion molecules such as ICAM, P-selectin and VCAM, all of which have been implicated in the development of atherosclerosis [50] and can be upregulated in endothelial cells following PCB exposure [10]. PARP inhibition has also been shown to protect endothelial cell barrier function [51], which again can be damaged following PCB exposure [11]. Therefore, there is a strong suggestion that PCB-mediated PARP activation in endothelial cells may account for the pro-atherosclerotic effects of these environmental contaminants.
Based on the results presented here we conclude that PCB exposure leads to increased oxidative stress and activation of PARP in vascular endothelial cells and impairment of vasorelaxation predisposing individuals to development of hypertension. This vascular function impairment coupled with the previously reported pro-inflammatory effects of PCBs on endothelial cells is likely the cause of the increased incidence of cardiovascular disease in populations exposed to environmental contaminants such as PCBs.
Acknowledgments
SGH was supported by a Heart Research UK intercalated B.Sc. scholarship (RG2550/07/08).
References
- 1.Carpenter DO. Polychlorinated biphenyls (PCBs): routes of exposure and effects on human health. Rev Environ Health. 2006;21(1):1–23. doi: 10.1515/reveh.2006.21.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Turrio-Baldassarri L, Abate V, Alivernini S, Battistelli CL, Carasi S, Casella M, et al. A study on PCB, PCDD/PCDF industrial contamination in a mixed urban-agricultural area significantly affecting the food chain and the human exposure. Part I. Soil and feed. Chemosphere. 2007;67(9):1822–30. doi: 10.1016/j.chemosphere.2006.05.124. [DOI] [PubMed] [Google Scholar]
- 3.Warner M, Eskenazi B, Patterson DG, Clark G, Turner WE, Bonsignore L, et al. Dioxin-like TEQ of women from the Seveso, Italy area by ID-HRGC/HRMS and CALUX. J Expo Anal Environ Epidemiol. 2005;15(4):310–8. doi: 10.1038/sj.jea.7500407. [DOI] [PubMed] [Google Scholar]
- 4.Hennig B, Reiterer G, Majkova Z, Oesterling E, Meerarani P, Toborek M. Modification of environmental toxicity by nutrients: implications in atherosclerosis. Cardiovasc Toxicol. 2005;5(2):153–60. doi: 10.1385/ct:5:2:153. [DOI] [PubMed] [Google Scholar]
- 5.Hennig B, Meerarani P, Slim R, Toborek M, Daugherty A, Silverstone AE, et al. Proinflammatory properties of coplanar PCBs: in vitro and in vivo evidence. Toxicol Appl Pharmacol. 2002;181(3):174–83. doi: 10.1006/taap.2002.9408. [DOI] [PubMed] [Google Scholar]
- 6.Toborek M, Barger SW, Mattson MP, Espandiari P, Robertson LW, Hennig B. Exposure to polychlorinated biphenyls causes endothelial cell dysfunction. J Biochem Toxicol. 1995;10(4):219–26. doi: 10.1002/jbt.2570100406. [DOI] [PubMed] [Google Scholar]
- 7.Slim R, Toborek M, Robertson LW, Hennig B. Antioxidant protection against PCB-mediated endothelial cell activation. Toxicol Sci. 1999;52(2):232–9. doi: 10.1093/toxsci/52.2.232. [DOI] [PubMed] [Google Scholar]
- 8.Lim EJ, Majkova Z, Xu S, Bachas L, Arzuaga X, Smart E, et al. Coplanar polychlorinated biphenyl-induced CYP1A1 is regulated through caveolae signaling in vascular endothelial cells. Chem Biol Interact. 2008;176(2–3):71–8. doi: 10.1016/j.cbi.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slim R, Toborek M, Robertson LW, Lehmler HJ, Hennig B. Cellular glutathione status modulates polychlorinated biphenyl-induced stress response and apoptosis in vascular endothelial cells. Toxicol Appl Pharmacol. 2000;166(1):36–42. doi: 10.1006/taap.2000.8944. [DOI] [PubMed] [Google Scholar]
- 10.Eum SY, Rha GB, Hennig B, Toborek M. c-Src is the primary signaling mediator of polychlorinated biphenyl-induced interleukin-8 expression in a human microvascular endothelial cell line. Toxicol Sci. 2006;92(1):311–20. doi: 10.1093/toxsci/kfj194. [DOI] [PubMed] [Google Scholar]
- 11.Eum SY, Andras IE, Couraud PO, Hennig B, Toborek M. Pcbs and tight junction expression. Environ Toxicol Pharmacol. 2008;25(2):234–40. doi: 10.1016/j.etap.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee YW, Park HJ, Son KW, Hennig B, Robertson LW, Toborek M. 2,2′,4,6,6′-Pentachlorobiphenyl (PCB 104) induces apoptosis of human microvascular endothelial cells through the caspase-dependent activation of CREB. Toxicol Appl Pharmacol. 2003;189(1):1–10. doi: 10.1016/s0041-008x(03)00084-x. [DOI] [PubMed] [Google Scholar]
