

Abstract
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death worldwide and is influenced by both genetic determinants and smoking. We identified genomic regions from 56 lung-tissue gene-expression microarrays and used them to select 889 SNPs to be tested for association with COPD. We genotyped SNPs in 389 severe COPD cases from the National Emphysema Treatment Trial and 424 cigarette-smoking controls from the Normative Aging Study. A total of 71 autosomal SNPs demonstrated at least nominal significance with COPD susceptibility (p = 3.4 × 10−6 to 0.05). These 71 SNPs were evaluated in a family-based study of 127 probands with severe, early-onset COPD and 822 of their family members in the Boston Early-Onset COPD Study. We combined p values from the case-control and family-based analyses, setting p = 5.60 × 10−5 as a conservative threshold for significance. Three SNPs in the iron regulatory protein 2 (IREB2) gene met this stringent threshold for significance, and four other IREB2 SNPs demonstrated combined p < 0.02. We demonstrated replication of association for these seven IREB2 SNPs (all p values ≤ 0.02) in a family-based study of 3117 subjects from the International COPD Genetics Network; combined p values across all cohorts for the main phenotype of interest ranged from 1.6 × 10−7 to 6.4 × 10−4. IREB2 protein and mRNA were increased in lung-tissue samples from COPD subjects in comparison to controls. In summary, gene-expression and genetic-association results have implicated IREB2 as a COPD susceptibility gene.
Introduction
Chronic obstructive pulmonary disease (OMIM 606963) is a genetically complex human disease. The identification of COPD susceptibility genes in particular, and genes for complex diseases in general, has proceeded through both candidate-gene approaches and positional-cloning approaches, but many findings have failed subsequent replication. An important feature of COPD is that cigarette smoking, the key environmental risk factor, can be quantified and included as a covariate in genetic-association studies.
Genome-wide association methods may uncover new susceptibility genes for complex diseases, but multidisciplinary methods that include integrative genomics may be a complementary approach to identifying previously unreported genes for complex diseases. Integrative genomic approaches, combining gene-expression data from both murine lung development and human lung tissue with human linkage and association analysis, have been used successfully by our research group for the identification of SERPINE2 as a putative COPD susceptibility gene.1,2
We hypothesized that a comprehensive evaluation of genes demonstrating altered expression patterns in lung tissue from individuals with COPD may assist in the identification of COPD genes that would not have been suspected on the basis of the known pathophysiology of COPD. We investigated this hypothesis by assessing genes differentially expressed in lung tissue from individuals with varying degrees of airflow obstruction and COPD; we then evaluted the identified SNPs in genes and genomic regions for association in a case-control cohort, followed by family-based genetic-association testing in two additional, independent cohorts. Some of the data presented here have previously been presented in the form of an abstract.3
Material and Methods
Microarray Gene-Expression Methods
The microarray methods and identification of differentially expressed genes in human lung tissue have been published.4 In brief, RNA was extracted from 56 lung-tissue specimens obtained during solitary lung-nodule resection; all tissue for the array analysis was distant from the nodule and without histologic evidence of malignancy.4 All patients provided written informed consent, underwent preoperative spirometry, and completed a questionnaire before surgery. Preoperative prebronchodilator lung function measures were used for classification of subjects, because not all subjects had postbronchodilator values. Percentage-based predicted values for forced expiratory volume in 1 s (FEV1) were calculated with Crapo equations for Whites and Hankinson equations for African Americans. Cases (n = 15) were defined as subjects with FEV1 < 70% predicted and FEV1/forced vital capacity (FVC) < 0.7 (mean FEV1 of the cases was 43% predicted), and controls (n = 18) as subjects with FEV1 > 80% predicted and FEV1/FVC > 0.7 (mean FEV1 of the controls was 101% predicted).4 Gene expression was assessed with the Affymetrix HG-U133 Plus 2.0 array, which interrogates 54,675 probe sets covering 47,000 human transcripts.
Sixty-five probe sets were identified as significantly differentially expressed between cases and controls as previously described.4 After the exclusion of three X chromosome probe sets (probe set ID nos. 1558515, 230986, and 235810), we used the location of the remaining 62 probe sets to identify regions for linkage disequilibrium (LD) tagging and SNP selection. The expression results for COPD cases and controls are summarized in the original manuscript published by Bhattacharya and colleagues4 (as an online supplement); we have listed the probe set ID numbers used for selection of gene regions (Tables S1 and S6, available online).
Cohorts
We used a tiered approach to identify SNPs that may be associated with COPD in three cohorts (Table 1). Genetic-association analysis was first performed in a case-control cohort (National Emphysema Treatment Trial [NETT] COPD cases and Normative Aging Study [NAS] controls), followed by replication in the Boston Early-Onset COPD (BEOCOPD) Study, an extended-pedigree family-based cohort. We combined the p values from the NETT-NAS and BEOCOPD association analyses to define genes to follow up in a third population, a family-based cohort with a broader range of FEV1 values (International COPD Genetics Network [ICGN]) (Figure 1). The institutional review boards (IRBs) of participating institutions in these studies approved the study protocol. Informed consent for use of blood samples for genetic studies was obtained for subjects from NETT, BEOCOPD, and ICGN studies. Anonymized data sets were used for genetic analysis of blood samples from NAS participants.
Table 1.
Demographics
| NETT-NAS |
BEOCOPD |
ICGN |
||||
|---|---|---|---|---|---|---|
| NETT | NAS | Probands | Relatives | Probands | Relatives | |
| Subjects | 389 | 472 | 127 | 822 | 1132 | 1985 |
| Female | 140 (35.99%) | 0 | 95 (74.80%) | 458 (55.72%) | 453 (40.02%) | 960 (48.36%) |
| Age | 66.69 (5.77) | 69.79 (7.53) | 48.09 (4.70) | 46.25 (18.75) | 58.31 (5.43) | 57.87 (9.42) |
| FEV1a | 0.81 (0.26) | 3.03 (0.50) | 0.65 (0.28) | 2.84 (1.03) | 1.12 (0.41) | 2.55 (1.01) |
| FEV1 % predicted | 28.02 (7.37) | 99.96 (13.12) | 21.86 (8.44) | 87.22 (20.27) | 36.08 (12.93) | 83.13 (26.08) |
| FEV1/FVCa | 0.32 (0.06) | 0.79 (0.05) | 0.31 (0.10) | 0.73 (0.13) | 0.37 (0.12) | 0.64 (0.14) |
| Pack-years of smoking | 66.33 (30.38) | 38.59 (26.66) | 38.86 (21.87) | 18.96 (25.04) | 52.16 (29.14) | 39.06 (25.00) |
| Current smokers | 0 | 31 (6.57%) | 16 (12.60%) | 248 (30.17%) | 369 (32.60%) | 1000 (50.38%) |
Values are shown as n (%) or mean (SD).
