

Abstract
Rapid intracellular transport and secretion of cytotoxic granules through the immunological synapse requires a balanced interaction of several proteins. Disturbance of this highly regulated process underlies familial hemophagocytic lymphohistiocytosis (FHL), a genetically heterogeneous autosomal-recessive disorder characterized by a severe hyperinflammatory phenotype. Here, we have assigned FHL-5 to a 1 Mb region on chromosome 19p by using high-resolution SNP genotyping in eight unrelated FHL patients from consanguineous families. Subsequently, we found nine different mutations, either truncating or missense, in STXBP2 in twelve patients from Turkey, Saudi Arabia, and Central Europe. STXBP2 encodes syntaxin binding protein 2 (Munc18-2), involved in the regulation of vesicle transport to the plasma membrane. We have identified syntaxin 11, a SNARE protein mutated in FHL-4, as an interaction partner of STXBP2. This interaction is eliminated by the missense mutations found in our FHL-5 patients, which leads to a decreased stability of both proteins, as shown in patient lymphocytes. Activity of natural killer and cytotoxic T cells was markedly reduced or absent, as determined by CD107 degranulation. Our findings thus identify a key role for STXBP2 in lytic granule exocytosis.
Introduction
Patients with hemophagocytic lymphohistiocytosis (HLH) present with episodes of prolonged fever, hepatosplenomegaly, and cytopenia in at least two lineages associated with high values of triglycerides and ferritin, an elevated number of sCD25 molecules, and high IFN-γ levels in the serum. Highly activated natural killer (NK) cells and cytotoxic T lymphocytes (CTL) show a markedly reduced or absent killing of (virus-infected) target cells. This high and lasting stimulation of the immune system is accompanied with an ineffective immune response and may lead to death unless an adequate immunosuppressive treatment is begun.1,2 Initial therapeutic strategies always lead to a marked suppression of the severe hyperinflammatory phenotype, but nevertheless, long-term cure can only be achieved by stem cell transplantation (SCT) from an allogeneic donor. HLH can be aquired or familial; therefore, not all HLH cases require SCT as an appropriate treatment. Age at onset, familiarity, and severity of the disease must be taken into account for the initiation of the transplantation procedure.
Familial hemophagocytic lymphohistiocytosis (FHL [MIM 267700]) is a rare, autosomal recessively inherited disorder with a clinical manifestation during infancy in 70%–80% of the patients.3,4 Previous genetic studies in FHL revealed an extended genetic heterogeneity. A first mapping approach included four consanguineous families of Pakistani origin and identified a 7.8 cM region on chromosome 9q21.3-q22, but the underlying defect is still unknown (FHL-1 [MIM 267700]).5 A second report described linkage of 10 out of 17 investigated families to a region on chromosome 10q21-q22,6 thus indicating genetic heterogeneity. Mutations in the perforin-1 gene (PRF1 [MIM 170280]) were then described at this locus (FHL-2 [MIM 603553]).7 Twenty to fifty percent of the patients have mutations in PRF1, with a strong geographical bias in the overall frequency.8–11 Additional genetic defects associated with FHL affect proteins involved in transport, membrane fusion, or exocytosis of perforin-containing lytic granules such as Munc13-4 (MIM 608897; FHL-3 [MIM 608898]) and syntaxin 11 (MIM 605014; FHL-4 [MIM 603552]).12,13 Again, the overall frequency of patients with a known genetic defect is strongly associated with their geographical origin. In the remaining 30%–70% of the familial cases, the genetic basis of the disease is still unknown.14,15
In order to identify additional candidate genes, we have screened samples of FHL patients for mutations in PRF1, UNC13D, or STX11. We identified 15 unrelated Turkish and Saudi Arabian patients from consanguineous families without mutations in any of these genes. After whole-genome homozygosity mapping in samples from these patients, we identified mutations in STXBP2 encoding syntaxin binding protein 2 (Munc18-2 [MIM 601717]), a protein involved in regulation of intracellular trafficking and control of SNARE complex assembly and disassembly.
Subjects and Methods
Patients and Diagnosis
All patients included in the study had a proven family background of hemophagocytic lymphohistiocytosis and/or were diagnosed according to the diagnostic criteria of the Histiocyte Society.16 Age at onset of the FHL-5 patients ranged from 1.5 mo to 3 yrs and 8 mo. During the course of the disease, CNS involvement was observed in seven of the patients. Eleven patients have been treated according the current protocols (HLH-94 and 2004), and nine patients have undergone SCT. Familial appearance was reported in five of our patients on the basis of the presence of affected siblings. Additional clinical and laboratory data are shown in Table 1. Some of our FHL patients have been clinically described before in the literature. Patient 2290 was described by Sparber-Sauer et al.17 as patient no. 185. A detailed clinical history of patient 0879 was recently published by our group as pertaining to patient P12.18 The study was conducted in accordance with the standards of the Declaration of Helsinki and those of the participating clinics. All families gave their written informed consent to the genetic analysis.
Table 1.
Summary of Clinical, Laboratory, and Mutation Data of the FHL-5 Patients
| Patient | Cs | Origin | Familial | Onset: Month of Age | HLH Criteriaa | HSCT/Month of Age | Outcome/FU (mo) | CNS | Cyto-toxicityb | Mutationsc |
|---|---|---|---|---|---|---|---|---|---|---|
| 0870 | – | German | – | 28 | 5/5 | no | d | + | —–/– | p.[Leu209Pro]+[SP] |
| 0879 | + | Turkish | + | 28 | 5/5 | no | e | – | —/– | p.[SP]+[SP] |
| 0884 | + | Turkish | – | 2 | 5/5 | MMUD/11 | NAD/84 | + | n.t. | p.[Arg292His]+[Arg292His] |
| 1155 | + | Turkish | – | 2 | 5/5 | no | death | + | n.t. | p.[Ile232 del]+[Ile232 del] |
| 1875 | + | Saudi Arabian | + | 4 | 5/8 | MSD | n.a. | n.a. | n.t. | p.[Pro477Leu]+[Pro477Leu] |
| 1945 | + | Turkish | + | 1.5 | 5/5 | MSD/4.5 | NAD/108 | + | n.t. | p.[Ile232 del]+[Ile232 del] |
| 1948 | + | Turkish | – | 9 | n.a. | n.a. | n.a. | – | n.t. | p.[Arg405Trp]+[Arg405Trp] |
| 1949f | + | Turkish | + | 3 | 7/8 | MUD/6 | NAD/20 | + | n.t. | p.[Arg405Glu]+[Arg405Glu] |
| 1976-1g | + | Saudi Arabian | + | 6 | 5/8 | MMUD/12 | NAD/23 | – | —/—– | p.[Pro477Leu]+[Pro477Leu] |
| 1976-2g | + | Saudi Arabian | + | 1.5 | 8/8 | MUD/3 | death of VOD/4 | + | —–/— | p.[Pro477Leu]+[Pro477Leu] |
| 2289 | – | German | – | 44 | 4/5 | MUD/192 | death/144 | – | —–/– | p.[SP]+[SP] |
| 2290 | – | German | – | 15 | n.a. | MUD/84 | death/1.5 | n.a. | n.t. | p.[Leu87ArgfsX32]+[SP] |
| 2291 | – | Czech | – | 15 | 6/8 | MMUD/28 | NAD/15 | + | n.t. | p.[SP]+[SP] |
Abbreviations are as follows: Cs, consanguinity; HSCT, hematopoeitic stem cell transplantation; MMUD, mismatched unrelated donor; MUD, matched unrelated donor; MSD, matched sibling donor; FU, follow up since diagnosis; NAD, no active disease; VOD veno-occlusive disease; CNS, central nervous system involvement, observed at any time of disease; n.a., not available; n.t., not tested.