- 13.Choi W, Eum SY, Lee YW, Hennig B, Robertson LW, Toborek M. PCB 104-induced proinflammatory reactions in human vascular endothelial cells: relationship to cancer metastasis and atherogenesis. Toxicol Sci. 2003;75(1):17–56. doi: 10.1093/toxsci/kfg149. [DOI] [PubMed] [Google Scholar]
- 14.Kreiss K, Zack MM, Kimbrough RD, Needham LL, Smrek AL, Jones BT. Association of blood pressure and polychlorinated biphenyl levels. J Am Med Assoc. 1981;245(24):2505–9. [PubMed] [Google Scholar]
- 15.Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR., Jr Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia. 2007;50(9):1841–51. doi: 10.1007/s00125-007-0755-4. [DOI] [PubMed] [Google Scholar]
- 16.Everett CJ, Mainous AG, 3rd, Frithsen IL, Player MS, Matheson EM. Association of polychlorinated biphenyls with hypertension in the 1999–2002 National Health and Nutrition Examination Survey. Environ Res. 2008;108(1):94–7. doi: 10.1016/j.envres.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7(1):108–13. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 18.Soriano FG, Pacher P, Mabley J, Liaudet L, Szabo C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ Res. 2001;89(8):684–91. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 19.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002;9(6):659–64. [PubMed] [Google Scholar]
- 20.Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. Br J Pharmacol. 2002;135(6):1347–50. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300(3):862–7. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 22.Soriano FG, Liaudet L, Szabo E, Virag L, Mabley JG, Pacher P, et al. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17(4):286–92. doi: 10.1097/00024382-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Samai M, Hague T, Naughton DP, Gard PR, Chatterjee PK. Reduction of para-quat-induced renal cytotoxicity by manganese and copper complexes of EGTA and EHPG. Free Radic Biol Med. 2008;44(4):711–21. doi: 10.1016/j.freeradbiomed.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Lai KP, Wong MH, Wong CK. Modulation of AhR-mediated CYP1A1 mRNA and EROD activities by 17beta-estradiol and dexamethasone in TCDD-induced H411E cells. Toxicol Sci. 2004;78(1):41–9. doi: 10.1093/toxsci/kfh045. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 26.Mabley JG, Suarez-Pinzon WL, Hasko G, Salzman AL, Rabinovitch A, Kun E, et al. Inhibition of poly (ADP-ribose) synthetase by gene disruption or inhibition with 5-iodo-6-amino-1,2-benzopyrone protects mice from multiple-low-dose-streptozotocin-induced diabetes. Br J Pharmacol. 2001;133(6):909–19. doi: 10.1038/sj.bjp.0704156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura J, Asakura S, Hester SD, de Murcia G, Caldecott KW, Swenberg JA. Quantitation of intracellular NAD (P)H can monitor an imbalance of DNA single strand break repair in base excision repair deficient cells in real time. Nucleic Acids Res. 2003;31(17):e104. doi: 10.1093/nar/gng105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachmann K, Sanyal G, Potter J, Schiavone R, Loch J. In vivo evidence that theophylline is metabolized principally by CYP1A in rats. Pharmacology. 1993;47(1):1–7. doi: 10.1159/000139071. [DOI] [PubMed] [Google Scholar]
- 29.Jagtap P, Soriano FG, Virag L, Liaudet L, Mabley J, Szabo E, et al. Novel phenan-thridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit Care Med. 2002;30(5):1071–82. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 30.Jensen AA. Background levels in humans. In: Kimbrough RD, Jensen AA, editors. Halogenated biphenyls, terphenyls, naphthalenes, dibenzodioxins and related products. Elsevier Science Publishers; 1989. pp. 345–64. [Google Scholar]
- 31.Wassermann M, Wassermann D, Cucos S, Miller HJ. World PCBs map: storage and effects in man and his biologic environment in the 1970s. Ann N Y Acad Sci. 1979;320:69–124. doi: 10.1111/j.1749-6632.1979.tb13137.x. [DOI] [PubMed] [Google Scholar]
- 32.Lotti M. Pharmacokinetics and blood levels of polychlorinated biphenyls. Toxicol Rev. 2003;22(4):203–15. doi: 10.2165/00139709-200322040-00003. [DOI] [PubMed] [Google Scholar]