Spirometry measures presented in the table include postbronchodilator measures for BEOCOPD and ICGN and prebronchodilator measures for NETT and NAS; for ICGN, the ratio presented is FEV1 / Vital Capacity.
Figure 1.
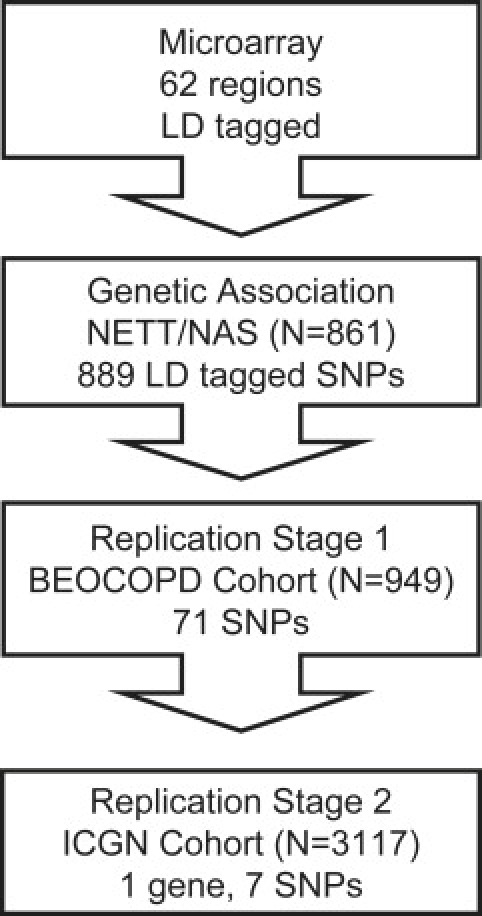
Workflow of Tiered Replication Approach: SNP Selection and Replication of SNP Associations
We performed LD tagging of 62 autosomal regions identified by microarray analysis on human lung tissue. LD tagging of the regions (plus or minus 5 kb from the start and stop locations) resulted in 889 SNPs tested for association in the NETT-NAS cohorts. We carried forward 71 SNPs associated at p ≤ 0.050 for attempted replication, using a family-based approach, in the BEOCOPD cohort. Finally, we performed a gene-based replication for IREB2 variants in the ICGN cohort, testing all seven SNPs that were associated in the NETT-NAS cohort.
Case-Control Cohort
NETT. The NETT was a multicenter randomized treatment trial for investigation of outcomes of medical therapy versus lung-volume-reduction surgery for the management of advanced COPD.5 A subset of individuals were subsequently enrolled in the NETT Genetics Ancillary Study and had blood samples for DNA available, as previously described.1 The current analysis included 389 non-Hispanic white cases with severe COPD. All subjects had FEV1 of less than or equal to 45% predicted and the presence of emphysema on high-resolution CT scan (performed at enrollment).5 Cigarette-smoke exposure was quantified from responses to questionnaire data.
NAS. We included 424 non-Hispanic white controls were from the NAS, a longitudinal study of aging conducted by the Veterans Administration.6 All control subjects had FEV1 measures greater than 80% of predicted, FEV1/FVC greater than 90% of predicted, and at least 10 pack-years of smoking. Cigarette-smoke exposure was quantified from responses to questionnaire data.
Family-Based Cohorts
BEOCOPD. The BEOCOPD cohort is an extended-pedigree study of genetic susceptibility to COPD; the ascertainment of probands and the recruitment of families have been reported previously.7 In brief, probands were ascertained on the basis of the presence of severe COPD, defined as FEV1 < 40% predicted before age 53 in the absence of severe alpha-1 antitrypsin deficiency. All family members were invited to participate, resulting in 949 total subjects included from 127 families. Each subject completed a modified version of the American Thoracic Society-Division of Lung Diseases questionnaire.8 Most subjects completed both pre- and postbronchodilator spirometry; postbronchodilator spirometry was performed approximately 15 min after the administration of albuterol. For the analysis of quantitative phenotypes, we focused on the absolute volume results for pre- and postbronchodilator FEV1 with covariate adjustments. Cigarette-smoke exposure was quantified from responses to questionnaire data. Given that the initial association step was for COPD susceptibility (presence or absence of COPD) in the NETT-NAS case-control analysis, we also performed an analysis of the presence or absence of moderate to severe COPD; for this analysis, a postbronchodilator FEV1 less than 60% predicted in the presence of FEV1/FVC less than 90% predicted defined moderate to severe COPD.1
ICGN. ICGN was a multicenter, multinational study of COPD, with probands ascertained on the basis of COPD as defined as postbronchodilator FEV1 < 60% predicted and FEV1/vital capacity (VC) < 90% predicted, age between 45 and 65, ≥ 5 pack-year smoking history, and at least one eligible sibling with ≥ 5 years of smoking.2 Eligible siblings were classified as having COPD if they had postbronchodilator FEV1 < 80% predicted and FEV1/VC < 90% predicted. Individuals with other lung diseases, such as lung cancer, tuberculosis, and pulmonary fibrosis, as well as subjects with severe alpha-1 antitrypsin deficiency, were excluded from the study. For the analysis of quantitative phenotypes, we focused on the absolute volume results for pre- and postbronchodilator FEV1 with covariate adjustments. Cigarette-smoke exposure was quantified from responses to questionnaire data.
Phenotypes and Covariates
Our tiered approach (Figure 1) included an initial assessment of genetic association of 889 SNPs with COPD susceptibility in the NETT-NAS case-control cohort. Our primary COPD quantitative phenotype in both family-based cohorts was FEV1, an important COPD intermediate phenotype that provides a quantitative assessment of COPD severity. Because association analysis of quantitative traits has increased power in comparison to qualitative phenotypes and because FEV1 is the primary measure of COPD severity, we analyzed FEV1 in the family-based cohorts and combined the resulting p values with those from the case-control association tests in the NETT-NAS screening cohort. For all subjects, cigarette-smoking intensity was calculated as: (number of cigarettes smoked per day / 20) / (age stopped smoking − age started smoking). For current smokers, the calculation of pack-years was the same, except that current age was used in the denominator instead of age stopped smoking being used. Covariates for family-based association testing of raw FEV1 values included age, height, gender, current smoking (yes or no), and pack-years of cigarette smoking.
SNP Selection and Genotyping
In the NETT-NAS case-control study, we genotyped 889 LD tagging SNPs in 62 regions identified after the addition of 5 kb upstream and downstream from the probe set location. We performed LD tagging with the Tagger program9 to capture an r2 of ≥ 0.8. Nonsynonymous SNPs were also genotyped if not captured by the LD tagging approach. Hardy-Weinberg equilibrium (HWE) was evaluated in the NAS control subjects. Subsequently, 71 associated SNPs (p ≤ 0.050) from the NETT-NAS experiment were genotyped in the BEOCOPD study, and seven SNPs in the Iron-Responsive Element-Binding Protein 2 gene (IREB2 [MIM 147582]) were genotyped in the ICGN cohort with the Sequenom iPLEX or TaqMan 5′ exonuclease assays (Applied Biosystems, Foster City, CA, USA). All SNP locations are referenced to UCSC human genome browser (hg18, March 2006, dbSNP build 129). Gene names provided in Tables 2, 3, and 4, and in Tables S1, S2, S3, and S6 represent the HUGO gene names (verified June 2009) of the gene closest to the studied SNP.