According to Henter et al.16
NK cells or CTLs; —– indicates absent, — indicates severely reduced, – indicates lower normal range.
Mutations in STXBP2; SP, splice defect caused by c.1247-1G>C.
Recurrent reactivations with spontaneous remission or response to steroids.
Recurrent reactivations responsive to steroids.
An affected sister (1949-2) showed the same mutation.
Affected brothers 1976-1, 1976-2.
Genotyping and Linkage Analysis
DNA was extracted from EDTA blood samples via standard procedures. DNA samples were genotyped with the Affymetrix GeneChip Human Mapping 250K Sty Array (Affymetrix). Genotypes were called by the GeneChip DNA analysis software (GDAS v3.0, Affymetrix). Data were checked with the program Graphical Representation of Relationships (GRR).19 Parametric linkage analysis was done with the programs Allegro20 and MERLIN,21 allowing for marker clustering as compensation for linkage disequilibrium (LD) between adjacent markers. Haplotypes were constructed with the program Allegro. All data handling was done with the use of the graphical user interface ALOHOMORA.22
Mutation analysis
PCR products of the coding exons included the adjacent intronic sequences for identification of splice-site variants. Primer sequences are listed in Table S1, available online. Products were directly sequenced with BigDye Terminator v1.1 (Applied Biosystems) and run on an ABI PRISM 3100 genetic analyzer.
Quantitative PCR
RNA was either purchased (human tissue total RNA, Clontech) or isolated from cells obtained by fluorescence-activated cell sorting (FACS) from human peripheral blood samples via the Trizol (Invitrogen) method. One microgram of total RNA was reverse transcribed with the Revert Aid kit (Fermentas) and random hexamers. The STXBP2 and STX11 transcript levels were determined by relative quantitative PCR (qPCR). qPCR was performed in a total reaction volume of 24 μl for each well containing 12 μl Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen), 10μl cDNA 1:50 prediluted, and 2 μl primer mix (5 μM each; primer sequences are given in Table S2). Each sample was analyzed in triplicate and amplified on a real-time PCR instrument (Applied Biosystems 7500). Relative mRNA levels were quantified via the comparative CT method (7500 System Software, Applied Biosystems). STXBP2 and STX11 mRNA values were normalized against the average values of GAPDH, ACTB, 18S rRNA, and TFRC as endogenous controls. For relative expression analysis, human granulocytes were used as calibrator.
Immunofluorescence Analysis
For immunofluorescence analysis, FACS-sorted cells from two independent donors were plated on poly-L-lysine-coated (Sigma) coverslips in RPMI1640 supplemented with 20% fetal calf serum (FCS), 2 mM glutamine, and 1 μg/ml Concanavalin A (Sigma) for 30 min. For stimulation, cells were plated after 24 hr on poly-L-lysine-coated coverslips for 30 min and stimulated with 1 nM interleukin 2 for 5 min. Cells were fixed in 4% (w/v) paraformaldehyde and permeabilized with 0.1% (w/v) saponin. Primary antibody labeling was done with anti-STXBP2 (Calbiochem) and anti-STX11 (BD Biosciences) in 3% (w/v) bovine serum albumin in PBS (BSA/PBS) for 3 hr at 4°C. Coverslips were washed with 1× PBS and incubated with fluorescently labeled secondary antibodies (anti-mouse IgG Alexa 555 and anti-rabbit Alexa 488, Molecular Probes) in 3% (w/v) BSA/PBS for 1 hr at 4°C. Nuclear DNA was stained with 1 μg/ml DAPI in PBS after secondary antibody incubation. Coverslips were washed again with 1× PBS and mounted on glass slides with Fluoromount G (Electron Microscopy Sciences). All images were acquired with a Zeiss LSM 510 confocal microscope. Colocalizations were analyzed with AxioVision software (Zeiss).
NK Cell and T Cell Cytotoxicity and Degranulation Assays
NK cell cytotoxicity was analyzed in standard 51Chromium-release assays. In brief, 51Cr-labeled K562 target cells were incubated with peripheral blood mononuclear cell (PBMC) effectors directly after Ficoll isolation for 4.5 hr at six different effector-to-target ratios in 96-well U-bottom plates. CTL cytotoxicity was analyzed in a redirected lysis assay. Phytohaemagglutinin (PHA) blasts were cultured for 6 to 8 days, labeled for 2 hr with anti-CD3 (clone UCHT-1; BD PharMingen), and used directly as effector cells on 51Cr-labeled L1210 target cells for 4.5 hr. The amount of 51Cr release into the supernatant was determined with a Packard Cobra II Auto Gamma (Perkin-Elmer). NK-to-target and CTL-to-target ratios were calculated by multiplying the respective effector-to-target-ratio with the percentage of NK cells and CTLs, respectively, as determined by flow cytometry.
NK cell degranulation was examined by analyzing the expression of the lysosomal marker protein CD107 on activated NK cells. For this, freshly isolated PBMCs were incubated with K562 cells at a ratio of 1:1 for 2 hr and then stained with anti-CD3-PerCP, anti-CD56-PE, anti-NKp46-APC, and anti-CD107a/b-FITC mAbs (all mAbs were obtained from BD PharMingen). For study of CTL degranulation, day 2 PHA blasts were stimulated with anti-CD3/anti-CD28 beads (Dynal Biotech) in the presence of antibodies against CD107a/b (clones H4A3 and H4B4; BD PharMingen). Monensin A (Golgistop; BD PharMingen) was added after 1 hr, and cells were incubated for an additional 4 hr to 5 hr. The cells were analyzed on a FACScan cytometer with Cellquest Pro software (BD Biosciences).
Cloning and Coimmunoprecipitation
Full-length human STXBP2 was cloned into the eukaryotic expression vector pEGFP-C2 (BD Bioscience) with the use of the endonuclease restriction sites EcoRI and KpnI, generating an N-terminal Enhanced Green Fluorescence Protein (EGFP) fusion protein. Full-length human STX11 (wild-type [WT]) was cloned in the expression vector pIRESneo2 (Clontech) as an N-terminal FLAG-tagged fusion protein.
