

Abstract
Severe acute respiratory syndrome (SARS) is an infectious disease caused by a novel coronavirus that cost nearly 800 lives. While there have been no recent outbreaks of the disease, the threat remains as SARS coronavirus (SARS‐CoV) like strains still exist in animal reservoirs. Therefore, the development of a vaccine against SARS is in grave need. Here, we have designed and produced a prototypic SARS vaccine: a self‐assembling polypeptide nanoparticle that repetitively displays a SARS B‐cell epitope from the C‐terminal heptad repeat of the virus’ spike protein. Biophysical analyses with circular dichroism, transmission electron microscopy and dynamic light scattering confirmed the computational design showing α‐helcial nanoparticles with sizes of about 25 nm. Immunization experiments with no adjuvants were performed with BALB/c mice. An investigation of the binding properties of the elicited antibodies showed that they were highly conformation specific for the coiled‐coil epitope because they specifically recognized the native trimeric conformation of C‐terminal heptad repeat region. Consequently, the antisera exhibited neutralization activity in an in vitro infection inhibition assay. We conclude that these peptide nanoparticles represent a promising platform for vaccine design, in particular for diseases that are characterized by neutralizing epitopes with coiled‐coil conformation such as SARS‐CoV or other enveloped viruses.
Keywords: coiled‐coils, peptide nanoparticles, protein design, repetitive antigen display, SARS coronavirus, SARS spike glycoprotein, subunit vaccines
Severe acute respiratory syndrome (SARS) first appeared in 2002 in southern China. According to the World Health Organization, the disease rapidly spread to 29 countries resulting in over 8000 people infected with 774 deaths. The causative agent of this atypical pneumonia was identified as a novel coronavirus (SARS‐CoV) (1) which had recently adapted to human–human transmission (2). Ferret badgers, civets and raccoon dogs present in live animal markets in China were all found to be infected with SARS‐CoV like viruses (3). However, the natural animal reservoir of SARS‐CoV like viruses was identified as bats (4). The re‐emergence of SARS is possible because SARS‐CoV like strains still exist in animal reservoirs. Thus, control measures such as development of safe and effective vaccines are needed.
Severe acute respiratory syndrome‐CoV’s attachment and subsequent entry into target cells is mediated by the spike (S) glycoprotein on the virion surface, which is also a major inducer of neutralizing antibodies (5). The spike protein is composed of two subunits: S1 and S2 (Figure 1A). The receptor‐binding domain (RBD) in the S1 subunit recognizes the host‐cell receptor human angiotensin‐converting enzyme 2. The S2 subunit is responsible for membrane fusion and has a fusion peptide (FP) sequence followed by two hydrophobic heptad repeat regions or coiled‐coils (HRN and HRC) (6) separated by a large interhelical domain or loop, a transmembrane (TM) domain and a cytoplasmic tail (Figure 1A). Once the spike protein is bound to the host‐cell receptor, a structural switch within the heptad repeat regions of S2 allows the FP and the TM domain to move near each other thus facilitating fusion of viral and cellular membrane, and allowing the nucleocapsid to enter the cell, the structural switch of HRN and HRC is a refolding of their trimeric states (7) to form a six‐helix bundle (6), in which three HRN helices are arranged into a central parallel, triple stranded, α‐helical coiled‐coil, and packed on the exterior of this core is an outer layer of three anti‐parallel HRC strands (8).
Figure 1.

(A) Schematic of full length SARS‐CoV S protein, residues 1–1255, which is divided into S1 (1–770) and S2 (771–1185) domains. The S1 domain contains the receptor‐binding domain (RBD). The S2 domain contains the predicted fusion peptide (FP), the N‐terminal heptad repeat region (HRN), interhelical domain (IHD), the C‐terminal heptad repeat region (HRC) and the transmembrane domain (TM). (B) Nanoparticle sequence (top) and HRC1 nanoparticle sequence (bottom): in black, the signaling sequence and the His‐tag used for purification, in green the pentameric coiled‐coil sequence, in blue the trimeric coiled‐coil sequence and in red the HRC1 epitope sequence. Alanines (shown in black) in the f position of the heptad repeat of the coiled‐coils are used to optimize interhelical contacts. (C) From top to bottom: HRC1 epitope sequence used in the nanoparticle immunogen (21); native HRC sequence (1150–1185) and schematic of maltose‐binding protein (MBP) fusion construct (MBP‐HRC‐GCN4). MBP was used as expression tag and purification tag, modified GCN4 sequence to stabilize the HRC sequence as a trimeric coiled‐coil and maintain the construct as a trimer (36). The GCN4 sequence is shown in purple.
Because the S proteins of coronaviruses are the most important antigenic determinants to induce neutralizing antibodies, SARS vaccine studies have focused on the S protein (9, 10, 11, 12, 13). Recently, Lokugamage et al. (14) reported that a chimeric coronavirus‐like particle carrying SARS‐CoV S protein and mouse hepatitis virus M, N and E proteins protected mice against challenge with SARS‐CoV. Also, Liniger et al. (15) showed that neutralizing antibodies were induced when SARS‐CoV S protein expressed by recombinant measles virus in infected Vero cells was injected into mice.