Table 2.
Association Results for the Top 17 Significant Autosomal SNPs Tested in the NETT-NAS Cohort
| Chr | HUGO Gene Name | SNP | dbSNP Location (Build 129) | p Value Genotypic Trend Test |
|---|---|---|---|---|
| 9 | PTCH1 | rs16909859 | 97244613 | 3.40 × 10−6 |
| 15 | IREB2 | rs2656069 | 76532762 | 1.03 × 10−5 |
| 15 | IREB2 | rs10851906 | 76561731 | 2.46 × 10−5 |
| 15 | IREB2 | rs2568494 | 76528019 | 4.40 × 10−5 |
| 15 | SEMA6D | rs7167021 | 45833273 | 1.92 × 10−4 |
| 1 | SYDE2 | rs7511739 | 85395045 | 3.15 × 10−4 |
| 15 | SEMA6D | rs17388803 | 45814496 | 3.25 × 10−4 |
| 15 | SEMA6D | rs16960030 | 45838944 | 4.58 × 10−4 |
| 15 | IREB2 | rs1964678 | 76541055 | 1.61 × 10−3 |
| 15 | IREB2 | rs965604 | 76576278 | 1.81 × 10−3 |
| 15 | IREB2 | rs13180 | 76576543 | 2.17 × 10−3 |
| 15 | SEMA6D | rs76739 | 45829438 | 2.76 × 10−3 |
| 15 | IREB2 | rs12593229 | 76552345 | 2.95 × 10−3 |
| 6 | PHACTR2 | rs9386042 | 144061147 | 3.44 × 10−3 |
| 3 | TIGIT | rs4682534 | 115516788 | 4.02 × 10−3 |
| 1 | ARHGAP29 | rs3789689 | 94458173 | 4.05 × 10−3 |
| 15 | SEMA6D | rs765 | 45822977 | 5.40 × 10−3 |
Table 3.
Pedigree-Based Association Test for Postbronchodilator FEV1 in BEOCOPD Families
| Chr | HUGO Gene Name | SNP | dbSNP Location (Build 129) | Minor Allele | MAF for BEOCOPD | Familiesa | NETT-NAS p Value | BEOCOPD p Valueb | Combined p Valuec |
|---|---|---|---|---|---|---|---|---|---|
| 15 | IREB2 | rs2568494d | 76528019 | A | 0.38 | 23 | 4.40 × 10−5 | 9.25 × 10−3 | 6.39 × 10−6 |
| 15 | IREB2 | rs2656069d | 76532762 | C | 0.19 | 10 | 1.03 × 10−5 | 0.35 | 4.93 × 10−5 |
| 15 | IREB2 | rs10851906 | 76561731 | G | 0.18 | 11 | 2.46 × 10−5 | 0.21 | 6.68 × 10−5 |
| 6 | PHACTR2 | rs9386042 | 144061147 | C | 0.25 | 10 | 3.44 × 10−3 | 0.07 | 2.37 × 10−3 |
| 21 | CYYR1 | rs222973 | 26800940 | T | 0.33 | 15 | 0.02 | 0.01 | 2.44 × 10−3 |
| 14 | DAAM1 | rs17833769 | 58725214 | A | 0.23 | 10 | 7.32 × 10−3 | 0.08 | 4.97 × 10−3 |
| 15 | SEMA6D | rs76739 | 45829438 | G | 0.41 | 22 | 2.76 × 10−3 | 0.28 | 6.24 × 10−3 |
| 15 | IREB2 | rs1964678 | 76541055 | A | 0.35 | 20 | 1.61 × 10−3 | 0.76 | 9.44 × 10−3 |
| 15 | IREB2 | rs965604 | 76576278 | G | 0.35 | 22 | 1.81 × 10−3 | 0.75 | 0.01 |
| 15 | IREB2e | rs13180 | 76576543 | C | 0.36 | 20 | 2.17 × 10−3 | 0.71 | 0.01 |
| 15 | SEMA6D | rs634939 | 45858069 | C | 0.41 | 16 | 0.01 | 0.18 | 0.01 |
| 6 | PHACTR2 | rs9321939 | 144096465 | C | 0.20 | 14 | 0.01 | 0.20 | 0.01 |
Abbreviations are as follows: Chr, chromosome; MAF, minor allele frequency.
Informative families for PBAT analysis for BEOCOPD.
BEOCOPD p value for postbronchodilator FEV1.
Fisher combined p value; same direction of effect for the risk alleles.
Met Fisher combined p value threshold of < 5.60 × 10−5.
Genome-wide association study data from British 1958 Birth Cohort Study for FEV1 p = 3 × 10−2.
Table 4.
Pedigree-Based Association Test for BEOCOPD Families for Presence or Absence of Moderate to Severe COPD
| Chr | HUGO Gene Name | SNP | dbSNP Location (Build 129) | Minor Allele | MAF for BEOCOPD | Informative Families for PBAT Analysis for BEOCOPD | NETT-NAS p Value | BEOCOPD p Value for Moderate to Severe COPD | Combined p Value: Fisher Methodc |
|---|---|---|---|---|---|---|---|---|---|
| 15 | IREB2 | rs2568494 a | 76528019 | A | 0.38 | 23 | 4.40 × 10−5 | 0.01 | 9.01 × 10−6 |
| 15 | IREB2 | rs2656069a | 76532762 | C | 0.19 | 10 | 1.03 × 10−5 | 0.27 | 3.88 × 10−5 |
| 15 | IREB2 | rs10851906a | 76561731 | G | 0.18 | 11 | 2.46 × 10−5 | 0.17 | 5.51 × 10−5 |
| 6 | PHACTR2 | rs9386042 | 144061147 | C | 0.25 | 10 | 3.44 × 10−3 | 6.22x10−3 | 2.52 × 10−4 |
| 15 | SEMA6D | rs11855291 | 45858167 | A | 0.23 | 13 | 8.28 × 10−3 | 0.08 | 5.39 × 10−3 |
| 21 | CYYR1 | rs222973 | 26800940 | T | 0.33 | 15 | 0.02 | 0.04 | 6.37 × 10−3 |
| 15 | IREB2 | rs965604 | 76576278 | G | 0.35 | 22 | 1.81 × 10−3 | 0.50 | 7.25 × 10−3 |
| 15 | IREB2 | rs1964678 | 76541055 | A | 0.35 | 20 | 1.61 × 10−3 | 0.57 | 7.40 × 10−3 |
| 15 | IREB2b | rs13180 | 76576543 | C | 0.36 | 20 | 2.17 × 10−3 | 0.44 | 7.60 × 10−3 |
| 1 | EIF2C4 | rs727005 | 36053923 | G | 0.09 | 10 | 0.04 | 0.02 | 8.02 × 10−3 |
| 12 | AMIGO2 | rs2898002 | 45762494 | G | 0.29 | 22 | 0.01 | 0.15 | 0.01 |
| 15 | IREB2 | rs12593229 | 76552345 | T | 0.35 | 21 | 2.95 × 10−3 | 0.55 | 0.01 |
| 15 | SEMA6D | rs76739 | 45829438 | G | 0.41 | 22 | 2.76 × 10−3 | 0.72 | 0.01 |
MAF denotes minor allele frequency.
