

Abstract
Fragile X syndrome (FXS) results from a CGG-repeat expansion that triggers hypermethylation and silencing of the FMR1 gene. FXS is referred to as the most common form of inherited intellectual disability, yet its true incidence has never been measured directly by large population screening. Here, we developed an inexpensive and high-throughput assay to quantitatively assess FMR1 methylation in DNA isolated from the dried blood spots of 36,124 deidentified newborn males. This assay displays 100% specificity and 100% sensitivity for detecting FMR1 methylation, successfully distinguishing normal males from males with full-mutation FXS. Furthermore, the assay can detect excess FMR1 methylation in 82% of females with full mutations, although the methylation did not correlate with intellectual disability. With amelogenin PCR used for detecting the presence of a Y chromosome, this assay can also detect males with Klinefelter syndrome (KS) (47, XXY). We identified 64 males with FMR1 methylation and, after confirmatory testing, found seven to have full-mutation FXS and 57 to have KS. Because the precise incidence of KS is known, we used our observed KS incidence as a sentinel to assess ascertainment quality and showed that our KS incidence of 1 in 633 newborn males was not significantly different from the literature incidence of 1 in 576 (p = 0.79). The seven FXS males revealed an FXS incidence in males of 1 in 5161 (95% confidence interval of 1 in 10,653–1 in 2500), consistent with some earlier indirect estimates. Given the trials now underway for possible FXS treatments, this method could be used in newborn or infant screening as a way of ensuring early interventions for FXS.
Introduction
Fragile X syndrome (FXS [MIM 300624]) is caused by mutations of the X-linked FMR1 (MIM 309550) gene, which results in the functional absence of the gene product, FMRP.1–3 The most common mutation of FMR1 is the expansion of a CGG trinucleotide repeat in the 5′ untranslated region of the gene. Expansions of more than 200 CGG repeats, known as the full mutation, result in FMR1 hypermethylation and chromatin condensation, which lead to transcriptional silencing.4–7 Expansion to the full mutation occurs when premutation alleles (from 55 up to 200 CGG repeats) are transmitted maternally to offspring. Initially, carriers of premutation alleles were believed to lack a clinical phenotype, but we now appreciate that approximately 25% of female premutation carriers have fragile X-associated primary ovarian insufficiency (FXPOI [MIM 311360]).8 Moreover, fragile X-associated tremor/ataxia syndrome (FXTAS [MIM 300623]) is often encountered in older men who are carriers of the FMR1 premutation.9 These adult-onset disorders found in premutation carriers are distinct from FXS and, at least for FXTAS, are believed to be RNA-mediated, not due to the reduction or absence of FMRP.10
Initial prevalence of FXS estimates, based on induction of the Xq27.3 fragile site (FRAXA) as a cytogenetic marker for the disorder, described the prevalence of FXS to be 1 in 1000 to 1 in 2610 males.11,12 After the discovery of the FMR1 gene in 1991 and the eventual molecular diagnostic test for FXS, these initial prevalence estimates were revised to ∼1 in 4000 males.13 Studies since that time have estimated the prevalence to range from 1 in 3717 to 1 in 8918 in the general population of European descent (summarized in Crawford et al.).14 The largest of these studies identified 20 males with FXS among 3738 boys in special education classes in the United Kingdom, yielding a general population prevalence of 1 in 5530.15 Another study by Crawford et al., surveying children with special education needs in the metropolitan area of Atlanta, GA, USA, estimated the prevalence of FXS at 1 in 3717 among males of European descent and 1 in 2545 among African American males.16 Among the 39 published studies, the average sample size was only 405 individuals (range: 53–3738), leading to exceptionally large confidence intervals (CIs). In addition, all of these estimates are indirect, given that they are extrapolations from different phenotypically defined patient populations, each surveying a small number of individuals. Furthermore, most of these studies have focused on populations of European descent, and there is limited information on the frequency of FXS in other racial or ethnic groups. For a more accurate definition of the incidence of FXS both in the general population and in racial or ethnic subgroups, a large screen of the general population, such as a newborn screen, is needed.
One requirement of a population screen for FXS is an inexpensive assay that is also highly sensitive and accurate. Although southern blot analysis of the CGG repeat is definitive, it is also expensive, low-throughput, and technically challenging in newborn screening environments. PCR across the CGG repeat is another possible assay, but in general, its sensitivity declines as the repeat lengthens into the full-mutation range, often resulting in the undesirable situation of a positive outcome being the absence of signal. Recent technical improvements, such as capillary southern analysis17 and a PCR method using a chimeric primer that randomly targets the CGG repeat, producing a smear after agarose gel electrophoresis,18 have been shown to detect CGG repeat expansions in the full-mutation range in DNA samples isolated from whole blood. Fernandez-Carvajal et al. recently used a two-tiered screening strategy of standard PCR amplification of the CGG repeat followed by the chimeric primer assay to screen 5267 dried blood spot samples from males from Spain.19 Of the 5267 samples, two failed to produce PCR products by the standard CGG repeat PCR. Follow-up testing of these two samples with the use of the chimeric primer assay suggested that these individuals had full-mutation expansions. Fernandez-Carvajal et al. estimate a FXS incidence of 1 in 2633 males, but with rather broad CIs (95% CI 1 in 10,000 to 1 in 714).
One of the epigenetic changes concomitant with CGG repeat expansion is DNA methylation.4 Full-mutation alleles are densely methylated, not only at the CpGs located within the CGG tract but also at CpGs in the flanking sequence, including the FMR1 promoter. In male FXS patients with the full mutation, as well as mosaic males with FXS, virtually every cytosine in each CpG in the promoter of FMR1 becomes methylated, whereas there is a complete absence of methylation in unaffected males.20 Thus, FMR1 DNA methylation can be used as a marker for FXS in males.
The traditional method for assessing DNA methylation is by southern analysis with the use of methylation-sensitive restriction enzymes, which provides information about CGG repeat length as well as methylation. Alternatively, sodium bisulfite conversion of DNA coupled with methylation-sensitive PCR (MSP)21 or methylation-specific multiplex ligation-dependent probe amplification (MLPA)22 can also assess DNA methylation. MSP is less labor intensive than southern analysis and, along with CGG repeat sizing by PCR amplification, is used in clinical testing for FXS in males. Importantly, MSP is amenable to high-throughput analysis, allowing for the assessment of FMR1 DNA methylation in a large cohort of samples.
In normal females, one of the two copies of FMR1 is methylated because of X chromosome inactivation.23 Therefore, normal females have a 1:1 ratio of methylated to unmethylated FMR1 DNA. In females who carry a full mutation, without the skewing of X chromosome inactivation, the ratio of methylated to unmethylated FMR1 DNA will shift from 1:1 (50%:50%) to 3:1 (75%:25%). Therefore, the quantification of FMR1 methylation could also lead to the identification of females who carry the full mutation. However, unlike males with the full mutation, not all females who carry a full mutation have intellectual disability. In a study by Rousseau et al., 41% of full-mutation carrier females had no degree of intellectual disability, in comparison to 100% of full-mutation males being intellectually disabled.24 However, it is important to recognize that intellectual disability is not the only expression of the full mutation. For example, a study by Freund et al.25 examined the prevalence of psychiatric disorders among a cohort of 17 females who carry full mutations and found that there was an increase in the prevalence of several psychiatric diagnoses, such as avoidant personality disorder, mood disorder, and stereotypy/habit disorder, in the full-mutation-carrying females as compared to controls. Although the study is limited in size, if these disorders other than intellectual disability are truly due to the full mutation, then disease penetrance in full-mutation-carrying females may approach 100%.