Generation of STXBP2 Mutants
The STXBP2 mutants were generated by use of the QuickChange II Site-Directed Mutagenesis Kit (Stratagene) in accordance with the manufacturer's protocol. Specific primers containing the respective mutations were designed by use of the web-based QuickChange Primer Design Program.
Expression of EGFP-STXBP2 and FLAG-Syntaxin11 in 293 HEK Cells
Human Embryonic Kidney (HEK) 293 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% FCS. 293 HEK cells were cotransfected with EGFP-STXBP2-containing (WT and mutants) vector and the FLAG-syntaxin11-containing vector, respectively, with the use of OptiMEM (Invitrogen) and Lipofectamine 2000 (Invitrogen), in accordance with the manufacturer's protocol. After 48 hr of transfection, EGFP expression was confirmed by fluorescence microscopy for the determination of transfection efficiency. Cotransfected 293 HEK cells were washed twice in ice-cold 1× PBS (Invitrogen) and lysed on ice in lysis buffer containing 50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 (Sigma Aldrich), and Complete Protease Inhibitor Cocktail Tablet (Roche). Cell debris were sedimented by centrifugation (14,000 rpm, 10 min, 4°C). Fresh total cell lysates were directly used for coimmunoprecipitation (Co-IP) analysis or stored at 4°C.
Coimmunoprecipitation Experiments
Protein-G PLUS agarose beads (Santa Cruz Biotechnology) were preincubated with ANTI-FLAG M2 murine IgG1 monoclonal antibody (Sigma Aldrich) in a rotator for 3 hr at 4°C. Beads were washed once with ice-cold 1× PBS before usage. Cotransfected fresh total 293 HEK cell lysates were diluted in 1× PBS and incubated with anti-FLAG-coupled Protein-G PLUS agarose beads in a rotator overnight at 4°C. Each Co-IP approach was washed four times with ice-cold 1× PBS followed by elution in 1× Laemmli buffer and boiled for 7 min at 95°C for SDS-PAGE analysis.
SDS-PAGE and Immunoblot Analysis
SDS-PAGE analysis was performed with self-casted SDS gels containing 10% acrylamide at 25 mA. Transfer of the proteins to a polyvinylidene fluoride (PVDF) membrane was performed in 10 mM CAPS buffer, pH 11 containing 10% methanol for 3 hr at 4°C, 400 mA. Blotted PVDF membranes were blocked at least 30 min in 5% dry, nonfat milk solved in 1× TBS. Incubation with the appropriate primary antibodies (anti-GFP [B2], Santa Cruz Biotechnology; monoclonal anti-FLAG M2 murine and polyclonal anti-FLAG rabbit, Sigma Aldrich) was performed in 2.5% dry, nonfat milk solved in 1× TBS over night at 4°C. The PVDF membranes were washed three times in 1× TBS followed by incubation with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (purchased from Dako). After three additional washing steps, the PVDF membranes were incubated with enhanced chemiluminescence (ECL) (0.1 M Tris-HCl [pH 8.6], 50 mg Luminol, 11 mg coumaric acid) for 4 min and exposed on X-ray films. For endogenous STXBP2, syntaxin 11, and actin detection, cell lysates were run on 4%–12% precast gels (Invitrogen), transferred onto nitrocellulose, and probed with rabbit anti-STXBP2 (a gift from V. Olkkonen), raised against a fragment of canine STXBP2 encoding amino acids 248–593, which comprise the central and C-terminal parts of STXBP2,23 anti-syntaxin 11 (a gift from R. Prekeris), and mouse anti-actin (Sigma).
Results
Gene Mapping and Mutation Analysis
In the present study, we have initially screened 48 unrelated patients from Turkey (n = 47) or Saudi Arabia (n = 1) with a consanguineous family background for mutations in PRF1, UNC13D, or STX11. Eighteen of these had mutations in PRF1 (38%), seven had mutations in UNC13D (15%), and eight patients had mutations in STX11 (17%), thus identifying 15 patients without mutations in any of these genes. With samples of these single patients, we performed a genome-wide linkage analysis and homozygosity mapping approach, using chip-based SNP genotyping. LOD score analysis with the use of 20,000 SNP markers under the assumption of heterogeneity and full penetrance revealed a maximum HLOD score of 5.9 on chromosome 19p (Figure 1A). Refined calculations with 3700 SNP markers on chromosome 19 gave a heterogeneity LOD (HLOD) score of 8.3 at rs624968. Detailed genotype analysis identified an overlapping region of homozygosity in seven of the 15 cases (Figure 1B). The remaining eight patients were heterozygous in this region and were disregarded in the following investigations. In addition, we included a previously screened extended consanguineous FHL pedigree from Saudi Arabian origin (1875 in Figure 1C) comprising two affected and three unaffected siblings and their parents. Whole-genome analysis of this family showed three homozygous regions with LOD scores over 2.0. Interestingly, one of these overlapped with the interval on chromosome 19p found in the analysis of the single FHL patients. The common region of homozygosity was 1040 kb in length and contained 36 genes (Figure 1C).
Figure 1.
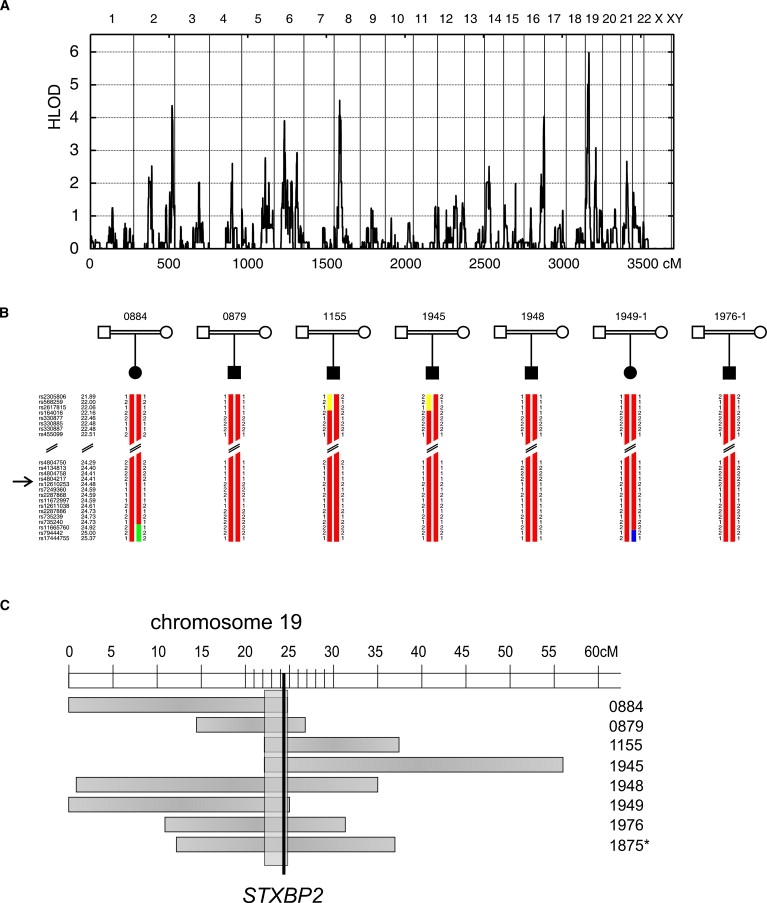
Identification of Mutations in STXBP2 Underlying FHL Type 5
(A) A genome-wide linkage scan was performed in 15 single patients with FHL from consanguineous families. LOD scores assuming locus heterogeneity (HLOD scores) were plotted for all autosomes, corresponding to approximately 3500 cM in total length, and revealed a locus on chromosome 19 with a maximum LOD score of 5.9.