Met Fisher combined p value threshold of < 5.60x10−5.
Genome-wide association study data from British 1958 Birth Cohort Study for FEV1 p = 3x10−2.
Same direction of effect for the risk alleles.
Statistical Methods
NETT-NAS Case-Control Analysis
SNPs were analyzed for association with COPD susceptibility in the NETT-NAS study with the use of an additive genetic model as implemented in SAS Genetics. HWE in the control subjects was evaluated for each SNP via a goodness-of-fit test. Association between SNPs and COPD case-control status was evaluated with the use of 2×3 contingency tables for investigation of allelic additive effects with the Cochran-Armitage trend test; 71 SNPs with an association p value ≤ 0.050 from the trend test were carried forward to the next step of replication in the BEOCOPD cohort.
Pedigree-Based Association Test
The quantitative phenotypes of pre- and postbronchodilator FEV1 were analyzed in the BEOCOPD and ICGN family-based studies; qualitative phenotypes for presence or absence of COPD were also investigated in the BEOCOPD cohort. We utilized the extension of the TDT as implemented in PBAT (UNIX version 3.5). We analyzed pre- and postbronchodilator FEV1 in both the BEOCOPD and the ICGN cohorts. The analyses for FEV1 were adjusted for age, height, sex, current smoking (yes or no), and pack-years of cigarette smoking. Analysis results were evaluated for only those SNPs with at least ten informative families in the BEOCOPD and ICGN studies. All PBAT models were run under the assumption of an additive mode of inheritance.
We used Fisher's method for combining p values from the case-control and family-based association tests. The direction of effect of the risk allele in all cohorts needed to be in the same direction to be considered acceptable for combining. For the combination of the case-control and BEOCOPD p values, a stringent threshold of significance, based on a Bonferroni correction for all SNPs which ignores LD between SNPs, was set at a combined p value less than 5.6 × 10−5 (∼0.05 / 889; a 4 degree of freedom [df] test). The combination of p values from all three cohorts was a 6 df test with the use of the Fisher method.
Evaluation of IREB2 Protein and mRNA in Human Lung
All human lung tissues for use in the protein expression experiments were obtained from either lung-transplantation explant tissues or tissues from consenting patients undergoing thoracic surgery. These lung sections were processed immediately after resection, and the isolated lung tissues were snap frozen and stored at −80°C. Lung homogenates from these frozen tissues were subjected to immunoblot analyses. Tissue from 22 COPD subjects and 18 controls were available for IREB2 protein expression studies. We isolated mRNA from these same lung tissues and performed RT-PCR with the use of human primers (Taqman human Hs_00386293-m1) for IREB2; RT-PCR data are referenced to β-glucuronidase (GusB).
IREB2 antibodies were obtained from Alpha Diagnostic International (San Antonio, TX, USA), and immunoblot analysis was performed via standard methods. The Student's t test was used for statistical comparisons.
Immunohistochemistry
Frozen sections from human lung were available. Control lung samples were obtained during lobectomy for lung nodules; lung tissue for cases consisted of tissues from lungs explanted during lung transplantation for COPD. Samples were hydrated and blocked with 3% hydrogen peroxide in methanol for 30 min. After blocking with 10% normal goat serum, tissues sections were immunoreacted overnight with IREB2 antibodies (Alpha Diagnostic, no. IRP21A, or Santa Cruz, no. sc-33682; 1:100). Bound primary antibodies were visualized with diaminobenzidine staining with the use of ABC kits (Vector Laboratories), and nuclei were counterstained with hematoxylin (Sigma). Both antibodies have shown similar potency and immunoreactivity. A probed tissue without primary antibody served as a negative control.
Results
The relevant demographics of all three study cohorts used in genetic association analysis are described in Table 1.
We tested 889 SNPs for association in the NETT-NAS cohort. Of these SNPs, 17 were significant at p ≤ 5.0 × 10−3(Table 2); a total of 71 SNPs demonstrated nominal significance in the case-control analysis at p ≤ 0.050 (Table S1), with 34 SNPs significant at p ≤ 0.01. The 71 SNPs significant at p ≤ 0.050 were carried forward for attempted replication via a family-based association approach in the BEOCOPD study. Of note, six SNPs were out of HWE in the NAS controls (Table S1), but we did opt to carry forward these SNPs for further assessment in our family-based cohort.Association results for all 889 tested SNPs are included in Table S6.
Family-Based Analysis
For the BEOCOPD analysis of pre- and postbronchodilator FEV1, of the 71 SNPs tested, 43 SNPs met the threshold for having at least ten informative families contributing to the PBAT analysis; these data are presented in Table 3 for postbronchodilator FEV1 for those SNPs with combined p ≤ 0.01 (results for pre- and postbronchodilator FEV1 with p ≤ 0.05 are in Tables S2 and S3). Two SNPs had p values less than 0.05 for both pre- and postbronchodilator FEV1 (IREB2 rs2568494, CYYR1 rs222973). We combined all p values from the BEOCOPD with the results from the NETT-NAS analysis, using a stringent threshold value for significance (0.05 / 889; 5.6 × 10−5). With the use of Fisher's method, two SNPs in IREB2 met this threshold for postbronchodilator FEV1 (rs2568494, p = 6.4 × 10−6; rs2656069, p = 4.9 × 10−5) (Table 3, Table S2), and one additional SNP in the IREB2 gene (rs10851906) met this stringent p value threshold for prebronchodilator FEV1 (Table S3). The combined p values for association for these three IREB2 SNPs were all less than 5.6 × 10−5 for the qualitative phenotype of moderate to severe COPD (Table 4).
We carried forward the three globally significant IREB2 SNPs (rs2568494, rs2656069, and rs10851906), as well as the four other LD tagging IREB2 SNPs significant in NETT-NAS analysis (rs1964678, rs12593229, rs965604, and rs13180), to perform gene-based replication testing in the ICGN cohort. All seven IREB2 SNPs were associated with pre- and postbronchodilator FEV1 in the ICGN cohort (Table 5 and Table S4). The combined p values (6 df test) across all three cohorts ranged in magnitude from 10−4 to 10−7 (Table 5 and Table S4).