Here, we describe a sensitive, rapid, low-cost, and quantitative methylation-sensitive PCR (Q-MSP) method that is amenable to high-throughput analysis for detection and quantification of FMR1 DNA methylation directly from newborn dried blood spots. Q-MSP can detect FMR1 at exceptionally low levels of FMR1 methylation—less than 1% methylated FMR1 DNA—easily detecting full-mutation and mosaic males with FXS. The sensitivity of the assay allows for a sample pooling strategy, thereby reducing the cost of population screening. To test the feasibility of using Q-MSP to screen for FXS and to provide a more accurate estimate of FXS incidence, we screened the dried blood spots of 36,124 deidentified males from the Georgia Public Health Laboratory Newborn Screening Program for FMR1 DNA methylation.
Material and Methods
Dried Blood Spot Samples
Dried blood spots were collected from the Georgia Public Health Laboratory Newborn Screening program from April 2006 to September 2008. Each sample was collected sequentially after the state had completed all newborn screening. The samples were deidentified, with only the gender and the race or ethnicity being recorded. Three-millimeter dried blood spot punches were collected in triplicate in 96-well plates with the use of a Wallac DBS Puncher (Perkin Elmer) and stored at room temperature. The Emory University and the Georgia Department of Human Resources institutional review boards approved this study.
Sodium Bisulfite Treatment of Genomic DNA
Sodium bisulfite treatment was performed essentially as described previously.26 In brief, 1 μg of genomic DNA isolated from peripheral blood lymphocytes was diluted in 25 μl dH2O. The DNA was denatured by the addition of 2.75 μl of 2 N NaOH, final concentration of 0.2 N NaOH, and incubated at 37°C for 10 min. After denaturation, 15 μl of freshly prepared 10 mM hydroquinone (Sigma catalog no. H9003) and 260 μl of 3.6 M sodium bisulfite pH∼5.0 (Sigma catalog no. 243973) were added and the reaction layered with mineral oil and incubated 4–16 hr at 54°C. A modification of the protocol for the Wizard SV Genomic DNA Clean-Up System (Promega A2361) was used for isolation of the DNA after sodium bisulfite treatment. In brief, 300 μl of a 1:1 mix of SV lysis buffer and 95% ethanol was mixed with the ∼300 μl of sodium bisulfite reaction. This mixture was transferred to a spin column and centrifuged for 1 min, binding the DNA to the resin within the column. The sample was washed two times with 600 μl of SV wash buffer. The column was centrifuged one more time without wash buffer for the removal of residual buffer and transferred to a new 1.5 ml microfuge tube. The DNA was eluted from the column by the addition of 50 μl of H2O, followed by centrifugation for 1 min. The DNA eluted from the column was desulfonated by the addition of 5.5 μl 3N NaOH and incubation at room temperature for 5 min. The DNA was ethanol precipitated, washed once with 75% ethanol, and suspended in 50 μl of 10 mM Tris-HCl (pH 8.0).
Sodium Bisulfite Treatment of Dried Blood Spots
Individual Samples
Dried blood spot punches in a 96-well plate were boiled in 60 μl of 1% SDS for 10 min for lyse of the cells and release of the DNA. After boiling, the samples were centrifuged and 25 μl of the supernatant transferred to a fresh 96-well plate. This extract was then treated with sodium bisulfite as described above.
Pooled Samples
Dried blood spot punches were pooled into groups of 44 individuals. There were two 3 mm punches per individual, for a total of 88 dried blood spot punches per pool. DNA was extracted from the pooled sample with the Wizard SV Genomic DNA Purification System (catalog no. A2361). In brief, 1500 μl of Wizard Nuclei Lysis Buffer was added to the dried blood spots, followed by 300 μl of 0.5 M ethylenediaminetetraacetate (EDTA) (pH 8.0). The sample was heated at 90°C for 10 min and then cooled to room temperature, and 150 μl of 25 mg/ml proteinase K solution was added. The sample was incubated overnight at 55°C. The next day, the sample was centrifuged, and 680 μl of supernatant was removed. An equal volume of Wizard SV Lysis Buffer was added to the supernatant and mixed. The sample was centrifuged at 14,000 rpm in microfuge for 1 min at room temperature. The supernatant was transferred to the 96-well binding column plate, and a vacuum was applied, pulling the sample through the column. The sample was washed four times with the Wizard SV 96 wash solution and dried with the vacuum for 10 min. The 96-well binding column plate was transferred to a new 96-well plate, and the DNA was eluted by the addition of 80 μl of water preheated to 65°C and application of a vacuum.
For pooled dried blood samples, sodium bisulfite treatment was performed with the EpiTect 96 Bisulfite Kit (QIAGEN) in accordance with the manufacturer's protocol. This procedure was found to result in a higher yield of DNA after bisulfite treatment than the traditional homebrew method described above. In brief, 20 μl of extracted DNA was treated with 85 μl of bisulfite solution and 35 μl of DNA protect buffer. The sample was incubated on a thermal cycler under the following conditions: initial denaturation at 99°C for 5 min, followed by 60°C for 25 min, 99°C for 5 min, 60°C for 85 min, 99°C for 5 min, and 60°C for 175 min. After sodium bisulfite treatment, 560 μl of BL solution with 10 mg/ml of carrier RNA was added, and the sample was transferred to the 96-well binding plate and pulled through the columns with a vacuum. The columns were washed with 500 μl Wash Buffer. The bisulfite-treated DNA was desulfonated on the column by the addition of 250 μl of BD solution and incubation at room temperature for 15 min. The desulfonated DNA bound to the column was washed two times with Wash Buffer followed by one wash with 95% ethanol. The columns were dried for 10 min under the vacuum. The 96-well binding plate was transferred to a collection plate, and 70 μl of Buffer EB and 10 μl of Top Elute were added to each column. A vacuum was applied for 1 min for elution of the DNA. The bisulfite-treated DNA was stored at −80°C.
Real-Time TaqMan Methylation-Sensitive PCR
Primer and Probe Design for FMR1
All primers for the TaqMan MSP were synthesized by Integrated DNA Technologies. The amplification primers used in the real-time TaqMan methylation-sensitive PCR reaction were designed to avoid CpG dinucleotides in the sense strand within the promoter of the FMR1 gene (Figure 1). The FMR1 amplification primers are: FMR1F 5′-GYGTTTTTAGGTTATTTGAAGAGAGAGGG-3′ and FMR1R 5′-CRACCCRCTACRAATATAAACACTAAAACC-3′ (Y = T or C; R = G or A). The TaqMan probes FMR1M2 (methylated DNA-specific probe) and FMR1U3 (unmethylated FMR1 DNA-specific probe) target a sequence from positions −97 to −72, relative to the transcription initiation site for FMR1. FMR1M2 (5′-CGGGGTCGAGGGGTTGAGTTCGCG-3′) is 5′-end labeled with Fam and is quenched by the addition of Black Hole Quencher 1 to the 3′ end of the oligonucleotide. FMR1U3 (5′ - TGGGGTTGAGGGGTTGAGTTTGTGGG - 3′) is 5′ end-labeled with Hex and quenched by the addition of Black Hole Quencher 1 to the 3′ end of the oligonucleotide. Bisulfite-treated DNAs were subjected to TaqMan PCR amplification in a 25 μl volume with the above primers and probes with the use of Invitrogen Platimum Taq DNA polymerase with the manufacturer's suggested buffer and 1.5 mM of MgCl2. The primer concentrations were 1 μM and the probe concentrations were 150 nM. The PCR conditions were an initial 95°C denaturation for 3 min followed by amplification cycles consisting of 95°C for 10 s, 67°C for 30 s, and 72°C for 60 s for 40 cycles.