- 33.Lin CH, Lin PH. Induction of ROS formation, poly (ADP-ribose) polymerase-1 activation, and cell death by PCB126 and PCB153 in human T47D and MDA-MB-231 breast cancer cells. Chem Biol Interact. 2006 doi: 10.1016/j.cbi.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Szabo C, Dawson VL. Role of poly (ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19(7):287–98. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 35.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh T, Donnelly T, Lyons D. Impaired endothelial nitric oxide bioavailability: a common link between aging, hypertension, and atherogenesis? J Am Geriatr Soc. 2009;57(1):140–5. doi: 10.1111/j.1532-5415.2008.02051.x. [DOI] [PubMed] [Google Scholar]
- 37.Yaghoubi M, Oliver-Krasinski J, Cayatte AJ, Cohen RA. Decreased sensitivity to nitric oxide in the aorta of severely hypercholesterolemic apolipoprotein E-deficient mice. J Cardiovasc Pharmacol. 2000;36(6):751–7. doi: 10.1097/00005344-200012000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Ding H, Hashem M, Wiehler WB, Lau W, Martin J, Reid J, et al. Endothelial dysfunction in the streptozotocin-induced diabetic apoE-deficient mouse. Br J Pharmacol. 2005;146(8):1110–8. doi: 10.1038/sj.bjp.0706417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benko R, Pacher P, Vaslin A, Kollai M, Szabo C. Restoration of the endothelial function in the aortic rings of apolipoprotein E deficient mice by pharmacological inhibition of the nuclear enzyme poly (ADP-ribose) polymerase. Life Sci. 2004;75(10):1255–61. doi: 10.1016/j.lfs.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Hans CP, Zerfaoui M, Naura AS, Catling A, Boulares AH. Differential effects of PARP inhibition on vascular cell survival and ACAT-1 expression favouring atherosclerotic plaque stability. Cardiovasc Res. 2008;78(3):429–39. doi: 10.1093/cvr/cvn018. [DOI] [PubMed] [Google Scholar]
- 41.Pacher P, Szabo C. Role of the peroxynitrite-poly (ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173(1):2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oumouna-Benachour K, Hans CP, Suzuki Y, Naura A, Datta R, Belmadani S, et al. Poly (ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation. 2007;115(18):2442–50. doi: 10.1161/CIRCULATIONAHA.106.668756. [DOI] [PubMed] [Google Scholar]
- 43.Hans CP, Zerfaoui M, Naura A, Troxclair D, Strong JP, Matrougui K, et al. Thieno[2,3-c]isoquinolin-5-one (TIQ-A), a potent poly (ADP-ribose) polymerase inhibitor, promotes atherosclerotic plaque regression in high fat diet-fed ApoE-deficient mice: effects on inflammatory markers and lipid content. J Pharmacol Exp Ther. 2009 doi: 10.1124/jpet.108.145938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. 2002;75(1):119–25. doi: 10.1093/ajcn/75.1.119. [DOI] [PubMed] [Google Scholar]
- 45.Oliver FJ, M-dM J, Nacci C, Decker P, Andriantsitohaina R, Muller S, et al. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18(16):4446–54. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andreone TL, O’Connor M, Denenberg A, Hake PW, Zingarelli B. Poly (ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170(4):2113–20. doi: 10.4049/jimmunol.170.4.2113. [DOI] [PubMed] [Google Scholar]
- 47.Hasko G, Mabley JG, Nemeth ZH, Pacher P, Deitch EA, Szabo C. Poly (ADP-ribose) polymerase is a regulator of chemokine production: relevance for the pathogenesis of shock and inflammation. Mol Med. 2002;8(5):283–9. [PMC free article] [PubMed] [Google Scholar]
- 48.Piconi L, Quagliaro L, Da Ros R, Assaloni R, Giugliano D, Esposito K, et al. Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly (ADP-ribose) polymerase. J Thromb Haemost. 2004;2(8):1453–9. doi: 10.1111/j.1538-7836.2004.00835.x. [DOI] [PubMed] [Google Scholar]
- 49.von Lukowicz T, Hassa PO, Lohmann C, Boren J, Braunersreuther V, Mach F, et al. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc Res. 2008;78(1):158–66. doi: 10.1093/cvr/cvm110. [DOI] [PubMed] [Google Scholar]
- 50.Giddings JC. Soluble adhesion molecules in inflammatory and vascular diseases. Biochem Soc Trans. 2005;33(Pt 2):406–8. doi: 10.1042/BST0330406. [DOI] [PubMed] [Google Scholar]
- 51.Lenzser G, Kis B, Snipes JA, Gaspar T, Sandor P, Komjati K, et al. Contribution of poly (ADP-ribose) polymerase to postischemic blood-brain barrier damage in rats. J Cereb Blood Flow Metab. 2007;27(7):1318–26. doi: 10.1038/sj.jcbfm.9600437. [DOI] [PubMed] [Google Scholar]