Table 5.
Pedigree-Based Association Test for IREB2 SNPs with Postbronchodilator FEV1 in ICGN Families
| SNP | Minor Allele | MAF for ICGN | Risk Allele | Informative Families for PBAT Analysis for ICGN | ICGN p Value for Postbronchodilator FEV1 | NETT-NAS p Value for Case-Control Status | BEOCOPD p Value for Postbronchodilator FEV1 | Combined p Value: Fisher Method a |
|---|---|---|---|---|---|---|---|---|
| rs2568494 | A | 0.42 | A | 479 | 1.64 × 10−3 | 4.40 × 10−5 | 9.25x10−3 | 1.64 × 10−7 |
| rs2656069 | C | 0.19 | T | 386 | 0.02 | 1.03 × 10−5 | 0.35 | 1.03 × 10−5 |
| rs1964678 | A | 0.35 | G | 484 | 5.84 × 10−3 | 1.61 × 10−3 | 0.76 | 5.94 × 10−4 |
| rs12593229 | T | 0.35 | G | 481 | 4.68 × 10−3 | 2.95 × 10−3 | 0.87 | 9.21 × 10−4 |
| rs10851906 | G | 0.19 | A | 384 | 0.02 | 2.46 × 10−5 | 0.21 | 1.65 × 10−5 |
| rs965604 | G | 0.35 | A | 483 | 4.71 × 10−3 | 1.81 × 10−3 | 0.75 | 5.42 × 10−4 |
| rs13180 | C | 0.36 | T | 467 | 5.07 × 10−3 | 2.17 × 10−3 | 0.71 | 6.42 × 10−4 |
MAF denotes minor allele frequency.
Same direction of effect for the risk alleles.
As we previously reported,4 microarray analysis used for selecting genes of interest identified differential IREB2 expression patterns in COPD cases compared to controls. The microarray results4 suggested decreased IREB2 expression in COPD cases. qPCR-defined gene expression patterns from this published cohort were significantly correlated with the array data, but they did not confirm statistically significant differences for IREB2 expression between cases and controls. Given our replicated genetic association data for IREB2 SNPs, we performed additional studies examining IREB2 protein and mRNA levels in human COPD lung tissues to clarify the direction of mRNA and protein levels between COPD case subjects and control subjects. We observed significantly higher IREB2 protein levels in emphysematous lung tissue (n = 22) in comparison to controls (n = 18) (p < 0.01) (Figure 2). RNA was isolated from these same lung tissues, and PCR demonstrated increased IREB2 mRNA in cases (n = 20) versus controls (n = 11) (Figure 2, p < 0.05). IREB2 protein localized to airway epithelial cells, endothelial cells, and smooth muscle cells in the blood vessels and to immune cells (macrophages) in the lung (Figure 3). In the airway epithelial cells, staining was pronounced at the cilial surface (Figure 4).
Figure 2.
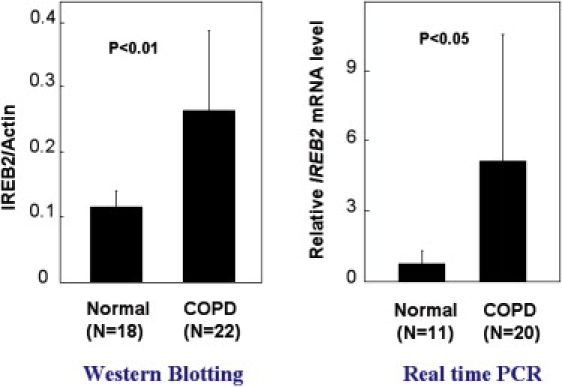
Increased IREB2 Protein and mRNA Expression in Human COPD Lung Tissues
Human lung-tissue homogenates from control (n = 18) and COPD (n = 22) subjects were analyzed for IREB2 protein expression by immunoblot analysis. Total IREB2 protein expression was normalized to β-actin, and the quantified results are depicted by graph. Data represent mean ± SD (n = 18 normal and n = 22 COPD patients), p < 0.01, comparing COPD lung tissue with normal lung. mRNA expression was extracted from the same lung tissues and quantified with RT-PCR (n = 11 normal and n = 20 COPD patients), p < 0.05, comparing COPD lung tissue with normal lung.
Figure 3.
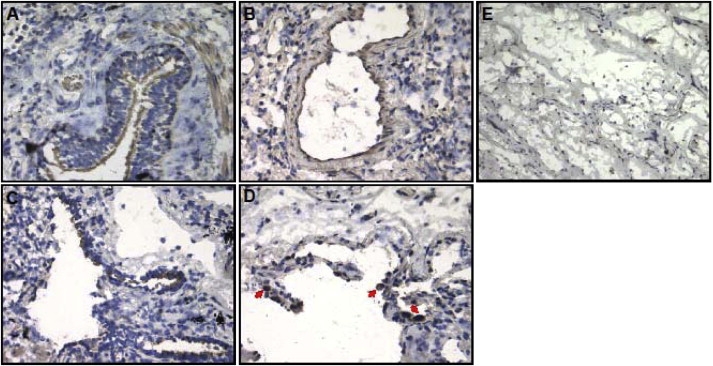
IREB2 Localization in Normal and Emphysematous Human Lung Tissue
(A–D) IREB2 protein expression by DBA staining (brown) and counterstaining by nuclei staining (blue). IREB2 expression in control tissue localized to (A) bronchial epithelial cells and (B) vascular endothelial cells; IREB2 expression in emphysematous tissue localized to (C) bronchial epithelial cells and (D) macrophages (red arrows).
(E) Negative control without primary antibody in control tissue. Magnification is 200×.
Figure 4.
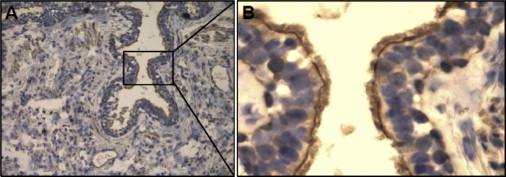
IREB2 Localizes to Human Epithelial Cell Cilial Surface
Localization of IREB2 protein expression along the cilial border in human lung epithelial cells from normal lung tissue
(A) 200× magnification.
(B) Enlarged image of (A) at 400× magnification.