Figure 1.
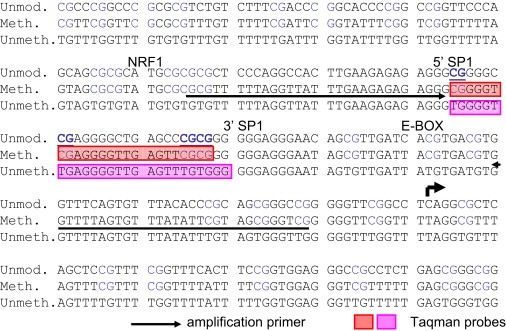
FMR1 Promoter Sequence Targeted for DNA Methylation Analysis
The sequence of the FMR1 promoter from position −192 to position +59 is shown. The top line is the genomic reference (Ref.). The second line represents the sequence after sodium bisulfite treatment and PCR amplification if every cytosine in each CpG is methylated (Meth.). The third line represents the sequence after sodium bisulfite treatment and PCR amplification if every cytosine in each CpG is unmethylated (Unmeth.). The amplification primers are underlined, and the TaqMan probes are indicated by the shaded boxes. The CpGs targeted by the TaqMan probes are underlined and in bold. The transcription start site is indicated by the arrow.
Amelogenin PCR
Male samples determined to have methylated FMR1 DNA were tested for verification of the gender via amelogenin PCR amplification. The amelogenin loci on the X and Y chromosomes carry homologous sequences that, when targeted by PCR, yield two differently sized products: a 542 base pair (bp) product from the X chromosome and a 358 bp product from the Y chromosome. The amelogenin primers used were AmelF (5′-CTCTGATGGTTGGCCTCAAG-3′) and AmelR (5′-ACCTTGCTCATATTATACTTGACAAAG-3′). PCR amplification was done with the use of Invitrogen Platinum Taq DNA polymerase with the manufacturer's recommended buffer and 3.5 mM MgCl2 and primer concentrations of 333 nM. After an initial denaturation step of 95°C for 3 min, the cycling conditions were 95°C for 30 s, 58°C for 45 s, and 72°C for 45 s for 35 cycles, followed by a final extension step of 72°C for 7 min. The PCR products were resolved by electrophoresis on a 1.5% agarose gel.
CGG Repeat Analysis
CGG repeat length was determined by PCR amplification with the use of the forward primer FXCFOR (5′-AGGCGCTCAGCTCCGTTTCGGTTTCACTTC-3′) and the Fam-labeled FXX3 reverse primer (5′-FAMGTGGGCTGCGGGCGCTCGAGG-3′). The PCR reaction conditions were as follows: 42% Q-Solution (QIAGEN), 2% DMSO, 1× PCR Buffer II (Roche), 2 mM MgCl2, 150 μM 7-deaza-dGTP, 48 μM dGTP,192 μM dATP, dTTP, cCTP, and 1.25 units of Taq DNA polymerase (Roche). The cycling parameters, after an initial denaturation step of 10 min at 95°C, were as follows: 95°C for 90 s, 67°C for 60 s, and 72°C for 3 min for 24 cycles, followed by a final extension at 72°C for 10 min. The PCR products were separated by capillary electrophoresis with an ABI 3100 Genetic Analyzer (ABI).
Southern Analysis
Southern analysis of males confirmed by amelogenein PCR was done with the use of genomic DNA isolated from the dried blood spot card. Residual dried blood from the newborn screening card was collected and boiled with 1 ml of 1% SDS for 10 min. The sample was centrifuged and the supernatant collected. Five hundred micrograms of proteinase K was added to the supernatant and incubated overnight at 55°C. The sample was phenol extracted three times, followed by one chloroform extraction. The DNA was precipitated by the addition of 1/10 volume of 3M sodium acetate and 2.5 volumes of ethanol, followed by incubation at −80°C from 1 to 16 hr. The DNA was pelleted by centrifugation, washed with 70% ethanol, and eluted in 10 μl TE.
Approximately 4 μg of isolated genomic DNA was digested in a 50 μl reaction with 40 units of EcoRI and 40 units of XhoI (New England Biolabs) with the use of the manufacturer's recommended buffer for approximately 16 hr. After 16 hr, 1 μl (10 units) of both EcoRI and XhoI were spiked into the reaction, and the digestion continued for an additional 4 hr. The digested DNAs were separated by electrophoresis on a 1% agarose gel. The DNA was transferred to a Hybond N+ nylon membrane (Amersham) by capillary transfer with the use of the TurboBlotter system (Whatman). After transfer, the membrane was baked for 2 hr at 80°C so that the DNA was fixed to the membrane. Prehybridization of the membrane was performed at 65°C with a hybridization solution (7% SDS, 1.5× SSC, 100 mg/ml polyethylene glycol [PEG] 8000, 250 μg/ml heparin) containing 430 μg/ml of salmon sperm DNA. Hybridization was performed with the use of the FMR1 probe A, a PCR product generated from the FMR1 promoter. The probe was labeled with alpha 32P-dATP with the High Prime Reaction kit (Roche). 1 × 107 cpm of labeled FMR1 probe A in 10 mls of hybridization solution containing 400 μg/ml of salmon sperm DNA was added to the membrane and rotated at 65°C for 16 hr. After hybridization, the membrane was washed once with Wash Buffer I (0.1% SDS, 2× SSC) at room temperature and once at 65°C, followed by one wash with Wash Buffer II (0.5% SDS, 0.1× SSC) at 65°C. The membrane was sealed in plastic wrap and exposed to film and/or Storm Phosphorimager screen (Molecular Dynamics).
Results
We have developed a Q-MSP assay that can be used for the detection of aberrant FMR1 methylation as a screen for FXS. First, we present the validation of detecting FMR1 methylation in isolated male genomic DNA, as well as in male dried blood samples. Second, we test whether quantification of FMR1 methylation by Q-MSP can be used for the identification of full-mutation carrier females and whether the detected amount of FMR1 methylation in a full-mutation carrier female correlates with the phenotype. Third, we examine the sensitivity of Q-MSP with respect to pooling male dried blood samples for efficient screening for FXS. Finally, to demonstrate the utility of the method for newborn screening in males, we provide results from screening 36,124 deidentified male dried blood samples for FMR1 methylation with respect to the identification of males with FXS and with KS.
Development and Validation of Q-MSP as a Screen for FXS
Detection and Quantification of Methylated FMR1 DNA in Males
To verify that Q-MSP can be used for detection of methylated FMR1 DNA, we took genomic DNAs isolated from blood from 25 clinically documented FXS males (22 full-mutation carriers and three mosaic) and treated them with sodium bisulfite, then subjected the treated DNAs to Q-MSP targeting FMR1. We also included three clinically documented KS males, because these males would also show FMR1 methylation because of their inactive X chromosome. As shown in Figure 2, Q-MSP can easily detect FMR1 DNA methylation in males with both FXS and KS. As expected, the proportion of methylated FMR1 DNA is considerably less in KS males, who carry both methylated and unmethylated FMR1 DNA on the inactive and active X chromosomes, respectively, as opposed to males with FXS, whose single copy of the FMR1 gene is completely or predominantly methylated.
Figure 2.