(B) SNP genotypes of seven FHL patients with homozygosity around STXBP2. Names of markers and their genetic localization are shown on the left. STXBP2 is located between rs4804217 and rs12610253.
(C) Eight patients showed overlapping regions of homozygosity on chromosome 19 (horizontal boxes). The minimal region (vertical box) was 1 Mb in length and contained the gene STXBP2. Pedigree 1875 (asterisk) was mapped independently and is not included in the LOD score analysis summarized in (A).
In a search for potential candidate genes, we focused on genes with a potential role in immune cells, the most promising of which was STXBP2. By direct sequencing, we then identified mutations in STXBP2 in all eight patients who mapped to the critical region (Figure 2A, Table 1, and Table 2). The gene encompasses 19 exons and codes for syntaxin binding protein 2, also known as Munc18-2, consisting of 593 amino acids. When screening other patients from nonconsanguineous FHL families, we identified mutations in STXBP2 in four additional patients, three from Germany and one from the Czech Republic (Table 1). In total, we identified five different missense mutations, one 3 bp deletion, two frameshift deletions, and a mutation at position −1 (c.1247-1G>C) of the splice acceptor site of exon 15 (Figure 2A and Table 2). In patient 0879, homozygous for the splice-site mutation, analysis of RNA revealed a deletion of exon 15 resulting in a frameshift and 126 new codons as a major event (p.Val417LeufsX126) but also a variety of other, including in-frame, products (Figure S1).
Figure 2.
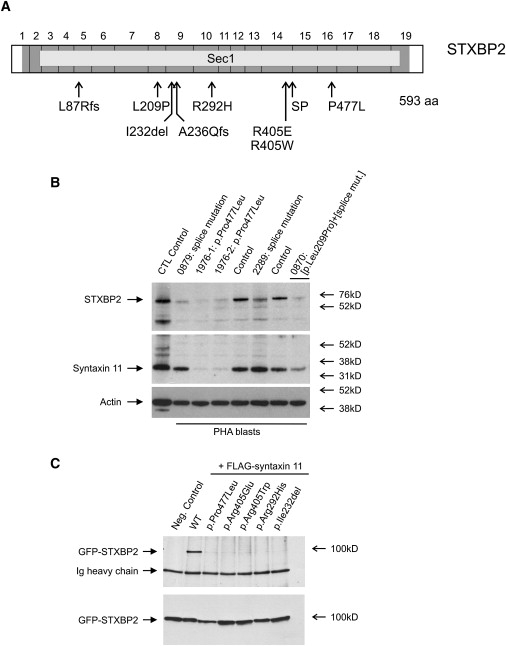
Structure of STXBP2, Protein Stability, and Interaction
(A) STXBP2 consists of 19 exons and includes 593 codons (dark gray). It encodes syntaxin binding protein 2 (STXBP2), which contains a large Sec1 domain (light gray). We identified nine different mutations distributed throughout the gene. SP, splice mutation c.1247-1G>C.
(B) Lysates from PHA blasts obtained from FHL-5 patients and control persons were analyzed for presence of STXBP2 and syntaxin 11. Patients showed reduced amounts of STXBP2. Moreover, patients with the homozygous STXBP2 missense mutation p.Pro477Leu (1976-1, 1976-2) showed clearly decreased amounts of syntaxin 11. Patient 0870, compound heterozygous for the splice-site mutation c.1247-1G>C and the missense mutation p.Leu209Pro, revealed an intermediate pattern.
(C) Coimmunoprecipitation demonstrated shared binding sites between STXBP2 and syntaxin 11. The ability to bind to syntaxin 11 is lost in all STXBP2 mutants with missense mutations and the one-codon deletion identified in our patients with FHL-5 (upper panel). All of the STXBP2 mutants were evenly detectable (lower panel).
Table 2.
Summary of Mutations Identified in STXBP2 in Patients with FHL
| Mutation | Exon | Protein | ConSeqa | Patient | Genotypeb |
|---|---|---|---|---|---|
| c.260delT | 5 | p.Leu87ArgfsX32 | – | 2290 | comp het |
| c.626T>C | 8 | p.Leu209Pro | 5 | 0870 | comp het |
| c.693_695delGAT | 9 | p.Ile232del | 8 | 1155 | homo |
| 1945 | homo | ||||
| c.706delG | 9 | p.Ala236GlnfsX24 | – | 2291 | comp het |
| c.875G>A | 10 | p.Arg292His | 8 | 0884 | homo |
| c.1213C>T | 14 | p.Arg405Trp | 9 | 1948 | homo |
| c.1214G>A | 14 | p.Arg405Glu | 9 | 1949d | homo |
| c.1247-1G>Cc | 15 | p.Val417LeufsX126c | – | 0879 | homo |
| 2290 | comp het | ||||
| 0870 | comp het | ||||
| 2291 | comp het | ||||
| 2289 | homo | ||||
| c.1430C>T | 16 | p.Pro477Leu | 7 | 1976e | homo |
| 1875 | homo |
The score for the evolutionary conservation of amino acids24 is included for residues changed as a result of missense mutations. The score is between grades 1 (variable) and 9 (conserved).
Abbreviations are as follows: homo, homozygous; comp het, compound heterozygous.
Splice mutation; only the major consequence is included. See text for details.
An affected sister (1949-2) showed the same mutation.
An affected brother (1976-2) showed the same mutation.
All missense mutations affect amino acids that are highly conserved among 40 proteins of the Sec/Munc family of syntaxin binding proteins from various species. This was also demonstrated by the ConSeq score that goes up to 9 for the highest degree of conservation (Table 2).24 Interestingly, we found the same homozygous mutation in the two Saudi Arabian patients. However, analysis of microsatellites and SNPs around STXBP2 did not show a conserved haplotype, excluding at least a closer relationship between the patients. Corresponding heterozygous mutations were detected in all of the parents, where available, but not in 210 chromosomes from ethnically matched control persons. We identified homozygous missense mutations in 5 out of 12 patients and a 3 bp deletion (c.693_695delGAT) in 2 of the 12 patients. These seven patients had early-onset disease first diagnosed before one year of age. The remaining five patients developed HLH after the first year of life. These patients either had a homozygous splice site mutation (c.1247-1G>C) or were compound heterozygous for two mutations, including the splice-site mutation.