Discussion
Integrative genomics approaches can be complementary to genome-wide association studies for facilitating the identification of previously unreported candidate genes for complex human diseases. Using a tiered integrative genomics approach, we identified the strongest genetic association of COPD-related phenotypes with variants in IREB2 (chromosome 15q25.1). We have demonstrated that IREB2 may be a COPD susceptibility gene, identified through the integration of human lung microarray gene expression data and replicated genetic association studies in three cohorts. Interestingly, IREB2 falls into the nucleic acid binding and metabolic process GO pathways identified in the microarray studies by Bhattacharya and colleagues.4 We subsequently went on to observe that IREB2 protein expression was higher in the lung tissue samples from subjects with COPD. IREB2 variants are in tight LD with SNPs associated with nicotine addiction. However, our data from human gene expression, genetic, and lung-tissue studies, as well as prior studies suggesting a role for lung iron imbalance in pulmonary inflammation,10,11 support a role for IREB2 in the pathogenesis of COPD.
The region that includes IREB2 has recently been significantly associated with COPD in a genome-wide association study12. This region on 15q, which includes several genes with variants in tight LD, including components of the nicotinic acetylcholine receptor, has also been identified in genome-wide association studies of lung cancer, a pulmonary disease also significantly influenced by cigarette smoking.13–15 Our integrative genomics and functional results suggest that IREB2 may be at least one of the key genes influencing COPD development in this region.
The regulation of iron uptake and iron distribution throughout the human body is under tight homeostatic control by iron regulatory proteins (IRPs). The protein product of IREB2 (also known as IRP2) is an RNA binding protein that, together with IREB1, is involved in maintaining human cellular iron metabolism. The IRPs sense cytosolic iron levels, and in response to iron, they modulate the expression of those proteins relevant to iron uptake, export, and sequestration.16 In the setting of systemic iron depletion, IRPs decrease iron storage and increase iron uptake.17 Hypoxemia is a common occurrence in COPD; important features of IREB2 are that it is suggested to be active at lower oxygen tensions16 and has been observed to be posttranslationally regulated by hypoxia.18 The IREB2 knockout mouse has been observed to develop microcytic anemia and neurodegeneration; this neurodegeneration is probably due to dysregulation of iron in the brain.19
Regional variation of iron and iron binding proteins has been demonstrated in the lungs of smokers20. Nelson and colleagues investigated whether concentrations of iron, ferritin, and transferrin varied in upper versus lower lobes of smokers. Bronchoalveolar lavage (BAL) samples from the upper lobes of smokers had higher concentrations of iron in comparison to the lower lobes, whereas BAL fluid levels from nonsmokers did not vary. They concluded that the distribution of lung iron and iron binding proteins may lead to varying burdens of oxidative injury in the lung and may be relevant to the pathogenesis of emphysema and lung cancer, both of which occur more commonly in the upper lobes of the lungs. Subsequently, this same research group observed regional variation in alveolar iron and IL1Beta, suggesting that the burden of iron has an impact upon regional inflammation in the lung.10 We have previously demonstrated association with xenobiotic metabolizing enzymes with emphysema distribution in the NETT cohort, and we speculated that there may be gradients of gene and protein expression in the lung that contribute to COPD. Although we did not investigate regional differences in IREB2 expression in human lung tissue, there was a trend for association for five IREB2 SNPs (rs1964678 p = 0.05; rs2656069 p = 0.07; rs12593229, rs965604, rs13180 p = 0.09) with upper lobe percentage of emphysema in a subset of 282 NETT subjects who had densitometric quantification of emphysema; there was no association with a phenotype that measured emphysema distribution (p > 0.05, data not shown). More research on regional distribution of IREB2 gene expression and IREB2 protein levels in the lung is required for clarification of these findings.
The only previous candidate gene association study of IREB2 variants with human disease is for Alzheimer disease,21 a disease associated with human aging. Features of COPD recapitulate characteristics of the lung that may occur with aging. Deposition and accumulation of iron is associated with cellular senescence,22 and iron in the lung has been noted to increase with age in humans and rats.23 Cigarette smoking has been associated with higher accumulated iron levels in the lung.11,24,25 Ghio and colleagues recently demonstrated that healthy smokers and smokers with COPD had higher concentrations of iron in the lung and systemically, suggesting that the disruption of iron homeostasis associated with smoking may be one contributing factor for the development of COPD long after smoking cessation.11 Thus, polymorphic variation in IREB2, as a major regulator of iron homeostasis, may further affect COPD when coupled with the increased levels of iron accumulated through exposure to cigarette smoke.
Increases in cell autophagy have been associated with smoking-related lung injury and COPD.26 Interestingly, we have recently observed that iron is required for autophagy, as treatment with the iron chelator desferoxamine inhibits autophagy and administering iron induces autophagy in both epithelial and endothelial cells (A.M.K.C., unpublished data). Whether an effect of IREB2 on COPD is through regulating autophagy is speculative but an important question that future studies should address.
Lung cancer and COPD probably share common genetic and environmental susceptibility factors.27 Three independent groups performing genome-wide association studies have identified the chromosome 15q25 region (that includes IREB2) as associated with lung cancer.13–15 Our independent identification of IREB2 via an integrative genomics approach suggests that this may be an important region for consideration with regard to combined susceptibility to COPD and lung cancer. In our study, the two IREB2 SNPs identified in the associated region in the lung cancer genome-wide association study by Hung and colleagues15 (rs17484235 and rs2656052) were associated with COPD case-control status (p = 1.16 × 10−3, 1.81 × 10−5 respectively) in NETT-NAS and with postbronchodilator FEV1 (all p < 0.01) in our BEOCOPD family-based cohort, for combined p values that range in magnitude from 10−4 to 10−6 for these SNPs (Table S5). Thus, IREB2 might be a gene relevant to combined susceptibility to COPD and lung cancer, or this gene might be independently associated with smoking behavior (although we did not detect an independent association for these SNPs with pack-years of cigarette smoking; data not shown). Alternatively, our association findings in IREB2 may be the result of high LD with SNPs in the CHRNA3/5 gene cluster (Figure 5), and key functional variants may actually be in the CHRNA3/5 gene cluster. However, the fact that we independently identified the IREB2 region and not the CHRNA3/5 region via gene expression analysis, together with the functional studies of IREB2 in human lung tissue, suggest that IREB2 may harbor key functional variation, either in addition to or instead of the CHRNA3/5 gene cluster. The identification of functional variants will be required for clarification of these issues.
Figure 5.
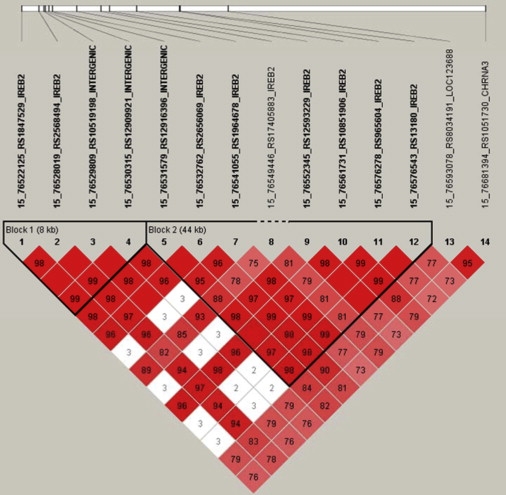
Linkage Disequilibrium Map for All IREB2 SNPs Genotyped in NETT-NAS Plus Two CHRNA3 Region SNPs Previously Associated with Lung Cancer and COPD
LD map of the 12 SNPs genotyped in IREB2 in NETT-NAS demonstrate high LD across the gene. Additionally, there is high LD in the NETT-NAS cohort between the IREB2 SNPs and two SNPs in the CHRNA3 region (rs8034191, rs1051730; association data published by Pillai et al.12) Red corresponds to r2 ≥ 0.8. Values for D′ are included in the text of boxes.