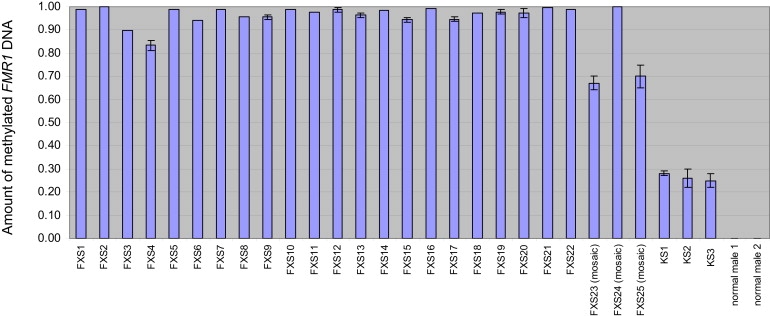
Proportion of Methylated FMR1 DNA Detected in 25 Different FXS Males and Three KS Males
Methylated FMR1 DNA was quantified by Q-MSP and is expressed as the percentage of methylated DNA (amount of methylated FMR1 DNA / amount of methylated FMR1 DNA + amount of unmethylated FMR1 DNA). The height of the bar corresponds to the mean amount of methylated FMR1 DNA from three measurements, with the error bars representing 1 SD.
To screen for FXS in the large cohort of samples necessary for a population-based study, we adapted this procedure to a 96-well format, using crude extracts from dried blood spots as starting material. To validate the assay, we screened 88 3 mm dried blood spots from males (86 normal males and two mosaic FXS males) simultaneously for FMR1 DNA methylation. The investigators were blinded to the location of the mosaic FXS dried blood spots within the 96-well plate. As shown in Figure 3, the two mosaic FXS male samples had significant amounts of FMR1 DNA methylation that was easily detected by Q-MSP, whereas the 86 normal males had no detectable FMR1 DNA methylation. Importantly, unmethylated FMR1 DNA was detected in all of the samples, verifying that the absence of signal for methylated FMR1 DNA in the negative samples was not due to any failure of PCR amplification.
Figure 3.
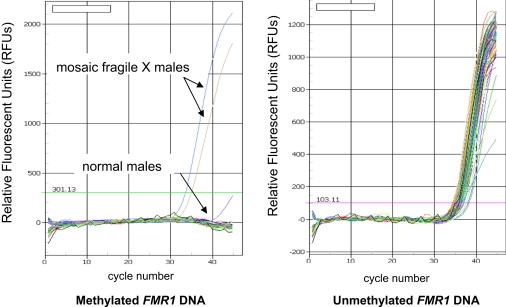
Amplification Plots for 88 Male Dried Blood Spot Samples Tested Simultaneously via Q-MSP for Methylated FMR1 DNA
Right panel: amplification plot for Fam-labeled TaqMan probe specific for methylated FMR1 DNA. Left panel: amplification plot for Hex-labeled TaqMan probe specific for unmethylated FMR1 DNA. Each line represents a single sample.
Quantification of Methylated FMR1 DNA in Females
In full-mutation females, skewing of X chromosome inactivation can alter the proportion of methylated FMR1 DNA from as low as 50% (completely favorably skewed; i.e., the inactive X chromosome always carries the mutated FMR1 gene) to 100% (completely unfavorably skewed; i.e., the active X chromosome always carries the mutated FMR1 gene). Therefore, a female with an elevated amount of methylated FMR1 DNA is at higher risk of carrying a full-mutation FMR1 allele.
To determine whether we could distinguish between normal females and full-mutation females, we used Q-MSP to quantify the proportion of methylated FMR1 DNA in a cohort of normal and full-mutation females (Figure 4). The mean proportion of methylated FMR1 DNA for 13 normal females was 0.59, with a standard deviation (SD) of 0.07. The slight increase from the predicted 0.50 is probably due to cross hybridization of the TaqMan probe specific for methylated DNA with unmethylated DNA. Assuming a normal distribution of methylation, we estimate from this data set that the percentage of methylated FMR1 DNA in 95% of normal females will be between 0.45 and 0.73. The mean percentage of methylated FMR1 DNA from a cohort of 33 females carrying the full mutation is 0.80, with a SD of 0.09. Thus, the percentage of methylated FMR1 DNA is significantly elevated among females who carry the full mutation (p = 5.4 × 109). We found 27 out of the 33 full-mutation females (82%) have an increased percentage of methylated FMR1 DNA of greater than 2 SD above the normal mean. Assuming a normal distribution, 2.5% of normal females would theoretically exceed this cutoff, resulting in a positive predictive value of 97% for detecting full-mutation females.
Figure 4.

Quantification of Methylated FMR1 DNA in 33 Females Who Carry the Full-Mutation Allele
The proportion of methylated FMR1 DNA was quantified by Q-MSP and is expressed as a percentage of total FMR1 DNA. Thirteen normal females with normal FMR1 alleles are represented by the green bars on the right-hand side of the graph. The 33 females carrying the full mutation are represented as follows: affected full mutation, red bars; unaffected full mutation, dark blue bars; and full mutation with no phenotypic information, white bars. The height of the bar corresponds to the mean amount of methylated FMR1 DNA from three measurements, with the error bars representing 1 SD. The normal range of methylation (normal female mean ± 2 SD) is represented by the shaded box.
The incomplete penetrance of the full mutation in females is believed to be due to the skewing of X chromosome inactivation, specifically in the brain. In this cohort of females carrying the full mutation, 24 had been clinically assessed, 19 of whom were classified as affected and five as unaffected. Of the 19 full-mutation females affected with FXS, 15 had elevated FMR1 DNA methylation of more than 2 SD above the mean (79%). Of the five full-mutation-carrying females unaffected by FXS, four had elevated FMR1 DNA methylation of greater than 2 SD above the mean (80%). These data indicate that the probability that an unaffected female carrying the full mutation has elevated FMR1 DNA methylation is approximately the same as the probability that an affected full-mutation carrier has elevated FMR1 DNA methylation. Therefore, although methylated FMR1 DNA can detect most full-mutation-carrying females, the test has little prognostic value.
Pooling of Male Dried Blood Spot Samples for FMR1 DNA Methylation Screening
Q-MSP is very sensitive, and in experiments in which genomic DNAs from FXS males and normal males are mixed, methylated FMR1 DNA can easily be detected when as little as 0.5% of a sample is from an FXS male (Supplemental Data, available online; Figure 1). In a population screen, the vast majority of males will test negative for the presence of methylated FMR1 DNA. Given the sensitivity of the assay, we reasoned that we could pool male samples together in a single tube, because only a limited number of pools would have methylated FMR1 DNA. If no FMR1 DNA methylation is detected, then all samples in that pool screen negative and no further testing is done; however, if FMR1 DNA methylation is detected in the pool, then the positive sample within the pool can be identified and tested individually so that the presence of the full mutation can be determined.
To test this idea, we added one dried blood spot from a mosaic FXS patient to 95 dried blood spots from normal males in a single tube. DNA was extracted from the pooled dried blood spots and treated with sodium bisulfite. As a control, we also extracted DNA from a pool of 96 normal males. We easily detected the methylated FMR1 DNA from the single mosaic FXS male sample among the 95 dried blood spots from normal males (Figure 5). Thus, up to ∼100 males could be screened simultaneously for methylated FMR1 DNA, at substantial labor and cost savings. In addition, as a quality control during the screening, we used pools of 43 dried blood spots from normal males that were spiked with a dried blood spot from either a single FXS or KS male, in addition to a control pool that contained dried blood spots from only normal males. Seven different spiked pools tested had FMR1 methylation detected by Q-MSP, whereas the pools that contained only normal males never had detectable FMR1 methylation (Supplemental Data; Figure 2). Moreover, of these seven independent spiked pools, each was also used multiple times as a control, for a total of 39 replications without discrepancy. These results demonstrate 100% sensitivity and 100% specificity for this method of detecting the full mutation in males.