Protein Stability
In order to determine the pathophysiologic relevance of different STXBP2 mutations, we generated T cell clones and/or PHA blasts from five patients. In cell lysates from patients with a homozygous missense mutation (1976-1, 1976-2), we found reduced amounts of STXBP2 in an immunoblot analysis (Figure 2B). In patients with a homozygous splice-site mutation (patients 0879 and 2289), slightly different signals of approximately the correct size were detected. Remarkably, we also observed markedly decreased levels of syntaxin 11 in the patients with a homozygous missense mutation in STXBP2 but not in patients with the homozygous splice-site mutation (Figure 2B). One patient with compound heterozygosity for a splice site and a missense mutation (0870) demonstrated an intermediate amount of syntaxin 11.
Protein Interaction Studies and Cellular Localization
To further corroborate the importance of the missense mutations, we then searched for potential interaction partners of STXBP2. Given that STXBP2 has previously been shown to interact with syntaxins 2 and 3 in epithelial and mast cells, we speculated that in CTL and NK cells, interactions between STXBP2 and syntaxin 11 could be important for cytotoxic granule release. By Co-IP, we demonstrated that STXBP2 and syntaxin 11 share common binding sites (Figure 2C). Using site-directed mutagenesis, we showed that all of the identified missense mutations in STXBP2 lead to a complete loss of the ability to bind to syntaxin 11 (Figure 2C). As expected, loss of binding to syntaxin 11 was not observed when STXBP2 was used with a polymorphic variant in a little conserved residue, p.Ala433Val (not shown).
Real-time quantitative PCR data revealed a high degree of correlation between the expression of STXBP2 and STX11 in different hematopoietic cells (Figure 3). Consistent with the supposed pathophysiology of FHL, a particularly high expression was observed in NK cells, monocytes, and T cells. Moreover, we have demonstrated colocalization of STXBP2 and syntaxin 11 in CD8+ T and NK cells via immunofluorescence analysis (Figure S2), which was weak only in B cells. Stimulation with interleukin 2 (IL-2) further enhanced the colocalization in CD8+ and NK cells. Taken together, these data suggest that syntaxin 11 is stabilized through a STXBP2/syntaxin 11 interaction, which is impaired in FHL-5 patients with a missense mutation in STXBP2.
Figure 3.
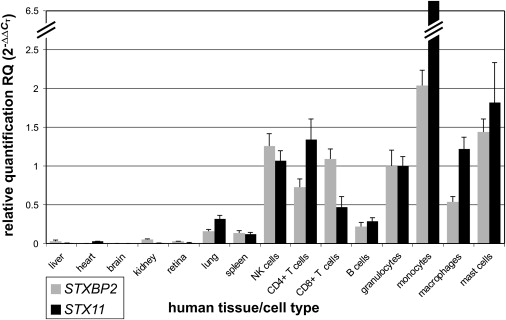
Expression of STXBP2 and STX11
Relative quantification of STXBP2 and STX11 in various human tissues by real-time PCR. Expression of both STXBP2 and STX11 is similar and particularly high in NK cells, T cells, monocytes, and other hematopoietic cells. Quantification was done with the use of ACTB, GAPDH, TFRC, and 18S rRNA as references ± SD.
Cytotoxicity and Degranulation
Recent findings have shown that patients with either FHL-3 or FHL-4 show a marked impairment of their capacity to secrete cytotoxic granules of either NK cells or T cells to their cell surface because of defective Munc13-4 and syntaxin 11, respectively.25,26 This finding also discriminates between patients with FHL-2 and FHL types 3 and 4. In order to assess the cytotoxic capacities in FHL-5 cases and assign them to a disease category, we analyzed degranulation and cytotoxicity of NK cells and CTLs in two early-onset and three late-onset patients with STXBP2 mutations. We measured NK cell and T cell degranulation after stimulation of intracellular granule exocytosis by extracellular staining of CD107 in four patients. CD107 was absent in three of the patients and in the lower normal range in one late-onset patient (Figure 4A). Ex vivo cytotoxicity on NK cell-sensitive K562 targets was impaired in all five patients tested, but one early-onset and one late-onset patient showed residual activity (Figure 4A). CTL degranulation was low in PHA blasts from all patients analyzed (Figure 4B). Additionally, CTL cytotoxicity was very low in two early-onset patients but in the lower normal range in three late-onset patients (Figure 4B). Similar to what is observed in patients with syntaxin-11 deficiency, the degranulation (not shown) and cytotoxicity defects could be at least partially reversed after in vitro stimulation with IL-2, indicating the activation of yet-undefined syntaxin-11/STXBP2 independent pathways under these conditions (Figure 4C).
Figure 4.
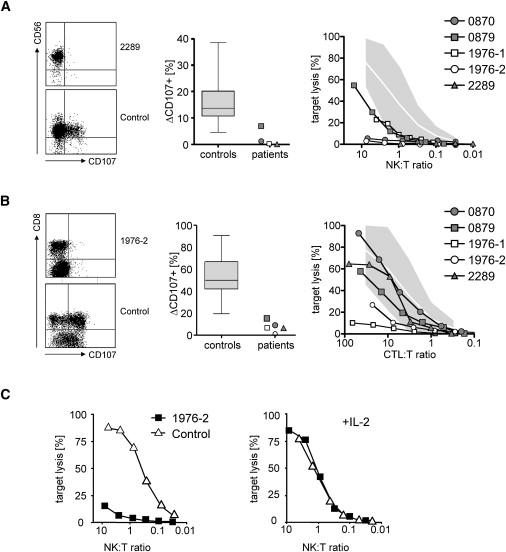
NK and T Cell Degranulation and Cytotoxicity of Patients with Mutations in STXBP2
(A and B) NK cell degranulation was examined by flow cytometry with the use of antibodies against CD107 after stimulation of PBMC with K562 cells (A). Plots are gated on CD3− NKp46+ cells (left panel). CTL degranulation was examined by flow cytometry with the use of antibodies against CD107 after stimulation with anti-CD3/anti-CD28 microbeads (B). Plots are gated on CD3+ cells. The middle panels show the percentage of CD107+ expression on stimulated NK cells and CTLs, respectively, of patients and 30 healthy controls (box, mean and 25%–75% percentile; whiskers, maximum and minimum value observed). The right panels show NK cell cytotoxicity against K562 target cells and CTL cytotoxicity against L1210 target cells, respectively. The shaded areas indicate the mean percentage of target cell lysis ± two standard deviations of 30 healthy controls. The NK:target and CTL:target ratios were determined by flow cytometry.
(C) Reconstitution of NK cell cytotoxicity after in vitro stimulation with IL-2. Left panel: Cytotoxicity of PBMC of patient 1976-2 and a healthy control person were assessed against K562 target cells. The NK cell to target ratio was determined by flow cytometry. Right panel: PBMC were cultured for 7 days in medium supplemented with 100 U/ml recombinant IL-2 before analysis of cytotoxicity on K562 target cells.