Nonetheless, a full‐length S protein should be used with caution. Kam et al. (16) reported that although a recombinant, trimeric SARS‐CoV S protein vaccine elicited a protective immune response in mice the anti‐S antibodies also mediated antibody‐dependent enhancement of viral entry into human B cells in vitro. In another study, ferrets vaccinated with SARS‐CoV full‐length S protein expressed by a recombinant modified vaccinia Ankara grown in BHK21 and Vero E6 cells (17) show enhanced virulence of hepatitis induced by SARS‐CoV. Furthermore, the use of a SARS S protein vaccine may lead to enhanced disease and immunopathology instead of protection as seen for feline coronavirus, feline infectious peritonitis virus (18). Given these concerns, the use of a SARS vaccine strategy in which the full‐length S protein is used may not be optimal for humans. Hence, the best approach would probably be to use small S protein epitopes that are major neutralization determinants. The use of the receptor‐binding domain of the SARS‐CoV spike protein induced highly potent neutralizing antibodies and long‐term protective immunity (19, 20). Furthermore, Tripet et al. (21) showed that antibodies directed to HRC of the S2 domain of SARS‐CoV spike protein inhibited SARS‐CoV replication in vitro.
For immunization purposes, it is of vital importance that the conformation of the B‐cell epitope in the vaccine is similar to that encountered in the native protein. For the SARS‐CoV spike protein in its pre‐fusogenic state, the HRC region is in an α‐helical trimeric coiled‐coil (7). Therefore, a peptide vaccine that would display HRC in its trimeric coiled‐coil conformation would be ideal. This requirement is met by the peptide nanoparticles developed in our laboratory. Raman et al. (22) described the design and production of a peptide that self‐assembles into a predicted regular icosahedral nanoparticle of about 16 nm of diameter. The monomeric peptide building block of the nanoparticle consists of a modified pentameric coiled‐coil from the cartilage oligomerization matrix protein (COMP) and a de novo designed trimeric coiled‐coil (23). If the trimeric coiled‐coil is extended by the HRC1 sequence described in Tripet et al. (residues 1156–1178 from the SARS S protein: ASVVNIQKEIDRLNEVAKNLNES), the native coiled‐coil conformation of the epitope should be maintained. In the study by Tripet et al. (21), the HRC1 epitope site was shown to be a more effective inhibitor of viral infectivity compared with another epitope site of HRC. Therefore, in our design the HRC1 region was selected to be in coiled‐coil register with the trimer sequence of the nanoparticle.
An important feature of the peptide nanoparticles is that a repetitive antigen display system is formed when the monomers self‐assemble. The repetitive display of the B‐cell epitope in the nanoparticle may lead to a strong humoral immune response. In addition, the peptide nanoparticles from Raman et al. (22) were assumed to have icosahedral symmetry, which mimics viral protein capsids. Furthermore, as these peptide nanoparticles would solely be a peptide‐based vaccine, the safety risks of live‐attenuated vaccines are avoided.
In this study, we designed a SARS subunit vaccine using a modified version of the peptide nanoparticle from Raman et al. (22). The modifications included replacement of cysteines by alanines (featured in black in Figure 1B) and addition of the SARS HRC1 epitope (Figure 1C) (21) at the C‐terminus to be repetitively displayed on the surface of the nanoparticles. It was expected that the antibodies directed against this B‐cell epitope will be conformation specific and hence be able to block fusion between cell and viral membrane by interacting with the C‐terminal heptad repeat on the viral spike glycoprotein thus preventing receptor‐induced conformation changes and virus entry. However, in this present design, no particular T‐helper epitope was engineered into the peptide sequence and neither the nanoparticle core nor the B‐cell epitope were predicted by the commonly available prediction programs to contain a specific T‐helper epitope for the haplotype of BALB/c mice.
Here, a biophysical characterization highlighting the correct self‐assembly of the peptide nanoparticle and its nanometer size range is presented. Immunization experiments showed that protective antibodies were elicited without the use of adjuvants and that they are conformation‐specific. These findings suggest that the modified peptide nanoparticles (denoted herein P6HRC1) are a promising platform for SARS vaccine design and probably also for other diseases that are characterized by B‐cell epitopes found in coiled‐coils of class I fusion protein in viruses, such as HIV, influenza and paramyxovirus (24).