Figure 5.
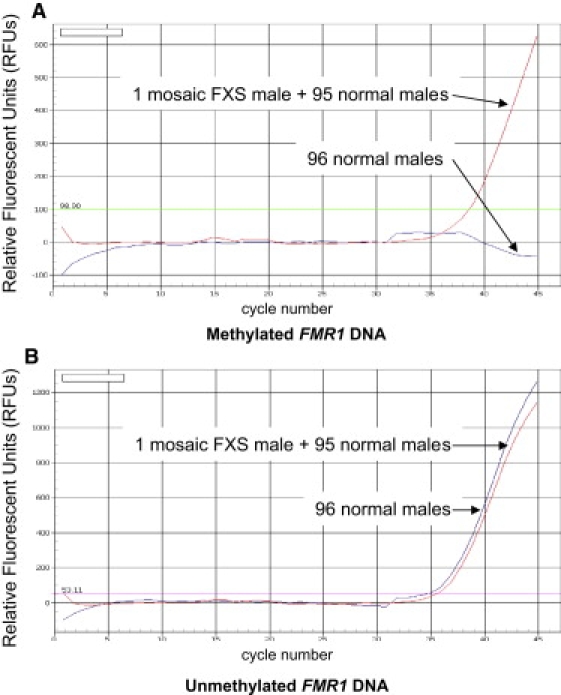
Amplification Plots of Pools of 96 Male Dried Blood Spot Samples with or without a Mosaic Male with FXS
The red lines represent the pooled sample containing one mosaic FXS male dried blood spot mixed with 95 dried blood spots from normal males. The blue lines represent the sample containing 96 dried blood spots from normal males.
(A) Amplification plot for TaqMan probe specific for methylated FMR1 DNA.
(B) Amplification plot for TaqMan probe specific for unmethylated FMR1 DNA.
Screening Deidentified Dried Blood Spots from the Georgia Public Health Laboratory
State-mandated newborn screening programs offer an opportunity to screen a large, unbiased collection of individuals from the general population. From the Georgia Public Health Laboratory, we collected 36,124 male dried blood samples after the completion of all newborn screening. The gender and racial or ethnic information from the demographic portion of the newborn screening card was recorded for each sample, and all other identifying information was discarded. The racial and ethnic diversity of these samples was as follows: 45% white; 30% African American; 15% Hispanic; 2% Asian; 2% multiracial; < 1% American Indian; and 5% unknown or unmarked. These percentages closely mirror the percentages reported by the Centers for Disease Control in the Vital Statistics Report for the state of Georgia in 2005, indicating that we have a random and accurate sampling of the newborns in the state.
Three-millimeter punches of dried blood samples were collected in triplicate in three 96-well plates, each plate containing 88 males with two punches each (a total of 176 punches). Samples from 44 males (88 dried blood spots) were pooled so that each plate contained two pools of 44 samples. If a pool contains no detectable methylated FMR1 DNA, then all 44 samples within that pool are called negative and no further testing is done. For pools that have detectable methylated FMR1 DNA, the replicate plates are used for identifying the positive individual by screening of a pool of four rows of 11 samples in one replicate plate and pooling of 11 columns of four samples in the other replicate plate. Thus, 15 reactions can identify the positive individual(s) within that pool of 44 samples.
The remainder of the dried blood spot cards from individuals with methylated FMR1 DNA were used for isolating genomic DNA for further testing. The gender of the individual was determined via amelogenin PCR. Males with FMR1 DNA methylation were tested individually by Q-MSP for verification of the presence of methylated FMR1 DNA. FMR1 DNA methylation-positive males were then tested for CGG repeat length by PCR amplification and by Southern blot analysis. A sample was considered to be from a KS male if two X chromosomes were detected by Southern analysis (as a typical female pattern), CGG repeat analysis (two different CGG repeat alleles detected), and/or semiquantitative amelogenin PCR. The remaining samples were tested by Southern blotting for FMR1 full mutations.
Of 821 pools screened, representing 36,124 newborns recorded as males, 650 pools had no detectable methylated FMR1 DNA and were called negative (28,600 newborns). Among the 171 pools with FMR1 DNA methylation, 177 newborns were identified, with several pools having multiple positives. Of these, 113 newborns (63%) were determined to be females via amelogenin PCR, and the majority of these probably reflect clerical errors on the part of the delivery hospitals in the state. Of the 64 males with methylated FMR1 DNA, seven were confirmed to carry the full mutation at FMR1, by Southern blot analysis, and 57 were found to have KS. A representative Southern blot of one of the FXS males identified by Q-MSP is shown in Figure 6. From these data, we estimate the incidence of FXS in the general population to be 1 in 5161 males (95% CI: 1 in 10,653–1 in 2500) and the incidence of KS to be 1 in 634 males (95% CI: 1 in 821–1 in 489).
Figure 6.
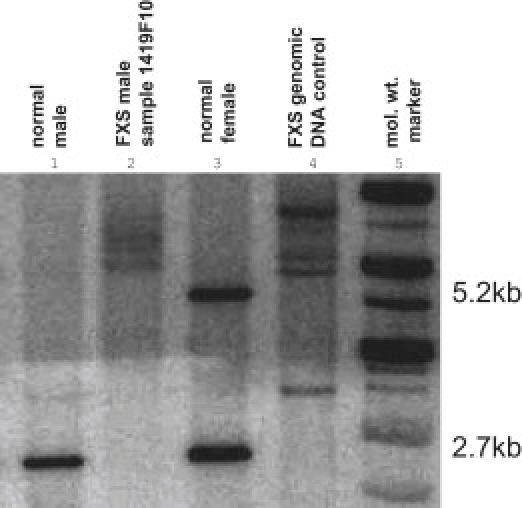
Southern Blot with the Use of DNA Isolated from the Dried Blood Spot Card from One of the FXS Males Identified in the Screen, Sample 1419F10
Lane 1. normal male control. Lane 2. sample 1419F10. Lane 3. normal female control. Lane 4. FXS genomic DNA control. Lane 5. molecular weight marker.
Using the reported racial and ethnic information for each sample, we calculated the incidence of FXS and KS among the various groups (Table 1). Of 16,252 samples from white males, four had FXS and 27 had KS. Of 10,979 samples from African American males, two had FXS and 20 had KS. Of 5396 samples from Hispanic males, one had FXS and three had KS. No FXS males were identified among the 848 Asian, 87 American Indian, and 779 multiracial males tested or among the samples identified as unknown or unmarked. Three KS males were identified among the 848 Asian male samples screened.
Table 1.