Discussion
Fast secretory granule exocytosis is often mediated by SNARE (soluble N-ethylmaleimide sensitive factor attachment protein [SNAP] receptor) complexes and represents a fundamental process for the release of neurotransmitter molecules in the neuronal system and for secretory lysosomes in immune cells.27,28 The analysis of genetic disorders characterized by defects in membrane trafficking in immune cells, particularly in patients with FHL, has decisively contributed to unravelling the secretion machinery.29–31 Here, we report that FHL type 5 (FHL-5) is caused by missense and truncating mutations in STXBP2. We used FHL patients from consanguineous families of Turkish and Saudi Arabian descent to search for unknown loci via autozygosity mapping. Because larger pedigrees with FHL patients are particularly rare, we collected samples of single cases for genome-wide screening, assuming that several of these would share a common region of homozygosity and that patients from the same area might exhibit a common founder haplotype. High-resolution chip-based SNP analysis did in fact allow us to identify a candidate region on chromosome 19p13 in 7 out of 15 patients with FHL and then led to the identification of mutations in STXBP2, the strongest functional candidate in this region.
In total, we detected nine different homozygous or compound heterozygous mutations in twelve independent patients of different geographic origins. We identified five missense mutations affecting highly conserved residues, one in-frame and two frameshift deletions, as well as a mutation at the splice acceptor site of exon 15. All of the mutations scattered over the whole gene. Although six of the patients descended from consanguineous Turkish and two from consanguineous Saudi Arabian families, we found the identical homozygous mutations only in two Turkish and in the two Saudi Arabian families, respectively. Marker analysis around STXBP2 excluded a founder mutation even in these families, further indicating allelic heterogeneity in FHL-5. FHL-5 is therefore neither restricted to any distinct ethnic or geographic origin, as is the case in FHL-4, nor caused by a hotspot mutation in STXBP2, such as mutation p.Trp374X in PRF1 in Turkish patients with FHL-2.14,15,32 This is underlined by our observation of mutations in STXBP2 in four FHL patients from Central Europe as well. Nevertheless, the exact frequency of FHL-5 has yet to be evaluated in a larger group of FHL patients.
The exact function of STXBP2 within the secretory machinery of immune cells is still not well understood. From previous studies, it is known that STXBP2 is important for controlling intracellular granule/membrane trafficking in polarized epithelial cells, neutrophils, and mast cells.33–36 Our expression studies confirmed this pattern and also demonstrated strong signals in other hematopoietic cells, such as T cells, NK cells, and monocytes. Interestingly, this expression is very similar to the expression of STX11, which has been shown before in various hematopoietic cells,25,37,38 and STXBP2 and syntaxin 11 colocalize in CD8+ T cells and NK cells. The increase in colocalization after stimulation with IL-2 is in line with the enhanced cytotoxic activity of NK cells induced by IL-2.39 STXBP2 appears in a granule-like pattern, as observed in mast cells, where it is redistributed into forming lamellipodia upon stimulation.34 Using coimmunoprecipitation experiments, we have demonstrated a direct interaction of STXBP2 with syntaxin 11. STXBP2 belongs to the Sec/Munc family of regulatory proteins involved in the assembly and disassembly of SNARE complexes and control of the specificity and timing of membrane fusion. Syntaxin 11 is an atypical member of the Q subfamily of SNARE proteins40 and is mutated in FHL-4.13 It has been reported that an interaction with syntaxin 3, another SNARE protein, could be disturbed by introducing missense mutations into STXBP2.33,35 One of these, p.Arg405Pro, which corresponds to the yeast mutant sec1-11, showed an almost complete loss of association with syntaxin 3, as observed with the use of in vitro and in vivo assays.33 The same highly conserved residue was mutated in two of our patients (patient 1948, p.Arg405Trp; patient 1949, p.Arg405Glu) and led to the loss of interaction with syntaxin 11. Experiments in mast cell lines also showed the importance of STXBP2 in granule exocytosis of these cells.34 Moreover, an interaction of STXBP2 and syntaxin 3 and a role for STXBP2 in granule exocytosis were recently shown in neutrophils.36 STXBP2 can thus contribute to the granule exocytosis machinery through the interaction with various SNARE proteins in different cell types.
If functional STXBP2 is absent as a result of missense mutations, syntaxin 11 seems to be destabilized. This indicates an interaction between the proteins and suggests a STXBP2/syntaxin-11 complex essential for protein stability. A similar effect was recently observed for complexes of Rab27a, which is mutated in patients with Griscelli syndrome type 2 (GS-2),41 with Slp1 and Slp2-a, respectively.42 Interestingly, GS-2 patients also develop HLH symptoms during the course of their disease. In contrast, syntaxin 11 appears to be present in normal amounts in patients with the homozygous splice-site mutation c.1247-1G>C in STXBP2. This may indicate a “leaky” splice mutation. Moreover, the cDNA analysis in one patient revealed the loss of exon 15 as a major event, which gives rise to a frameshift after codon 416 and a potential protein of almost the same size as STXBP2 and several other products. It is possible that some of these products, as detected in the immunoblot analysis, are effective for stabilization of syntaxin 11 and lead to the milder defect in CTL cytotoxicity observed here and the later onset of the disease in comparison to the severe cases with STXBP2 missense mutations.
The pronounced and sustained immune dysregulation in FHL is caused mainly by defects in cellular cytotoxicity, which is required not only for pathogen control but also for immune homeostasis. In the absence of a negative feedback signal, continuous stimulation of T cells and subsequent activation of macrophages lead to the characteristic hypercytokinemia and fatal organ infiltrations.29,43 Mutations in PRF1 (FHL-2), UNC13D (FHL-3), and STX11 (FHL-4) all affect this secretory pathway of NK cells and T cells, either through an inactivation of the granule content (perforin 1) or by impairing the delivery of such granules through the plasma membrane, caused by defects in Munc13-4 or syntaxin 11. Finally, target cells are recognized appropriately but not killed effectively, ending in the severe clinical picture of a hemophagocytic syndrome. Functional tests in NK cells and CTLs from our FHL-5 patients underline these findings. We showed a severe reduction of both the degranulation capacity and the capacity to kill target cells. Interestingly, the latter defect can be partially restored by short-term culturing with IL-2. Given that IL-2 upregulates the expression of perforin and granzymes44,45 and that the partial restoration of degranulation and cytotoxicity by NK cells was already described for patients with FHL-425 but was not seen in patients with FHL-3, we assume that the increase in protein production may allow secretion of these proteins via alternative routes that bypass the defective STXBP2/syntaxin-11 pathway.