Materials and Methods
Peptide nanoparticle synthesis
Oligonucleotides (purchased from IdtDNA) coding for the SARS HRC1 B‐cell epitope (residues 1156–1178 from the SARS S protein: ASVVNIQKEIDRLNEVAKNLNES) were annealed and ligated into a modified pPEP‐T vector that coded for the peptide monomer of the core particle. The resulting plasmid was transformed into the Eschericia Coli strain BL21(DE3)pLysS expression cells (Novagen, Madison, WI, USA). Bacterial growth was done at 37 °C in Luria broth medium in the presence of ampicillin (200 μg/mL) and chloramphenicol (30 μg/mL). Expression was induced with 1 mm isopropyl β‐d‐thiogalactopyranoside. After 3 h of expression, cells were harvested and centrifuged at 4000 × g for 10 min and frozen at −80 °C. Purification was done under denaturing conditions. Cell pellet was thawed in ice, resuspended in lysis buffer A which is composed of 9 m urea, 100 mm NaH2PO4, 10 mm Tris and 10 mmβ‐mercaptoethanol, pH 8 and lysed by sonication. The cell membranes were removed by centrifugation (45 min at 30 500 × g). The supernatant was then incubated with nickel beads (Qiagen, Valencia, CA, USA) for 1 h. Protein contaminants were washed from the column using a pH gradient. The first wash was done with lysis buffer A and the second and third washes at pH 6.3 and 5.9, respectively, with a buffer containing 9 m urea, 100 mm NaH2PO4, 20 mm sodium citrate, 10 mm imidazole and 10 mmβ‐mercaptoethanol. Elution was done at pH 5.0 again with 9 m urea, 100 mm NaH2PO4, 20 mm sodium citrate, 10 mm imidazole and 10 mmβ‐mercaptoethanol. Purity was verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Peptide identity was confirmed with mass spectrometry. Following purification, the denatured monomeric peptides were dialyzed against the refolding buffer 20 mm Tris pH 7.5, 150 mm NaCl and 10% glycerol containing 8 m urea followed by 6 m urea, 4 m urea, 2 m urea, 1 m urea and no urea in the same refolding buffer. The protein was filtered with a 0.1 μm polyvinylidene fluoride membrane filter before and after dialysis (Milipore #SLVV 033 RS, Millipore Billerica, MA, USA).
Circular dichroism
Circular dichroism (CD) experiments were performed in an Applied Photophysics Pi Star 180 spectrapolarimeter at 20 °C. Circular dichroism spectra were recorded from 190 to 250 nm in 20 mm sodium phosphate pH 7.5, 150 mm NaCl, 10% glycerol. Temperature denaturation profiles were obtained with a 1‐mm path length cell at 0.36 mg/mL protein concentration by following the change in molar ellipticity at 222 nm from 6 to 85 °C and a temperature increase rate of 1 °C/min.
Dynamic light scattering
Hydrodynamic diameter was obtained with a Malvern Zetasizer Nano S (Malvern, Worcestershire, UK) equipped with a 633‐nm laser. The measurements were done at 25 °C in a buffer containing 20 mm Tris pH 7.5, 150 mm NaCl, 10% glycerol.
Transmission electron microscopy
Samples were negatively stained with 1% uranyl acetate (SPI) at a peptide concentration of 0.07 mg/mL. Electron micrographs were taken with a Philips EM 300 (Philips Electron Optics, Eindhoven, The Netherlands) transmission electron microscope at an accelerating voltage of 80 kV. The micrographs were scanned at 600 dpi.
Analytical ultracentrifugation
Sedimentation velocity analysis was conducted at 20 °C and 36 000 rpm (104509 g) using interference optics with a Beckman‐Coulter XL‐I analytical ultracentrifuge (Beckman‐Coulter, Fullerton, CA, USA). Double sector synthetic boundary cells equipped with sapphire windows were used to match the sample and reference menisci. The rotor was equilibrated under vacuum at 20 °C and after a period of ∼1 h at 20 °C the rotor was accelerated to 36 000 rpm. Interference scans were acquired at 40‐second intervals for 1.5 h. Extinction coefficients, molecular masses, partial specific volumes and solvent densities were calculated using Sednterp (25). Initial analysis was performed using Sedfit (26) to obtain c(s) distributions and DcDt+ (27, 28, 29) to obtain g(s*) distributions.
Immunization protocol
Ten 6‐week‐old BALB/c mice (No. 1–10) were immunized interperitoneally. Primary immunization contained only 10 μg of sample P6HRC1 in 20 mm Tris pH 7.5, 150 mm NaCl, 10% glycerol buffer (0.05 mg/mL). Secondary, tertiary and one booster immunizations (at days 14, 28 and 42) also contained 10 μg of sample P6HRC1 in the same pH 7.5 buffer. Mice were killed and bled on day 56. As negative controls, P6 (nanoparticle without SARS HRC1 epitope) was used to immunize five mice (No. 21–25) with the same immunization procedure. Sera were prepared according to standard protocol.
ELISA
Ninety‐six‐well polystyrene plates were coated with diluted P6, P6HRC1, MBP‐HRC‐GCN4 (0.01 mg/mL), SARS‐CoV S protein and native HRC peptide (Figure 1) using 100 mm carbonate, pH 9.6 overnight at 4 °C. C‐terminal heptad repeat region in MBP‐HRC‐GCN4 construct was the native HRC sequence residues 1150–1178. After removing the coating solution and washing thrice with PBS, each well was blocked with 100 μL 5% BSA in PBS (37 °C, 1 h). The sera were diluted in 1% BSA in PBS and added to each well and incubated 37 °C, 1 h. The sample solution was removed and washed thrice by PBS with 0.1% Tween 20. Next, horseradish peroxidase conjugated anti‐mouse IgG (goat) (diluted 1:10000) was added to each well and incubated for 1 h at 37 °C. Sample solution was then removed and the wells were washed thrice by PBS with 0.1% Tween 20. 2,2′‐Azino‐di‐(3‐ethyl‐benthiazoline‐sulfonic acid) in 10 mm citrate, pH 4.2 with 0.1% H2O2 was added to each well and then the plates were read at 450 nm.