Racial and Ethnic Distribution of Identified Males with FXS and KS
| Number of Samples (Percentage of Population) | Positives | Incidence | 95% CI | |
|---|---|---|---|---|
| FXS | ||||
| White | 16,252 (45%) | 4 | 1/4063 | 1/10,477–1/1580 |
| African American | 10,979 (30%) | 2 | 1/5490 | 1/20,017–1/1506 |
| Hispanic | 5396 (15%) | 1 | 1/5396 | 1/30,567–1/953 |
| Asian | 847 (2%) | 0 | ||
| American Indian | 86 (< 1%) | 0 | ||
| Multiracial | 778 (2%) | 0 | ||
| Unknown | 343 (1%) | 0 | ||
| Unmarked | 1440 (4%) | 0 | ||
| TOTAL | 36,124 (100%) | 7 | 1/5161 | 1/10,653–1/2500 |
| KS | ||||
| White | 16,252 (45%) | 27 | 1/602 | 1/876–1/414 |
| African American | 10,979 (30%) | 20 | 1/549 | 1/848–1/356 |
| Hispanic | 5396 (15%) | 3 | 1/1799 | 1/5288–1/612 |
| Asian | 847 (2%) | 3 | 1/282 | 1/830–1/97 |
| American Indian | 86 (< 1%) | 0 | ||
| Multiracial | 778 (2%) | 0 | ||
| Unknown | 343 (1%) | 0 | ||
| Unmarked | 1440 (4%) | 4 | 1/360 | 1/925–1/140 |
| TOTAL | 36,124 (100%) | 57 | 1/634 | 1/821–1/489 |
Discussion
Here, we report a quantitative, methylation-sensitive PCR assay that is an effective and inexpensive method of population screening for FXS males. The assay is amenable for adaptation to a high-throughput format that can be used for detecting FMR1 methylation in individual male dried blood samples. Alternatively, the sensitivity of the method allows for pooling of male dried blood spot samples into large groups, reducing the labor and expense for screening. With pooling, we estimate the cost of the screening, including labor and reagents, to be under $3 per sample.
Our approach can also be used as a way of screening for full-mutation females. Approximately 82% of females who carry the full mutation have elevated FMR1 DNA methylation, defined as greater than 2 SD above the mean of normal methylation. Thus, the positive predictive value of identifying the full-mutation genotype in females is 97%; however, only about 41% of the full mutations in these females are destined to be penetrant.24 Relative to phenotype in full-mutation females, this approach did not easily distinguish penetrant from nonpenetrant females. Here, 80% of unaffected females with the full mutation had elevated FMR1 DNA methylation, indistinguishable from affected females. This could be problematic, because revealing a potential diagnosis to parents when the full mutation is destined to be nonpenetrant in their child could affect the parents' perception of their child's health, lead to parental stress, and affect the parent-child relationship, all of which have been well documented in cases when false-positive diagnoses have been made during newborn screening for metabolic disease, leading to “vulnerable child syndrome” or the “nocebo phenomenon,” when the expectation of sickness in an otherwise normal child leads to a parental emotional response that can itself lead to illness or distress.27 However, this outcome would need to be balanced by the view that by not identifying newborn girls with the full mutation, those girls who will be affected with symptoms of FXS may not be given either palliative early-intervention treatment or, if effective pharmaceuticals are developed for FXS, a significant chance of an improved outcome.
To test the feasibility of using this approach as a newborn screen for FXS, we screened 36,124 deidentified male dried blood spot samples from the state of Georgia's newborn screening program. Because the assay identifies males with KS as well, owing to the FMR1 methylation of their inactive X chromosome, we also identified 57 males with KS, yielding a general population incidence of 1 in 634 (95% CI: 1 in 821–1 in 489). A study by Nielsen et al., surveying 34,910 newborns over a 13 yr period in Denmark, estimated the incidence of KS to be 1 in 576 boys,28 statistically indistinguishable from our estimate (p = 0.79; Chi-square test with Yates correction). Other estimates of KS range from 1 in 1087 to 1 in 333 live male births.29–38 It has been suggested that the incidence of KS has increased in recent years, and the incidence estimate from these recent studies combined is 1.72 per 1000 (1 in 581) male births,36 similar to our estimate of 1 in 634. Given that the population incidence of KS has been more thoroughly studied than FXS and the amount of FMR1 DNA methylation is considerably lower in KS males than in FXS males (Figure 2), the detection of KS males was, in this study, an important internal control for FXS full-mutation screening, because KS incidence served as a sentinel suggesting that our ascertainment was unbiased.
Among the three main racial or ethnic groups represented in the state of Georgia, namely whites, African Americans, and Hispanics, the incidence of KS was approximately equal among whites (1 in 602) and African Americans (1 in 549) but was less frequent among Hispanics (1 in 1799). However, given the relatively small numbers of Hispanic KS males identified, the paucity of KS in this ethnic group suggested a lower incidence but was not significant from other ethnicities (p = 0.06; Chi-square test with Yates correction). To our knowledge, there have been no previous incidence estimates of KS specifically among Hispanics. Maternal age may be a contributing factor, given that the average maternal age among Hispanics in Georgia is about two years younger than the average maternal age among African Americans and about four years younger than that of white mothers (data taken from the National Down Syndrome Project).39 Additional studies with larger sample numbers are needed for verification that there is truly a decreased incidence of KS among Hispanics.
The incidences of FXS among the three main racial or ethnic groups in the state of Georgia were not statistically different, with an incidence of 1 in 4063 in whites, an incidence of 1 in 5490 in African Americans, and an incidence of 1 in 5396 in Hispanics. However, these sample sizes are too small to allow a definitive conclusion that there is not a significant difference in the incidence of FXS, and additional studies with larger cohorts are needed for determining whether FXS is truly equally prevalent among these groups. The overall population incidence estimate of 1 in 5161 from this study is similar to the prevalence estimate from the largest of the studies using targeted populations, which yielded an incidence estimate of 1 in 5530.15 Two large, direct, general population screens, both using a series of dried blood spot samples from Spain, identified two males with full mutations out of approximately 5000 samples screened. A study by Rife et al. identified two apparent full-mutation-carrying males by lack of amplification of the CGG repeat, yielding an incidence estimate of 1 in 2466.40 A more recent study by Fernandez-Carvajal et al. using dried blood spots from the northwest region of Spain, identified two FXS males out of 5267, yielding an incidence estimate of 1 in 2633 (95% CI: 1 in 10,000–1 in 714).19 However, given the relatively small sample size of both of these studies, the differences in incidence estimates between our study and the two Spanish studies were not statistically significant (p = 0.67 and p = 0.72, respectively; Chi-square test with Yates correction). Saul et al. conducted a prospective study of FXS screening, using 1459 samples that were ascertained immediately after birth from volunteers from two South Carolina hospitals.41 They identified two full-mutation-carrying males among this cohort. Because of the limited sample size and of how the samples were ascertained, the authors concluded that high prevalence of FXS among this cohort was probably a chance occurrence. Because the study reported here is by far the largest undertaken for FXS, and because the outcome compares favorably with the largest indirect study of a targeted population, our incidence of 1 in 5161 males is probably a close approximation of the true incidence of FXS.
FXS has been proposed as a prototype for population screening,42 and our methodology lends itself well to newborn or infant screening. Identifying FXS in the newborn period or in early infancy would enable early intervention for these children. In addition, if the pharmaceutical therapies currently in clinical trials demonstrate any efficacy, identifying children with FXS would allow them to be treated earlier, maximizing the possible benefits of these agents.43,44 Furthermore, identification of FXS children in a newborn screen would also prevent the stress and anxiety, in addition to the monetary costs, that parents incur in their “diagnostic odyssey,” which at present translates to three or four years spent in order to a diagnosis of FXS.45–47 Also, through cascade testing, identifying children with full mutations would lead to the identification of mothers with premutations and of clinically unrecognized full-mutation-carrying mothers. These women could be counseled appropriately about the risks of FXS in future pregnancies. In addition, these women, as well as other members of their family, could be counseled about their risks of premature ovarian failure and FXTAS. A limitation of the study is the detection of FXS among females. Although 82% of full-mutation-carrying females could be identified by Q-MSP, our assay could not distinguish those diagnosed with intellectual disabilities and those without.