After identification of the IREB2 gene as a candidate via gene expression studies, we observed association in three cohorts, with a combined p value threshold meeting a conservative threshold of at least 10−5. Limitations of our study included the fact that genes were identified through an expression data set of lung nodule resections, and this panel of genes may pick up signal relevant to lung neoplastic changes. However, a recent paper demonstrated increased mRNA of CHRNA5 but no change in IREB2 mRNA in lung adenocarcinomas, supporting the idea that our finding is independent of the gene expression patterns of the nodule tissue itself.28 Also of note is that the microarray data for the discovery phase (Bhattacharya et al.4) did not validate the direction of differential expression of IREB2 in the COPD cases versus controls, but the mRNA and protein data that we present (Figure 2) suggest an increase in IREB2 in subjects with COPD in comparison to controls. Of note, none of the subjects in the COPD genetic association study cohorts had known cancer, and the fact that our findings replicated for key intermediate COPD phenotypes is crucial. We do not know the IREB2 genotypes of the subjects from the microarray study; additionally, microarray data were considered from a small number of subjects that fit into stringent categories for COPD case (n = 15) versus control (n = 18) status.4 An additional consideration includes the differences in phenotypes used for gene selection (case-control status) versus replication of results (FEV1 for the family-based cohorts). The mean FEV1 for the cases in NETT and the BEOCOPD probands is not statistically different (p > 0.05); also, our findings for a qualitative phenotype association (for presence or absence of moderate to severe COPD) in BEOCOPD still identify IREB2 as the most highly associated gene for further investigation (p = 9.0 × 10−6; Table 4). Quantitative traits have increased power in comparison to qualitative traits, so we opted for attempted replication for FEV1 in our second family-based cohort, given the broader range of FEV1 in ICGN that captures a span of COPD severity. The functional relevance of this association study and true functional variants in IREB2, if they exist, are yet to be defined. However, the rs13180 SNP was significantly associated in the NETT-NAS case-control status and the ICGN analyses for FEV1. This SNP is reported to be in complete LD with a functional promoter SNP (rs2656070) that disrupts SP1 and AP1 binding sites21. Although the rs13180 association did not replicate in our BEOCOPD cohort, the combined p value across all three studies was robust and in the same direction. This SNP also has a nominal p value for association to the FEV1 phenotype in the British Birth Cohort, a genome-wide association data set (public access).Our findings may be due to spurious association, but independent replication in multiple cohorts suggests otherwise. We have demonstrated association with SNP variants in two family-based studies, suggesting that the role for potential population stratification is limited.
In conclusion, using an integrative genomics approach, we have identified IREB2 as a potential gene for COPD susceptibility. This approach has highlighted not only the iron regulation pathways as potentially important to COPD, but also the complementary role of integrative genomics with genome-wide association studies. The identification of IREB2 variants associated with COPD in a region associated with lung cancer suggests the potential for overlapping genetic determinants of these two smoking-related diseases. A role for IREB2 in the lung may be through influences on lung iron stores and subsequent oxidative stress or alterations in autophagy; these pathways need to be fully investigated in both animal models and human cohorts.
Supplemental Data
Supplemental Data include six tables and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
British 1958 Birth Cohort Study, http://www.b58cgene.sgul.ac.uk
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
University of California-Santa Barbara (UCSC) human genome browser, http://genome.ucsc.edu/
Acknowledgments
We acknowledge the NETT Genetics Ancillary Study co-investigator group, including Joshua Benditt, Gerard Criner, Malcolm DeCamp, Philip Diaz, Mark Ginsburg, Larry Kaiser, Marcia Katz, Mark Krasna, Neil MacIntyre, Barry Make, Rob McKenna, Fernando Martinez, Zab Mosenifar, Andrew Ries, Paul Scanlon, Frank Sciurba, and James Utz. We acknowledge the contribution of the International COPD Genetics Network (ICGN) investigators for the development of the ICGN cohort. In addition to E.S. and D.L. (co-authors), the ICGN investigators include A. Agusti, P. M.A. Calverley, C.F. Donner, R.D. Levy, B. J. Make, P.D. Paré, J. Vestbo, S.I. Rennard, and E. F.M. Wouters. Support for this project was provided by National Institutes of Health (NIH) grants HL072918, HL71885, and HL72303 and by a Doris Duke Clinical Scientist Award (D.L.D.). The NETT-NAS and BEOCOPD studies were supported by NIH grants HL075478, HL084323, and HL083069 (E.K.S.). Additionally the NETT was supported by the National Heart, Lung and Blood Institute (grants N01HR76101, N01HR76102, N01HR76103, N01HR76104, N01HR76105, N01HR76106, N01HR76107, N01HR76108, N01HR76109, N01HR76110, N01HR76111, N01HR76112, N01HR76113, N01HR76114, N01HR76115, N01HR76116, N01HR76118, N01HR76119), the Centers for Medicare and Medicaid Services, and the Agency for Healthcare Research and Quality. The Normative Aging Study is supported by the Cooperative Studies Program/Epidemiology Research and Information Center of the U.S. Department of Veterans Affairs and is a component of the Massachusetts Veterans Epidemiology Research and Information Center (MAVERIC), Boston, MA, USA. The development of the ICGN cohort was funded by GlaxoSmithKline. The collection of lung tissue at Temple Lung Center was supported by the Pennsylvania Department of Health (PA-DOH 02-70-02).
References
- 1.Demeo D.L., Mariani T.J., Lange C., Srisuma S., Litonjua A.A., Celedon J.C., Lake S.L., Reilly J.J., Chapman H.A., Mecham B.H. The SERPINE2 gene is associated with chronic obstructive pulmonary disease. Am. J. Hum. Genet. 2006;78:253–264. doi: 10.1086/499828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu G., Warren L., Aponte J., Gulsvik A., Bakke P., Anderson W.H., Lomas D.A., Silverman E.K., Pillai S.G. The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am. J. Respir. Crit. Care Med. 2007;176:167–173. doi: 10.1164/rccm.200611-1723OC. [DOI] [PubMed] [Google Scholar]
- 3.DeMeo, D. L., Mariani, T., Lange, C., Bhattacharya, S., Srisuma, S., Shapiro, S., Bueno, R., Silverman, E. K., and Reilly, J.R. (2007). Genomic and geneitc approaches identify IREB2 as a novel susceptibility gene for chronic obstructive pulmonary disease [abstract92]. Presented at the annual meeting of The American Society of Human Genetics, October 2007, San Diego, California Available from http://www.ashg.org/genetics/ashg07s/index.shtml.