Viral neutralization assays
The neutralization activity of the HRC nanoparticle antisera on SARS‐CoV infectivity of Vero E6 cells was assessed. Neutralization assays were performed in triplicate wells in 6‐well flat‐bottom plates in the biosafety level 3 laboratory. One hundred microliters of sera containing 2 or 20 μg/mL of antibody or buffer alone was mixed with 200 μL of SARS virus containing 2 × 104 plague forming units in Dulbecco’s Modified Eagles Medium (DMEM; Gibco Invitrogen, Carlsbad, CA, USA) containing 10% FBS and 2% penicillin, streptomycin and fungizone (PSF; Gibco Invitrogen) in 96‐well deep dish plates. The virus was incubated with the sera for 1 h at 37 °C and 5% CO2. 10× serial dilutions of the virus and sera mixture were made in DMEM with 10% FBS and 2% PSF and inoculated onto Vero E6 cells in 6‐well plates and incubated for 1 h at 37 °C and 5% CO2. The inocula were removed and the cells were overlaid with 2 mL of Seakem agar containing MEM (Gibco Invitrogen) and 5% FBS and 2% PSF. The plates were incubated for 72 h and then the cells were fixed and stained with crystal violet for 4 h. Plagues were counted and the inhibition of viral infectivity by antibody to nanoparticles alone or HRC1 nanoparticles were shown as a percentage of the titer of virus incubated with buffer alone.
Results and Discussion
Design of the peptide nanoparticle with HRC1 epitope
The protein shell of viruses is built up from many copies of one or a few polypeptide chains (30). In particular, the protein shells of many spherical viruses have icosahedral symmetry. In an attempt to mimic nature’s design of small virus’ capsids, Raman et al. (22) designed a single monomeric peptide building block that was predicted to form an icosahedral peptide nanoparticle. The idea behind the design of this building block is that the symmetry elements found in an icosahedron: fivefold, threefold and twofold rotational symmetry axes are also found in coiled‐coils. By using such peptidic coiled‐coil oligomerization domains with their respective symmetries, it was possible to design three‐dimensional building blocks that would form a peptide nanoparticle with icosahedral symmetry (Figure 2). Therefore, based on the design by Raman et al., our monomeric building blocks of the peptide nanoparticle consist of sequences than can form both, trimeric and pentameric coiled‐coils.
Figure 2.

(A) 3D monomeric building block of P6HRC1 composed of a modified pentameric coiled‐coil domain from COMP (green) and trimeric de novo designed coiled‐coil domain (blue) which is extended by the coiled‐coil sequence of SARS HRC1 (red). (B) Computer models of the complete peptide nanoparticle P6HRC1 with T = 1 (top) and T = 3 (bottom) icosahedral symmetry. The calculated diameters of these particles are about 23 and 28 nm and the molecular weight 757 and 2271 kDa, respectively.
As shown in the sequence in Figure 1B, the monomeric peptide is composed of a pentamerization domain, a short linker and a trimerization domain that is extended with the HRC1 SARS B‐cell epitope sequence. This epitope was shown to elicit a humoral immune response characterized by neutralizing antibodies (21). Moreover, this epitope is ideally suited to extend the de novo trimeric coiled‐coil as the B‐cell epitope itself is a trimeric coiled‐coil (6, 7).
During a stepwise dialysis, the monomers self‐assembled into peptide nanoparticles which may accommodate 60 peptide chains in the case of a T = 1 icosahedron (Figure 2B top), or bigger assemblies such as a T = 3 icosahedron with 180 peptide chains (Figure 2B bottom). Also, other self‐assembly states with an intermediate number of peptide chains may be possible; however, such nanoparticles will not have a regular icosahedral symmetry. In fact, the larger assemblies probably will not have exact T = 3 icosahedral symmetry, because this would require that the pentameric coiled‐coil would switch into a hexamer, which is unlikely for the COMP oligomerization domain.
To optimize interhelical contacts between the pentameric coiled‐coil and the trimeric coiled‐coil, their sequences have been modified. In the design by Raman et al. (22), the angle between the helices is constrained by a disulfide bridge (Figure 2A). Our experience, however, shows that peptide nanoparticles that contain cysteines tend to aggregate (data not shown). Therefore, we replaced the cysteines at positions f of the heptad repeat of the coiled‐coil close to the glycine linker with alanines (shown in black in Figure 1B). Such a modification should not abrogate the α‐helical character of the sequences because alanine has a higher α‐helical propensity than cysteine (31).
CD investigation of the peptide nanoparticle
To evaluate the secondary structure and the effect of thermal denaturation, CD spectroscopy was used. As depicted in Figure 3A, the peptide nanoparticle P6HRC1 shows double minima at 208 and 222 nm, which is characteristic of α‐helical structure and is consistent with the fact that the peptide nanoparticle is composed of coiled‐coils. The peptide nanoparticles seem to be thermally very stable as only ∼18% of the protein is unfolded at 85 °C (Figure 3B).
Figure 3.