In the majority of our FHL-5 patients, we found missense mutations in STXBP2 affecting highly conserved residues. This observation is somewhat surprising in comparison to the other FHL-related genes, in which the majority of mutations lead to a premature stop of the protein synthesis.46,47 This may underline the importance of STXBP2 in granule exocytosis. Interestingly, missense mutations also dominate in early infantile epileptic encephalopathy type 4 (MIM 612164), which is caused by mutations in STXBP1 (Munc18-1), the neuronal homolog of STXBP2.48 STXBP1 plays a major role in secretory vesicle release through the control of SNARE complex formation, and the protein itself seems to act as a “chief commander” to make exocytosis more efficent.27,49 However, the role of STXBP2 and its interaction with syntaxin 11 in lymphocyte cytotoxicity that have been elucidated by our findings in FHL-5 are proof of the crucial relevance of STXBP2 for intracellular vesicle trafficking and lytic granule exocytosis.
Supplemental Data
Supplemental Data include two figures and two tables and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Basic Local Alignment Search Tool (BLAST), http://blast.ncbi.nlm.nih.gov/Blast.cgi
Ensembl Genome Browser, http://www.ensembl.org/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov
QuickChange Primer Design Program, http://www.stratagene.com/qcprimerdesign/
University of California-Santa Cruz (UCSC) Genome Browser, http://genome.ucsc.edu/
HLH treatment protocols, http://www.histiocytesociety.org/
Acknowledgments
We are grateful to all of the patients and to their family members for their participation in this study. We thank the following physicians for their generous cooperation in this study: U. Kocak (Ankara, Turkey); A. Al Jefri (Ryadh, Saudi Arabia); S. Bielack (Stuttgart, Germany); M. Schündeln (Essen, Germany); H. Jürgens (Münster, Germany); A. Schulz (Ulm, Germany); and E. Mejstrikova (Prague, Czech Republic). We wish to thank Marc Nätebus, Ariane Ott, Stephanie Mewes, Katharina Michael, and Anette Peffekoven for excellent technical assistance. This work was supported by grants from the Histiocytosis Association of America (U.z.S.), the Fördergemeinschaft Kinderkrebszentrum Hamburg e.V. (U.z.S.), the Thyssen Foundation (S.E.), the Bundesministerium für Bildung und Forschung (grant 01 EO 0803 to S.E.), and the Ministry for Innovation, Science, Research, and Technology of the Land Nordrhein Westfalen (H.C.H.).
References
- 1.Henter J.I., Samuelsson-Horne A., Arico M., Egeler R.M., Elinder G., Filipovich A.H., Gadner H., Imashuku S., Komp D., Ladisch S. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 2.Stephan J.L., Donadieu J., Ledeist F., Blanche S., Griscelli C., Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993;82:2319–2323. [PubMed] [Google Scholar]
- 3.Arico M., Janka G., Fischer A., Henter J.I., Blanche S., Elinder G., Martinetti M., Rusca M.P. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203. [PubMed] [Google Scholar]
- 4.Janka G.E. Familial hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 1983;140:221–230. doi: 10.1007/BF00443367. [DOI] [PubMed] [Google Scholar]
- 5.Ohadi M., Lalloz M.R., Sham P., Zhao J., Dearlove A.M., Shiach C., Kinsey S., Rhodes M., Layton D.M. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3–22 by homozygosity mapping. Am. J. Hum. Genet. 1999;64:165–171. doi: 10.1086/302187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufourcq-Lagelouse R., Jabado N., Le Deist F., Stephan J.L., Souillet G., Bruin M., Vilmer E., Schneider M., Janka G., Fischer A. Linkage of familial hemophagocytic lymphohistiocytosis to 10q21–22 and evidence for heterogeneity. Am. J. Hum. Genet. 1999;64:172–179. doi: 10.1086/302194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stepp S.E., Dufourcq-Lagelouse R., Le Deist F., Bhawan S., Certain S., Mathew P.A., Henter J.I., Bennett M., Fischer A., de Saint Basile G. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 8.Goransdotter Ericson K., Fadeel B., Nilsson-Ardnor S., Soderhall C., Samuelsson A., Janka G., Schneider M., Gurgey A., Yalman N., Revesz T. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am. J. Hum. Genet. 2001;68:590–597. doi: 10.1086/318796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suga N., Takada H., Nomura A., Ohga S., Ishii E., Ihara K., Ohshima K., Hara T. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br. J. Haematol. 2002;116:346–349. doi: 10.1046/j.1365-2141.2002.03266.x. [DOI] [PubMed] [Google Scholar]
- 10.Molleran Lee S., Villanueva J., Sumegi J., Zhang K., Kogawa K., Davis J., Filipovich A.H. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J. Med. Genet. 2004;41:137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clementi R., zur Stadt U., Savoldi G., Varoitto S., Conter V., De Fusco C., Notarangelo L.D., Schneider M., Klersy C., Janka G. Six novel mutations in the PRF1 gene in children with haemophagocytic lymphohistiocytosis. J. Med. Genet. 2001;38:643–646. doi: 10.1136/jmg.38.9.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldmann J., Callebaut I., Raposo G., Certain S., Bacq D., Dumont C., Lambert N., Ouachee-Chardin M., Chedeville G., Tamary H. Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 13.zur Stadt U., Schmidt S., Kasper B., Beutel K., Diler A.S., Henter J.I., Kabisch H., Schneppenheim R., Nurnberg P., Janka G. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet. 2005;14:827–834. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 14.Zur Stadt U., Beutel K., Kolberg S., Schneppenheim R., Kabisch H., Janka G., Hennies H.C. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum. Mutat. 2006;27:62–68. doi: 10.1002/humu.20274. [DOI] [PubMed] [Google Scholar]
- 15.Horne A., Ramme K.G., Rudd E., Zheng C., Wali Y., al-Lamki Z., Gurgey A., Yalman N., Nordenskjold M., Henter J.I. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br. J. Haematol. 2008;143:75–83. doi: 10.1111/j.1365-2141.2008.07315.x. [DOI] [PubMed] [Google Scholar]
- 16.Henter J.I., Horne A., Arico M., Egeler R.M., Filipovich A.H., Imashuku S., Ladisch S., McClain K., Webb D., Winiarski J. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 17.Sparber-Sauer M., Honig M., Schulz A.S., Zur Stadt U., Schutz C., Debatin K.M., Friedrich W. Patients with early relapse of primary hemophagocytic syndromes or with persistent CNS involvement may benefit from immediate hematopoietic stem cell transplantation. Bone Marrow Transplant. 2009;44:333–338. doi: 10.1038/bmt.2009.34. [DOI] [PubMed] [Google Scholar]
- 18.Beutel K., Gross-Wieltsch U., Wiesel T., Stadt U.Z., Janka G., Wagner H.J. Infection of T lymphocytes in Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children of non-Asian origin. Pediatr. Blood Cancer. 2009;53:184–190. doi: 10.1002/pbc.22037. [DOI] [PubMed] [Google Scholar]