We must note that this methodology would not lead to the identification of premutation carriers in infant males, because premutation FMR1 DNA is not hypermethylated. Therefore, males who are at risk of developing FXTAS, an adult-onset disorder, would not be identified as infants. An alternative method, CGG repeat tract sizing, has also been proposed for newborn screening for FXS. This methodology would reveal premutation alleles, identifying infants who are at risk of developing FXTAS. The ethical consequences of screening newborns for adult-onset neurodegenerative diseases has been debated extensively, and two committees of the American Academy of Pediatrics have recommended against predictive testing for adult-onset disorders in persons under 18 years.48,49 However, identification of premutation males would identify a mother at risk for future full-mutation pregnancies, as well as families with pre- and full-mutation individuals. As with the debate over screening full-mutation-carrying females without being prognostic, screening for only full mutations or for premutations also deserves further consideration.
Both FMR1 DNA methylation analysis and CGG repeat tract sizing will lead to the identification of KS males in newborn or infant screening. The same arguments that are used to support screening for FXS, early intervention and preventing the diagnostic odyssey, also apply to KS. However, those with KS have relatively mild phenotypic features that may present clinically at many points during the course of their life,50 and they are often undiagnosed their entire lives, although this is due to variable expressivity or mosaicism rather than to nonpenetrance, because nearly all KS males are infertile.51 Whether or not revealing a diagnosis of KS from a newborn screen for FXS is appropriate merits further debate, similar to screening for premutations or for full mutations in females.
In summary, we describe here a highly sensitive and specific assay for methylated FMR1 DNA that is cost effective and carries sufficient high-throughput capability to serve as a population screening method for FXS in male newborns. Although the approach also identified full mutations in females, it is not prognostic in these females. We utilized this approach to screen 36,124 deidentified dried blood spots from newborn males and demonstrate the incidence of FXS to be 1 in 5161 males. Given the size of this screen, this probably represents the true male incidence of FXS in the United States.
Supplemental Data
Supplemental Data include two figures and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank Kasinathan Muralidharan for assistance in assay development, Elizabeth Franco and M. Ramachandran from the Georgia Public Health Laboratory Newborn Screening Program for their critical assistance, and Fuping Zhang for technical assistance. We also thank Michael Friez and Roger Stephenson for providing DNA samples from phenotyped full-mutation females. This work was supported by National Institutes of Health grants HD020521 and HD024064 and by Centers for Disease Control and Prevention grant AAMC MM-0937-06/06 (to S.T.W.).
References
- 1.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 2.Fu Y.H., Kuhl D.P., Pizzuti A., Pieretti M., Sutcliffe J.S., Richards S., Verkerk A.J., Holden J.J., Fenwick R.G., Warren S.T. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 3.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 4.Sutcliffe J.S., Nelson D.L., Zhang F., Pieretti M., Caskey C.T., Saxe D., Warren S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 5.Eberhart D.E., Warren S.T. Nuclease sensitivity of permeabilized cells confirms altered chromatin formation at the fragile X locus. Somat. Cell Mol. Genet. 1996;22:435–441. doi: 10.1007/BF02369435. [DOI] [PubMed] [Google Scholar]
- 6.Coffee B., Zhang F., Warren S.T., Reines D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999;22:98–101. doi: 10.1038/8807. [DOI] [PubMed] [Google Scholar]
- 7.Coffee B., Zhang F., Ceman S., Warren S.T., Reines D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 2002;71:923–932. doi: 10.1086/342931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allingham-Hawkins D.J., Babul-Hirji R., Chitayat D., Holden J.J., Yang K.T., Lee C., Hudson R., Gorwill H., Nolin S.L., Glicksman A. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study–preliminary data. Am. J. Med. Genet. 1999;83:322–325. [PMC free article] [PubMed] [Google Scholar]
- 9.Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B., Hagerman P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 10.Jin P., Zarnescu D.C., Zhang F., Pearson C.E., Lucchesi J.C., Moses K., Warren S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 11.Webb T.P., Bundey S., Thake A., Todd J. The frequency of the fragile X chromosome among schoolchildren in Coventry. J. Med. Genet. 1986;23:396–399. doi: 10.1136/jmg.23.5.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner G., Robinson H., Laing S., Purvis-Smith S. Preventive screening for the fragile X syndrome. N. Engl. J. Med. 1986;315:607–609. doi: 10.1056/NEJM198609043151002. [DOI] [PubMed] [Google Scholar]
- 13.Turner G., Webb T., Wake S., Robinson H. Prevalence of fragile X syndrome. Am. J. Med. Genet. 1996;64:196–197. doi: 10.1002/(SICI)1096-8628(19960712)64:1<196::AID-AJMG35>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 14.Crawford D.C., Acuna J.M., Sherman S.L. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet. Med. 2001;3:359–371. doi: 10.1097/00125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Youings S.A., Murray A., Dennis N., Ennis S., Lewis C., McKechnie N., Pound M., Sharrock A., Jacobs P. FRAXA and FRAXE: the results of a five year survey. J. Med. Genet. 2000;37:415–421. doi: 10.1136/jmg.37.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford D.C., Meadows K.L., Newman J.L., Taft L.F., Scott E., Leslie M., Shubek L., Holmgreen P., Yeargin-Allsopp M., Boyle C. Prevalence of the fragile X syndrome in African-Americans. Am. J. Med. Genet. 2002;110:226–233. doi: 10.1002/ajmg.10427. [DOI] [PubMed] [Google Scholar]
- 17.Strom C.M., Huang D., Li Y., Hantash F.M., Rooke J., Potts S.J., Sun W. Development of a novel, accurate, automated, rapid, high-throughput technique suitable for population-based carrier screening for Fragile X syndrome. Genet. Med. 2007;9:199–207. doi: 10.1097/gim.0b013e31803d3ac9. [DOI] [PubMed] [Google Scholar]
- 18.Tassone F., Pan R., Amiri K., Taylor A.K., Hagerman P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Carvajal I., Walichiewicz P., Xiaosen X., Pan R., Hagerman P.J., Tassone F. Screening for Expanded Alleles of the FMR1 Gene in Blood Spots from Newborn Males in a Spanish Population. J. Mol. Diagn. 2009;11:324–329. doi: 10.2353/jmoldx.2009.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoger R., Kajimura T.M., Brown W.T., Laird C.D. Epigenetic variation illustrated by DNA methylation patterns of the fragile-X gene FMR1. Hum. Mol. Genet. 1997;6:1791–1801. doi: 10.1093/hmg/6.11.1791. [DOI] [PubMed] [Google Scholar]