(A) Far UV circular dichroism (CD) spectra of P6HRC1. Spectra were recorded in 20 mm sodium phosphate pH 7.5, 150 mm NaCl, 10% glycerol. Peptide concentration was 0.36 mg/mL. (B) Temperature denaturation profile of the helical peptide nanoparticle P6HRC1. Denaturation was monitored by CD at 222 nm in 20 mm sodium phosphate pH 7.5, 150 mm NaCl, 10% glycerol.
Size distribution analysis of P6HRC1
Computer modeling predicts an average diameter of 23 nm for the P6HRC1 peptide nanoparticle with T = 1 icosahedral symmetry (Figure 2B top), similar to the size of small viruses. The particle size of an immunogen is very important as it directly affects its immunogenicity (32). In a study using antigen covalently coupled to inert nano‐beads of different sizes, the smaller particles (40–50 nm) were shown to elicit a more potent combined cellular and humoral responses than larger particles (33).
Transmission electron microscopy (TEM) revealed that our peptide chains self‐assembled into nanoparticles with spherical shapes and sizes ranging from 25 to 30 nm (Figure 4A) comparable with the value predicted by computer modeling. Nonetheless, the peptide nanoparticles were not perfectly spherical and their shapes might not be completely regular because of the presence of N‐terminal amino acids in the peptide sequence that do not form coiled‐coils such as the six histidines used for purification (Figure 1B). In a size per volume analysis, dynamic light scattering (Figure 4B) also showed nanoparticles sizes comparable with TEM results (Figure 4A). Together, these data indicate satisfactory self‐assembly of the monomeric peptide chains to form single nanoparticles of the expected size range. To further investigate this size/symmetry relationship, analytical ultracentrifugation analyses were performed.
Figure 4.
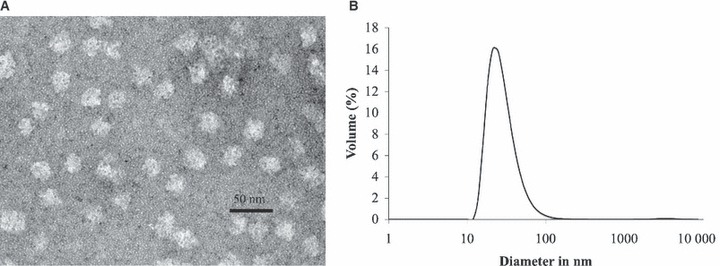
(A) Transmission electron microscopy image of P6HRC1 nanoparticles at 242 000×. The sample was negatively stained with 1% uranyl acetate. Sample concentration was 0.076 mg/mL and the buffer was 20 mm Tris pH 7.5, 150 mm NaCl, 10% glycerol. Nanoparticle size ranges from 25 to 30 nm. (B) Dynamic light scattering data of P6HRC1. Size distribution by volume shows a peak corresponding to a size of 26 nm. Sample concentration was 0.072 mg/mL and the buffer was 20 mm Tris pH 7.5, 150 mm NaCl, 10% glycerol.
From the computer modeling, it is expected that 60 monomers self‐assemble to form a T = 1 icosahedron. With the molecular weight obtained from sedimentation velocity experiments, it is possible to determine the number of monomers that constitute a peptide nanoparticle. Figure 5A displays a plot of the normalized sedimentation coefficient distribution, g(s*). The distributions peak near 29S. Using a calculated frictional ratio of 1.3, this sedimentation coefficient corresponds to nanoparticles with a molecular weight of about 1.4 MDa, that is ∼110 peptide chains per nanoparticle. Based on this, the largest fraction of the peptide nanoparticle seems to be in between a T = 1 and 3 icosahedral symmetry, models of which are shown in Figure 2B. This would imply that the angle between the pentamer and trimer is on average slightly smaller than shown for a T = 1 symmetry in Figure 2A. If this is the case, then a smaller angle would allow more than 60 monomers to co‐assemble into a single nanoparticle. However, it is clear from the g(s*) distributions and the tail of the distribution curve to the higher S values that the sample is not homogeneous but rather contains a mixture of different species, which agrees with the data obtained from DLS and TEM. Similarly, Padilla et al. (34) found different sized species for a designed self‐assembling protein cage which they attributed to a certain degree of flexibility or polymorphism in the assembled particles.
Figure 5.

(A) Sedimentation velocity ultracentrifugation of P6HRC1. The overlay shows the normalized sedimentation coefficient distribution g(s*) plots obtained from program DcDt+ for three different concentrations of P6HRC1. The distributions peak near 29S, which under the conditions used correspond to a molecular weight of 1.4 MDa or 110 monomers per nanoparticle. The buffer used was 20 mm Tris pH 7.5, 150 mm NaCl, 5% glycerol. (B) Sedimentation velocity ultracentrifugation of P6HRC1 showing a model of a continuous distribution of molecular masses, i.e. a c(M) analysis obtained using program Sedfit. The buffer used was 20 mm Tris pH 7.5, 150 mm NaCl, 5% glycerol. Molecular weight for P6HRC1 ranges from 700 kDa to 3 MDa.