- 19.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 20.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 21.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 22.Ruschendorf F., Nurnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 23.Riento K., Jantti J., Jansson S., Hielm S., Lehtonen E., Ehnholm C., Keranen S., Olkkonen V.M. A sec1-related vesicle-transport protein that is expressed predominantly in epithelial cells. Eur. J. Biochem. 1996;239:638–646. doi: 10.1111/j.1432-1033.1996.0638u.x. [DOI] [PubMed] [Google Scholar]
- 24.Berezin C., Glaser F., Rosenberg J., Paz I., Pupko T., Fariselli P., Casadio R., Ben-Tal N. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- 25.Bryceson Y.T., Rudd E., Zheng C., Edner J., Ma D., Wood S.M., Bechensteen A.G., Boelens J.J., Celkan T., Farah R.A. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–1915. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcenaro S., Gallo F., Martini S., Santoro A., Griffiths G.M., Arico M., Moretta L., Pende D. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13–4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108:2316–2323. doi: 10.1182/blood-2006-04-015693. [DOI] [PubMed] [Google Scholar]
- 27.Rizo J., Rosenmund C. Synaptic vesicle fusion. Nat. Struct. Mol. Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blott E.J., Griffiths G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002;3:122–131. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 29.Menasche G., Feldmann J., Fischer A., Basile Gde S. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol. Rev. 2005;203:165–179. doi: 10.1111/j.0105-2896.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 30.Stinchcombe J., Bossi G., Griffiths G.M. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004;305:55–59. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 31.Stinchcombe J.C., Griffiths G.M. Secretory mechanisms in cell-mediated cytotoxicity. Annu. Rev. Cell Dev. Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 32.Rudd E., Goransdotter Ericson K., Zheng C., Uysal Z., Ozkan A., Gurgey A., Fadeel B., Nordenskjold M., Henter J.I. Spectrum and clinical implications of syntaxin 11 gene mutations in familial haemophagocytic lymphohistiocytosis: association with disease-free remissions and haematopoietic malignancies. J. Med. Genet. 2006;43:e14. doi: 10.1136/jmg.2005.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riento K., Kauppi M., Keranen S., Olkkonen V.M. Munc18–2, a functional partner of syntaxin 3, controls apical membrane trafficking in epithelial cells. J. Biol. Chem. 2000;275:13476–13483. doi: 10.1074/jbc.275.18.13476. [DOI] [PubMed] [Google Scholar]
- 34.Martin-Verdeaux S., Pombo I., Iannascoli B., Roa M., Varin-Blank N., Rivera J., Blank U. Evidence of a role for Munc18–2 and microtubules in mast cell granule exocytosis. J. Cell Sci. 2003;116:325–334. doi: 10.1242/jcs.00216. [DOI] [PubMed] [Google Scholar]
- 35.Kauppi M., Wohlfahrt G., Olkkonen V.M. Analysis of the Munc18b-syntaxin binding interface. Use of a mutant Munc18b to dissect the functions of syntaxins 2 and 3. J. Biol. Chem. 2002;277:43973–43979. doi: 10.1074/jbc.M208315200. [DOI] [PubMed] [Google Scholar]
- 36.Brochetta C., Vita F., Tiwari N., Scandiuzzi L., Soranzo M.R., Guerin-Marchand C., Zabucchi G., Blank U. Involvement of Munc18 isoforms in the regulation of granule exocytosis in neutrophils. Biochim. Biophys. Acta. 2008;1783:1781–1791. doi: 10.1016/j.bbamcr.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 37.Prekeris R., Klumperman J., Scheller R.H. Syntaxin 11 is an atypical SNARE abundant in the immune system. Eur. J. Cell Biol. 2000;79:771–780. doi: 10.1078/0171-9335-00109. [DOI] [PubMed] [Google Scholar]
- 38.Arneson L.N., Brickshawana A., Segovis C.M., Schoon R.A., Dick C.J., Leibson P.J. Cutting edge: syntaxin 11 regulates lymphocyte-mediated secretion and cytotoxicity. J. Immunol. 2007;179:3397–3401. doi: 10.4049/jimmunol.179.6.3397. [DOI] [PubMed] [Google Scholar]
- 39.Trinchieri G., Matsumoto-Kobayashi M., Clark S.C., Seehra J., London L., Perussia B. Response of resting human peripheral blood natural killer cells to interleukin 2. J. Exp. Med. 1984;160:1147–1169. doi: 10.1084/jem.160.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang B.L., Low D.Y., Hong W. Syntaxin 11: a member of the syntaxin family without a carboxyl terminal transmembrane domain. Biochem. Biophys. Res. Commun. 1998;245:627–632. doi: 10.1006/bbrc.1998.8490. [DOI] [PubMed] [Google Scholar]
- 41.Menasche G., Pastural E., Feldmann J., Certain S., Ersoy F., Dupuis S., Wulffraat N., Bianchi D., Fischer A., Le Deist F. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 2000;25:173–176. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 42.Holt O., Kanno E., Bossi G., Booth S., Daniele T., Santoro A., Arico M., Saegusa C., Fukuda M., Griffiths G.M. Slp1 and Slp2-a localize to the plasma membrane of CTL and contribute to secretion from the immunological synapse. Traffic. 2008;9:446–457. doi: 10.1111/j.1600-0854.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janka G.E. Familial and acquired hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 2007;166:95–109. doi: 10.1007/s00431-006-0258-1. [DOI] [PubMed] [Google Scholar]
- 44.Liu C.C., Rafii S., Granelli-Piperno A., Trapani J.A., Young J.D. Perforin and serine esterase gene expression in stimulated human T cells. Kinetics, mitogen requirements, and effects of cyclosporin A. J. Exp. Med. 1989;170:2105–2118. doi: 10.1084/jem.170.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salcedo T.W., Azzoni L., Wolf S.F., Perussia B. Modulation of perforin and granzyme messenger RNA expression in human natural killer cells. J. Immunol. 1993;151:2511–2520. [PubMed] [Google Scholar]
- 46.Trizzino A., zur Stadt U., Ueda I., Risma K., Janka G., Ishii E., Beutel K., Sumegi J., Cannella S., Pende D. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J. Med. Genet. 2008;45:15–21. doi: 10.1136/jmg.2007.052670. [DOI] [PubMed] [Google Scholar]
- 47.Rudd E., Bryceson Y.T., Zheng C., Edner J., Wood S.M., Ramme K., Gavhed S., Gurgey A., Hellebostad M., Bechensteen A.G. Spectrum, and clinical and functional implications of UNC13D mutations in familial haemophagocytic lymphohistiocytosis. J. Med. Genet. 2008;45:134–141. doi: 10.1136/jmg.2007.054288. [DOI] [PubMed] [Google Scholar]
- 48.Saitsu H., Kato M., Mizuguchi T., Hamada K., Osaka H., Tohyama J., Uruno K., Kumada S., Nishiyama K., Nishimura A. De novo mutations in the gene encoding STXBP1 (MUNC18–1) cause early infantile epileptic encephalopathy. Nat. Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 49.Toonen R.F., Verhage M. Munc18–1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci. 2007;30:564–572. doi: 10.1016/j.tins.2007.08.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.