- 21.Frommer M., McDonald L.E., Millar D.S., Collis C.M., Watt F., Grigg G.W., Molloy P.L., Paul C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nygren A.O., Lens S.I., Carvalho R. Methylation-specific multiplex ligation-dependent probe amplification enables a rapid and reliable distinction between male FMR1 premutation and full-mutation alleles. J. Mol. Diagn. 2008;10:496–501. doi: 10.2353/jmoldx.2008.080053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirchgessner C.U., Warren S.T., Willard H.F. X inactivation of the FMR1 fragile X mental retardation gene. J. Med. Genet. 1995;32:925–929. doi: 10.1136/jmg.32.12.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rousseau F., Heitz D., Tarleton J., MacPherson J., Malmgren H., Dahl N., Barnicoat A., Mathew C., Mornet E., Tejada I. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am. J. Hum. Genet. 1994;55:225–237. [PMC free article] [PubMed] [Google Scholar]
- 25.Freund L.S., Reiss A.L., Abrams M.T. Psychiatric disorders associated with fragile X in the young female. Pediatrics. 1993;91:321–329. [PubMed] [Google Scholar]
- 26.Kubota T., Das S., Christian S.L., Baylin S.B., Herman J.G., Ledbetter D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997;16:16–17. doi: 10.1038/ng0597-15. [DOI] [PubMed] [Google Scholar]
- 27.Gurian E.A., Kinnamon D.D., Henry J.J., Waisbren S.E. Expanded newborn screening for biochemical disorders: the effect of a false-positive result. Pediatrics. 2006;117:1915–1921. doi: 10.1542/peds.2005-2294. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen J., Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum. Genet. 1991;87:81–83. doi: 10.1007/BF01213097. [DOI] [PubMed] [Google Scholar]
- 29.Sergovich F., Valentine G.H., Chen A.T., Kinch R.A., Smout M.S. Chromosome aberrations in 2159 consecutive newborn babies. N. Engl. J. Med. 1969;280:851–855. doi: 10.1056/NEJM196904172801602. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs P.A., Melville M., Ratcliffe S., Keay A.J., Syme J. A cytogenetic survey of 11,680 newborn infants. Ann. Hum. Genet. 1974;37:359–376. doi: 10.1111/j.1469-1809.1974.tb01843.x. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen J., Wohlert M., Faaborg-Andersen J., Hansen K.B., Hvidman L., Krag-Olsen B., Moulvad I., Videbech P. Incidence of chromosome abnormalities in newborn children. Comparison between incidences in 1969–1974 and 1980–1982 in the same area. Hum. Genet. 1982;61:98–101. doi: 10.1007/BF00274195. [DOI] [PubMed] [Google Scholar]
- 32.Hamerton J.L., Canning N., Ray M., Smith S. A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin. Genet. 1975;8:223–243. doi: 10.1111/j.1399-0004.1975.tb01498.x. [DOI] [PubMed] [Google Scholar]
- 33.Lin C.C., Gedeon M.M., Griffith P., Smink W.K., Newton D.R., Wilkie L., Sewell L.M. Chromosome analysis on 930 consecutive newborn children using quinacrine fluorescent banding technique. Hum. Genet. 1976;31:315–328. doi: 10.1007/BF00270861. [DOI] [PubMed] [Google Scholar]
- 34.Buckton K.E., O'Riordan M.L., Ratcliffe S., Slight J., Mitchell M., McBeath S., Keay A.J., Barr D., Short M. A G-band study of chromosomes in liveborn infants. Ann. Hum. Genet. 1980;43:227–239. doi: 10.1111/j.1469-1809.1980.tb01556.x. [DOI] [PubMed] [Google Scholar]
- 35.Hansteen I.L., Varslot K., Steen-Johnsen J., Langard S. Cytogenetic screening of a new-born population. Clin. Genet. 1982;21:309–314. doi: 10.1111/j.1399-0004.1982.tb01377.x. [DOI] [PubMed] [Google Scholar]
- 36.Morris J.K., Alberman E., Scott C., Jacobs P. Is the prevalence of Klinefelter syndrome increasing? Eur. J. Hum. Genet. 2008;16:163–170. doi: 10.1038/sj.ejhg.5201956. [DOI] [PubMed] [Google Scholar]
- 37.Idzelene I.P. Genetika. 1978;14:2193–2196. [PubMed] [Google Scholar]
- 38.Maeda T., Ohno M., Matsunobu A., Yoshihara K., Yabe N. A cytogenetic survey of 14,835 consecutive liveborns. Jinrui Idengaku Zasshi. 1991;36:117–129. doi: 10.1007/BF01876812. [DOI] [PubMed] [Google Scholar]
- 39.Freeman S.B., Allen E.G., Oxford-Wright C.L., Tinker S.W., Druschel C., Hobbs C.A., O'Leary L.A., Romitti P.A., Royle M.H., Torfs C.P. The National Down Syndrome Project: design and implementation. Public Health Rep. 2007;122:62–72. doi: 10.1177/003335490712200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rife M., Badenas C., Mallolas J., Jiménez L., Cervera R., Maya A., Glover G., Rivera F., Milà M. Incidence of fragile X in 5,000 consecutive newborn males. Genet. Test. 2003;7:339–343. doi: 10.1089/109065703322783725. [DOI] [PubMed] [Google Scholar]
- 41.Saul R.A., Friez M., Eaves K., Stapleton G.A., Collins J.S., Schwartz C.E., Stevenson R.E. Fragile X syndrome detection in newborns-pilot study. Genet. Med. 2008;10:714–719. doi: 10.1097/GIM.0b013e3181862a76. [DOI] [PubMed] [Google Scholar]
- 42.Bailey D.B., Skinner D., Davis A.M., Whitmarsh I., Powell C. Ethical, legal, and social concerns about expanded newborn screening: fragile X syndrome as a prototype for emerging issues. Pediatrics. 2008;121:e693–e704. doi: 10.1542/peds.2007-0820. [DOI] [PubMed] [Google Scholar]
- 43.Hagerman R.J., Berry-Kravis E., Kaufmann W.E., Ono M.Y., Tartaglia N., Lachiewicz A., Kronk R., Delahunty C., Hessl D., Visootsak J. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123:378–390. doi: 10.1542/peds.2008-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang S., Bray S.M., Li Z., Zarnescu D.C., He C., Jin P., Warren S.T. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 2008;4:256–263. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- 45.Bailey D.B., Skinner D., Hatton D., Roberts J. Family experiences and factors associated with the diagnosis of fragile X syndrome. J. Dev. Behav. Pediatr. 2000;21:315–321. doi: 10.1097/00004703-200010000-00001. [DOI] [PubMed] [Google Scholar]
- 46.Bailey D.B., Skinner D., Sparkman K.L. Discovering fragile X syndrome: family experiences and perceptions. Pediatrics. 2003;111:407–416. doi: 10.1542/peds.111.2.407. [DOI] [PubMed] [Google Scholar]
- 47.Carmichael B., Pembrey M., Turner G., Barnicoat A. Diagnosis of fragile-X syndrome: the experiences of parents. J. Intellect. Disabil. Res. 1999;43:47–53. doi: 10.1046/j.1365-2788.1999.43120157.x. [DOI] [PubMed] [Google Scholar]
- 48.Molecular genetic testing in pediatric practice: A subject review. Committee on Genetics. Pediatrics. 2000;106:1494–1497. doi: 10.1542/peds.106.6.1494. [DOI] [PubMed] [Google Scholar]
- 49.Nelson R.M., Botkjin J.R., Kodish E.D., Levetown M., Truman J.T., Wilfond B.S., Harrison C.E., Kazura A., Krug E., Schwartz P.A. Ethical issues with genetic testing in pediatrics. Pediatrics. 2001;107:1451–1455. doi: 10.1542/peds.107.6.1451. [DOI] [PubMed] [Google Scholar]
- 50.Visootsak J., Graham J.M. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet. J. Rare Dis. 2006;1:42. doi: 10.1186/1750-1172-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abramsky L., Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat. Diagn. 1997;17:363–368. doi: 10.1002/(sici)1097-0223(199704)17:4<363::aid-pd79>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.