Another useful analysis for characterizing the size distribution of a mixture of macromolecules from a sedimentation velocity run in analytical ultracentrifugation is the continuous distribution of molecular masses, c(M). The c(M) plot for the P6HRC1 sample with the highest concentration is shown in Figure 5B. The molecular masses range from a few hundred thousand Daltons to over 5 MDa, with the majority of the particles having molecular masses between ∼700 and ∼3 MDa, again corresponding to an average of about 110 peptide chains per particle.
Specificity of the antibodies obtained from immunizations
To evaluate the specificity of the antibodies elicited by immunization with nanoparticles or P6HRC1 nanoparticles qualitative ELISA was performed. Initially, as a control experiment, the ELISA plates were coated with nanoparticles without epitope to see whether the antibodies recognized the nanoparticle core. The antisera bound strongly to the nanoparticles as expected at 1:500 to 1:4000 dilution range (Figure 6A). In a logical sequence, the other control experiment was to coat the ELISA plates with the P6HRC1 peptide nanoparticle carrying the HRC1 epitope. As expected, the antibodies showed strong recognition of the P6HRC1 peptide nanoparticle with epitope (Figure 6B) in the same dilution range of 1:500 to 1:4000.
Figure 6.
(A) ELISA reactivity of nanoparticle mice antisera with nanoparticles (1 μg/well) coated on the plate. (B) ELISA reactivity of P6HRC1 nanoparticle mice antisera with P6HRC1 nanoparticles (1 μg/well) coated on the plate. (C) ELISA reactivity of P6HRC1 nanoparticle mice antisera with SARS‐CoV S protein (1–1180) (0.2 μg/well) coated on the plate. (D) ELISA reactivity of P6HRC1 nanoparticle mice antisera with MBP‐HRC‐GCN4 trimer (0.5 μg/well) coated on the plate. (E) ELISA reactivity of P6HRC1 nanoparticle mice antisera with native monomeric HRC peptide (0.5 μg/well) coated on the plate.
In terms of oligomeric state, the HRC peptide can be presented as a monomer or a trimer as in its native conformation (6, 7). We wanted to examine whether the antibodies obtained from immunization with P6HRC1 would bind to both forms of HRC or be specific to either the monomeric or the trimeric state. Therefore, to verify recognition of the native HRC, the ELISA plates were then coated with the SARS S protein in which the HRC sequence forms a weakly stable trimeric coiled coil. As seen in Figure 6C, antisera in the dilution range 1:100 to 1:800 bind to the peptide in its native form within the protein S which shows the strong immunogenic nature of the self‐assembling peptide nanoparticle.
To further investigate the specificity to the trimeric conformation of HRC, a construct which forces HRC into a trimeric state, was used to coat ELISA plates: MBP‐HRC‐GCN4 (35). In MBP‐HRC‐GCN4, maltose‐binding protein was used as an expression tag and purification tag and the modified GCN4 sequence maintained the construct as a trimer while also stabilizing the HRC sequence (Figure 1C) as a trimeric coiled‐coil (36). In the dilution range 1:50 to 1:400, the antisera bound the plated MBP‐HRC‐GCN4 trimeric construct (Figure 6D). On the other hand, when the plates were coated with the synthetic HRC peptide (Figure 1C), which on its own forms an unfolded monomer when bound to ELISA plate, there was little or no binding (Figure 6E). The results with trimeric HRC (as in MBP‐HRC‐GCN4) (Figure 6D) and monomeric HRC (Figure 6E) indicate that the antibodies produced are conformation‐specific which is expected from the nanoparticle design and confirms that this is an optimal platform for the display of conformation‐specific epitopes with α‐helical and in particular with coiled‐coil conformations. Similarly, in previous experiments with template‐based coiled‐coil antigens (7) the HRC1 sequence was found to elicit antibodies that were conformation‐specific for the trimeric pre‐fusogenic state of HRC and were not able to bind to the post‐fusion six‐helix bundle state (21). The peptide nanoparticle is designed in such a way that the de novo trimeric coiled‐coil is extended by the HRC1 trimeric coiled‐coil stabilizing trimeric coiled‐coil epitopes.
However, there is a 10‐fold difference in antibody reactivity with P6HRC1 (1:500 to 1:4000) and with MBP‐HRC‐GCN4 (1:50 to 1:400). This most likely reflects the fact that a considerable fraction of the antibodies are directed against the core of the nanoparticle which is also surface accessible to a substantial degree. Another reason for this could be that the trimeric HRC1 coiled‐coils in the peptide nanoparticle contain free C‐terminal α‐carboxyl groups which do not exist in MBP‐HRC‐GCN4 or the native HRC peptide (Cα‐amidated). We have previously shown that ionic interactions at the COOH terminus of a protein are very important in antigen–antibody interactions (37). Thus, antibodies made to the carboxyl termini of HRC1 would not necessarily recognize MBP‐HRC‐GCN4 or native HRC peptide.