- 4.Bhattacharya S., Srisuma S., Demeo D.L., Shapiro S.D., Bueno R., Silverman E.K., Reilly J.J., Mariani T.J. Molecular biomarkers for quantitative and discrete COPD phenotypes. Am. J. Respir. Cell Mol. Biol. 2009;40:359–367. doi: 10.1165/rcmb.2008-0114OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rationale and design of the National Emphysema Treatment Trial (NETT): A prospective randomized trial of lung volume reduction surgery. J. Thorac. Cardiovasc. Surg. 1999;118:518–528. doi: 10.1016/s0022-5223(99)70191-1. [DOI] [PubMed] [Google Scholar]
- 6.Bell B., Rose C.L., Damon A. The Veterans Administration longitudinal study of healthy aging. Gerontologist. 1966;6:179–184. doi: 10.1093/geront/6.4.179. [DOI] [PubMed] [Google Scholar]
- 7.Silverman E.K., Chapman H.A., Drazen J.M., Weiss S.T., Rosner B., Campbell E.J., O'Donnell W.J., Reilly J.J., Ginns L., Mentzer S. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1998;157:1770–1778. doi: 10.1164/ajrccm.157.6.9706014. [DOI] [PubMed] [Google Scholar]
- 8.Ferris B.G. Epidemiology Standardization Project (American Thoracic Society) Am. Rev. Respir. Dis. 1978;118:1–120. [PubMed] [Google Scholar]
- 9.de Bakker P.I., Yelensky R., Pe'er I., Gabriel S.B., Daly M.J., Altshuler D. Efficiency and power in genetic association studies. Nat. Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 10.O'Brien-Ladner A.R., Nelson S.R., Murphy W.J., Blumer B.M., Wesselius L.J. Iron is a regulatory component of human IL-1beta production. Support for regional variability in the lung. Am. J. Respir. Cell Mol. Biol. 2000;23:112–119. doi: 10.1165/ajrcmb.23.1.3736. [DOI] [PubMed] [Google Scholar]
- 11.Ghio A.J., Hilborn E.D., Stonehuerner J.G., Dailey L.A., Carter J.D., Richards J.H., Crissman K.M., Foronjy R.F., Uyeminami D.L., Pinkerton K.E. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am. J. Respir. Crit. Care Med. 2008;178:1130–1138. doi: 10.1164/rccm.200802-334OC. [DOI] [PubMed] [Google Scholar]
- 12.Pillai S.G., Ge D., Zhu G., Kong X., Shianna K.V., Need A.C., Feng S., Hersh C.P., Bakke P., Gulsvik A. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amos C.I., Wu X., Broderick P., Gorlov I.P., Gu J., Eisen T., Dong Q., Zhang Q., Gu X., Vijayakrishnan J. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorgeirsson T.E., Geller F., Sulem P., Rafnar T., Wiste A., Magnusson K.P., Manolescu A., Thorleifsson G., Stefansson H., Ingason A. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hung R.J., McKay J.D., Gaborieau V., Boffetta P., Hashibe M., Zaridze D., Mukeria A., Szeszenia-Dabrowska N., Lissowska J., Rudnai P. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 16.Rouault T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 17.Rouault T., Klausner R. Regulation of iron metabolism in eukaryotes. Curr. Top. Cell. Regul. 1997;35:1–19. doi: 10.1016/s0070-2137(97)80001-5. [DOI] [PubMed] [Google Scholar]
- 18.Hanson E.S., Foot L.M., Leibold E.A. Hypoxia post-translationally activates iron-regulatory protein 2. J. Biol. Chem. 1999;274:5047–5052. doi: 10.1074/jbc.274.8.5047. [DOI] [PubMed] [Google Scholar]
- 19.LaVaute T., Smith S., Cooperman S., Iwai K., Land W., Meyron-Holtz E., Drake S.K., Miller G., Abu-Asab M., Tsokos M. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 20.Nelson M.E., O'Brien-Ladner A.R., Wesselius L.J. Regional variation in iron and iron-binding proteins within the lungs of smokers. Am. J. Respir. Crit. Care Med. 1996;153:1353–1358. doi: 10.1164/ajrccm.153.4.8616566. [DOI] [PubMed] [Google Scholar]
- 21.Coon K.D., Siegel A.M., Yee S.J., Dunckley T.L., Mueller C., Nagra R.M., Tourtellotte W.W., Reiman E.M., Papassotiropoulos A., Petersen F.F. Preliminary demonstration of an allelic association of the IREB2 gene with Alzheimer's disease. J. Alzheimers Dis. 2006;9:225–233. doi: 10.3233/jad-2006-9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massie H.R., Aiello V.R., Banziger V. Iron accumulation and lipid peroxidation in aging C57BL/6J mice. Exp. Gerontol. 1983;18:277–285. doi: 10.1016/0531-5565(83)90038-4. [DOI] [PubMed] [Google Scholar]
- 23.Ghio A.J., Pritchard R.J., Dittrich K.L., Samet J.M. Non-heme (Fe3+) in the lung increases with age in both humans and rats. J. Lab. Clin. Med. 1997;129:53–61. doi: 10.1016/s0022-2143(97)90161-x. [DOI] [PubMed] [Google Scholar]
- 24.Thompson A.B., Bohling T., Heires A., Linder J., Rennard S.I. Lower respiratory tract iron burden is increased in association with cigarette smoking. J. Lab. Clin. Med. 1991;117:493–499. [PubMed] [Google Scholar]
- 25.Moreno J.J., Foroozesh M., Church D.F., Pryor W.A. Release of iron from ferritin by aqueous extracts of cigarette smoke. Chem. Res. Toxicol. 1992;5:116–123. doi: 10.1021/tx00025a020. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z.H., Kim H.P., Sciurba F.C., Lee S.J., Feghali-Bostwick C., Stolz D.B., Dhir R., Landreneau R.J., Schuchert M.J., Yousem S.A. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3:e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brody J.S., Spira A. State of the art. Chronic obstructive pulmonary disease, inflammation, and lung cancer. Proc. Am. Thorac. Soc. 2006;3:535–537. doi: 10.1513/pats.200603-089MS. [DOI] [PubMed] [Google Scholar]
- 28.Falvella F.S., Galvan A., Frullanti E., Spinola M., Calabro E., Carbone A., Incarbone M., Santambrogio L., Pastorino U., Dragani T.A. Transcription deregulation at the 15q25 locus in association with lung adenocarcinoma risk. Clin. Cancer Res. 2009;15:1837–1842. doi: 10.1158/1078-0432.CCR-08-2107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.