In vitro SARS‐CoV neutralizing activity of antibodies to SARS HRC1 nanoparticles
After determining by ELISA that anti‐P6HRC1 sera recognized the prefusion state of the SARS‐CoV S protein, we tested their neutralizing activities to SARS‐CoV. The sera from mice numbers 3, 4, 6 and 9 which were immunized with SARS HRC1 nanoparticles were combined and concentrated 10‐fold. These four sera had strong binding activity to both trimeric MBP‐GCN4‐HRC construct and the prefusion state of S protein. The sera from mice immunized by nanoparticles alone were similarly concentrated. Both undiluted and concentrated sera were then tested for their ability to neutralize SARS‐CoV virus infectivity. The results are shown in Figure 7. Sera from mice immunized with HRC1 nanoparticles showed a concentration‐dependent neutralization of SARS‐CoV infectivity. Sera from mice immunized with nanoparticles alone had no significant neutralization activity.
Figure 7.

In vitro SARS‐CoV neutralizing activities of antibodies to P6HRC1 nanoparticle. Neutralization is shown as percentage of virus plus buffer alone. HRC1 nanoparticle sera significantly inhibited SARS‐CoV infection of Vero E6 cells, whereas antisera to nanoparticles alone did not neutralize virus infectivity.
In conclusion, we have generated the desired conformation‐specific antibodies that were able to neutralize SARS‐CoV infectivity using the nanoparticle presentation system. In a future experiment to test conformation specificity, we plan to introduce a glycine and proline linker between the de novo trimeric coiled‐coil and the HRC1 epitope, which will disrupt the coiled‐coil heptad repeat between the two sequences. In such a design, we would expect to observe a loss of conformation specificity in the immune response because of a weaker trimeric coiled‐coil in the HRC epitope. We have also engineered the pan‐DR T‐cell epitope PADRE into the peptide sequence and will test the influence of this known T‐helper epitope on the immunogenicity of the nanoparticle constructs. If promising results are obtained, the next steps will then include a challenge of mice with the virus to test whether the animals recover from infection.
Conclusions
We have presented a platform for a novel SARS subunit vaccine. It has the advantages of a peptide‐based vaccine in its lack of infectivity, ease of protein expression, purification, high purity and stability. Furthermore, the antibodies against the HRC1 from the SARS spike protein are conformation‐specific, recognize the prefusion, trimeric conformation of HRC and have neutralization activity on SARS‐CoV. These anti‐SARS antibodies were obtained without adjuvants. The immunogenic effect of this system is because of the nanometer size of the particle, repetitive display of the epitope and a good mimicry of the epitope’s native conformation. Thus, this self‐assembling polypeptide nanoparticle has great potential to be employed in vaccines because of the versatility of the antigen‐presenting system and good humoral immune response that can be obtained.
Acknowledgments
The authors thank the National Analytical Ultracentrifugation Facility at the University of Connecticut, Dr Carolyn Teschke for use of CD instrumentation and Dr Marie Cantino and Mr Stephen Daniels for assistance with the transmission electron microscope. This work was supported by Program Project Grant AI059576 from the National Institutes of Health to R. S. H. and K.V.H. and the John Stewart Chair in Peptide Chemistry to R. S. H.
References
- 1. Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks‐Wilson A., Butterfield Y.S.N., Khattra J. et al. (2003) The genome sequence of the SARS‐associated coronavirus. Science;300:1399–1404. [DOI] [PubMed] [Google Scholar]
- 2. Nicholls J., Dong X., Jiang G., Peiris M. (2003) SARS: clinical virology and pathogenesis. Respirology;8:S6–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W. et al. (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science;302:276–278. [DOI] [PubMed] [Google Scholar]
- 4. Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H. et al. (2005) Bats are natural reservoirs of SARS‐like coronaviruses. Science;310:676–679. [DOI] [PubMed] [Google Scholar]
- 5. Zhou Z., Post P., Chubet R., Holtz K., McPherson C., Petric M., Manon C. et al. (2006) A recombinant baculovirus‐expressed S glycoprotein vaccine elicits high titers of SARS‐associated coronavirus (SARS‐CoV) neutralizing antibodies in mice. Vaccine;24:3624–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tripet B., Howard M.W., Jobling M., Holmes R.K., Holmes K.V., Hodges R.S. (2004) Structural characterization of the SARS‐coronavirus spike S fusion protein core. J Biol Chem;279:20836–20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McReynolds S., Jiang S., Guo Y., Celigoy J., Schar C., Rong L., Caffrey M. et al. (2008) Characterization of the prefusion and transition states of severe acute respiratory syndrome coronavirus S2‐HR2. Biochemistry;47:6802–6808. [DOI] [PubMed] [Google Scholar]
- 8. Xu Y., Lou Z., Liu Y., Pang H., Tien P., Gao G.F., Rao Z. et al. (2004) Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J Biol Chem;279:49414–49419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saif L.J. (1993) Coronavirus immunogens. Vet Microbiol;37:285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bisht H., Roberts A., Vogel L., Bukreyev A., Collins P.L., Murphy B.R., Subbarao K. et al. (2004) Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc Natl Acad Sci USA;101:6641–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bukreyev A., Lamirande E.W., Buchholz U.J., Vogel L.N., Elkins W.R., Claire M.S., Murphy B.R. et al. (2004) Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet;363:2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang J., Cao Y., Du J., Bu X., Ma R., Wu C. (2007) Priming with SARS CoV S DNA and boosting with SARS CoV S epitopes specific for CD4+ and CD8+ T cells promote cellular immune responses. Vaccine;25:6981–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kitabatake M., Inoue S., Yasui F., Yokochi S., Arai M., Morita K., Shida H. et al. (2007) SARS‐CoV spike protein‐expressing recombinant vaccinia virus efficiently induces neutralizing antibodies in rabbits pre‐immunized with vaccinia virus. Vaccine;25:630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lokugamage K.G., Yoshikawa‐Iwata N., Ito N., Watts D.M., Wyde P.R., Wang N., Newman P. et al. (2008) Chimeric coronavirus‐like particles carrying severe acute respiratory syndrome coronavirus (SCoV) S protein protect mice against challenge with SCoV. Vaccine;26:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liniger M., Zuniga A., Tamin A., Azzouz‐Morin T.N., Knuchel M., Marty R.R., Wiegand M. et al. (2008) Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine;26:2164–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kam Y.W., Kien F., Roberts A., Cheung Y.C., Lamirande E.W., Vogel L., Chu S.L. et al. (2007) Antibodies against trimeric S glycoprotein protect hamsters against SARS‐CoV challenge despite their capacity to mediate FcγRII‐dependent entry into B cells in vitro. Vaccine;25:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Czub M., Weingartl H., Czub S., He R., Cao J. (2005) Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine;23:2273–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corapi W.V., Olsen C.W., Scott F.W. (1992) Monoclonal antibody analysis of neutralization and antibody‐dependent enhancement of feline infectious peritonitis virus. J Virol;66:6695–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He Y., Zhou Y., Liu S., Kou Z., Li W., Farzan M., Jiang S. et al. (2004) Receptor‐binding domain of SARS‐CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem Biophys Res Commun;324:773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Du L., Zhao G., He Y., Guo Y., Zheng B., Jiang S., Zhou Y. et al. (2007) Receptor‐binding domain of SARS‐CoV spike protein induces long‐term protective immunity in an animal model. Vaccine;25:2832–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tripet B., Kao D.J., Jeffers S.A., Holmes K.V., Hodges R.S. (2006) Template‐based coiled‐coil antigens elicit neutralizing antibodies to the SARS‐coronavirus. J Struct Biol;155:176–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raman S., Machaidze G., Lustig A., Aebi U., Burkhard P. (2006) Structure‐based design of peptides that self‐assemble into regular polyhedral nanoparticles. Nanomed Nanotechnol Biol Med;2:95–102. [DOI] [PubMed] [Google Scholar]
- 23. Burkhard P., Meier M., Lustig A. (2000) Design of a minimal protein oligomerization domain by a structural approach. Protein Sci;9:2294–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrison S.C. (2008) Viral membrane fusion. Nat Struct Mol Biol;15:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laue T.M., Shah B.D., Ridgeway T.M., Pelletier S.L. (1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science. London: Royal Society of Chemistry;p. 90–125. [Google Scholar]
- 26. Schuck P. (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J;78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stafford W.F. (1992) Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal Biochem;203:295–301. [DOI] [PubMed] [Google Scholar]
- 28. Philo J.S. (2000) A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal Biochem;279:151–163. [DOI] [PubMed] [Google Scholar]
- 29. Philo J.S. (2006) Improved methods for fitting sedimentation coefficient distributions derived by time‐derivative techniques. Anal Biochem;354:238–246. [DOI] [PubMed] [Google Scholar]
- 30. Branden C., Tooze J. (1999) Introduction to Protein Structure, 2nd edn New York, NY: Garland Publishing. [Google Scholar]
- 31. Zhou N.E., Monera O.D., Kay C.M., Hodges R.S. (1994) α‐Helical propensities of amino acids in the hydrophobic face of an amphipathic α‐helix. Protein Pept Lett;1:114–119. [Google Scholar]
- 32. Xiang S.D., Scholzen A., Minigo G., David C., Apostolopoulos V., Mottram P.L., Plebanski M. et al. (2006) Pathogen recognition and development of particulate vaccines: does size matter? Methods;40:1–9. [DOI] [PubMed] [Google Scholar]
- 33. Fifis T., Gamvrellis A., Crimeen‐Irwin B., Pietersz G.A., Li J., Mottram P.L., McKenzie I.F.C. et al. (2004) Size‐dependent immunogenicity: therapeutic and protective properties of nano‐vaccines against tumors. J Immunol;173:3148–3154. [DOI] [PubMed] [Google Scholar]
- 34. Padilla J.E., Colovos C., Yeates T.O. (2001) Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci USA;98:2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harbury P.B., Kim P.S., Alber T. (1994) Crystal structure of an isoleucine‐zipper trimer. Nature;371:80–83. [DOI] [PubMed] [Google Scholar]
- 36. Harbury P., Zhang T., Kim P., Alber T. (1993) A switch between two‐, three‐, and four‐stranded coiled coils in GCN4 leucine zipper mutants. Science;262:1401–1407. [DOI] [PubMed] [Google Scholar]
- 37. Hodges R., Heaton R., Parker J., Molday L., Molday R. (1988) Antigen–antibody interaction. Synthetic peptides define linear antigenic determinants recognized by monoclonal antibodies directed to the cytoplasmic carboxyl terminus of rhodopsin. J Biol Chem;263:11768–11775. [PubMed] [Google Scholar]