

Abstract
Glaucoma, a main cause of blindness in the developed world, is characterized by progressive degeneration of retinal ganglion cells (RGCs), resulting in irreversible loss of vision. Although members of the neurotrophin gene family in various species are known to support the survival of numerous neuronal populations, including RGCs, it is less clear whether they are also required for survival and maintenance of adult neurons in humans. Here, we report seven different heterozygous mutations in the Neurotrophin-4 (NTF4) gene accounting for about 1.7% of primary open-angle glaucoma patients of European origin. Molecular modeling predicted a decreased affinity of neurotrophin 4 protein (NT-4) mutants with its specific tyrosine kinase receptor B (TrkB). Expression of recombinant NT-4 carrying the most frequent mutation was demonstrated to lead to decreased activation of TrkB. These findings suggest a pathway in the pathophysiology of glaucoma through loss of neurotrophic function and may eventually open the possibility of using ligands activating TrkB to prevent the progression of the disease.
Introduction
Gradual visual impairment due to progressive loss of retinal ganglion cells (RGCs), axons, and glial cells causing a typical form of atrophy of the optic nerve characterizes glaucoma.1–4 Elevated intraocular pressure (IOP) has been suggested as a risk factor, and as a result, all current treatments aim at lowering IOP as a means of slowing the progression of the degenerative process.5,6 However, patients often fail to show much improvement even after IOP reduction, whereas others develop glaucoma in the absence of elevated IOP. Patients showing no anatomical impairment of aqueous humor outflow in the chamber angle are referred to as having the open-angle form of the disease (POAG [MIM 137760]), which typically manifests itself after the age of 40.7 A family history of the disease has long been recognized as a risk factor, suggesting that specific gene defects contribute to disease pathogenesis of POAG, a condition that exhibits high clinical heterogeneity. In some families, glaucoma segregates as a Mendelian trait with reduced penetrance and variable expressivity, but most cases are sporadic, suggesting a multifactorial contribution in the etiology of the disease.8 Classical linkage approaches in POAG families has allowed the delineation of 14 loci (GLC1A-1N),9 and glaucoma-causing genes have been identified in three of them, including myocilin (MYOC/GLC1A [MIM 601652]);10 optineurin (OPTN/GLC1E [MIM 602432]);11 and WD repeat domain 36 (WDR36/GLC1G [MIM 609669]).12 Each of these genes is responsible for a small proportion of POAG cases (1%–5%). In addition, heterozygous loss-of-function mutations in CYP1B1 (MIM 601771) constitute a risk factor (odds ratio [OR] = 5.8) for development of POAG.13–16 Recessive loss-of-function CYP1B1 mutations are usually found in newborns with primary congenital glaucoma (MIM 231300).
Recent reports have suggested for neurotrophins and their receptors a protective role in RGCs.17–19 In particular, studies of animal models have shown an increase in retinal expression of neurotrophins and their receptors after exposure to high IOP,20 glutamate,21 and nitric oxide (NO).22 Given that elevated IOP, ischemia, release of cytotoxins such as glutamate and NO, and vascular dysfunction are all implicated in glaucoma, impaired neurotrophin signaling to retinal cells or lack of trophic support in the retina by neurotrophins can be postulated in the development of the disease. To our knowledge, however, genetic variation in neurotrophins or their receptors has not yet been investigated in glaucoma.
In this study, we focused on NT-4, a member of the neurotrophin protein family comprising four members in mouse and human.23,24 Like brain-derived neurotrophic factor (BDNF), NT-4 also activates the receptor TrkB on RGCs and prevents their death in vitro and after axotomy in animal models.25–29 In the absence of NT-4, the retina develops normally in rodents, allowing the role of NT-4 to be explored in pathological situations in the adult.30 NT-4 levels are upregulated in the retina after ischemic lesion, and in animals lacking NT-4, retinal damage is more severe than that in control animals.30 In addition, the human NTF4 gene is located on chromosome 19q13.33,24 which was previously identified as a putative glaucoma locus in a genome-wide linkage scan.31 NTF4 is translated as pre-pro-neurotrophin and cleaved to release the mature active protein, which acts in solution as a noncovalently linked homodimer triggering the phosphorylation of TrkB, thus transducing its trophic effects in neurons.32 We now report a mutation screen in NTF4 in three different large cohorts of POAG patients, two from Germany and one from the Netherlands, and their respective control groups. We found a strong association of NT-4 variants predicted to alter protein function in the patients. We have subsequently shown that the most frequent variant indeed impairs TrkB signaling. Our findings implicate impaired neuronal survival in the etiology of glaucoma.
Subjects and Methods
Patients and Control Groups
The study was approved by the ethical review board of the medical faculty of the University of Erlangen-Nuremberg, the University Hospital Tübingen, the University Hospital Würzburg, and Erasmus University Rotterdam, the Netherlands, and was in accordance with the tenets of the declaration of Helsinki. All subjects gave informed consent before entering the study.
The discovery group of glaucoma patients consisted of 399 subjects of German (European) origin. Of these, 270 had primary open-angle glaucoma (high-pressure POAG), 47 had juvenile open-angle glaucoma (JOAG), and 82 had normal-tension open-angle glaucoma (NTG). All individuals underwent standardized clinical examinations for glaucoma at the Ophthalmologic Department of the University of Erlangen-Nuremberg. The examinations comprised slit-lamp biomicroscopy, gonioscopy, automated visual field testing (Octopus G1, Interzeag, Switzerland), fundus photography (Zeiss-Fundus camera, Germany), optional laser scanning tomography (HRT I and II, Heidelberg Engineering, Germany) of the disc, and 24 hr Goldmann-applanation intraocular pressure (IOP) tonometry profile with five measurements. Manifest high-tension POAG was defined as the presence of glaucomatous optic disc damage (in at least one eye), visual field defects in at least one eye, and IOP higher than 21 mmHg in one eye without therapy. According to Jonas,33 stage 0 optic disc was defined as normal, stage I as vertical elongation of the cup and neuroretinal rim loss at the 12 o'clock and 6 o'clock position, stage II as focal rim loss, stage III and IV as advanced rim loss, and stage V as absolute optic disc atrophy.34 Disc area was measured with HRT or estimated with a Goldmann lens and a Haag-Streit slit lamp. A pathologic visual field was defined by a pathologic Bebie curve, three adjacent test points with more than 5 dB sensitivity loss, or at least one point with more than 15 dB sensitivity loss.
Primary melanin dispersion, pseudoexfoliation and previously raised IOP after trauma, a period of steroid administration, and uveitis were excluded as causes for secondary glaucoma. Glaucomatous optic nerve damage was defined as focal or diffuse loss of the neuroretinal rim or nerve fiber layer associated with a specific visual field defect. Patients who showed glaucomatous changes of the optic disc and visual field but no IOP elevation over 21 mmHg after 24 hr IOP measurement (sitting and supine body position) without therapy were diagnosed as having NTG. Additionally, in the NTG patient group, a neurological examination was performed for exclusion of intracerebral expansion or malperfusion. Stenosis of the aorta carotis interna was excluded by means of sonography. Patients were classified as JOAG when age of onset in the index case was below 40 years and no other ocular reason for open-angle glaucoma was visible. At the time of examination, the age of the patients ranged from 14 to 96 years, with a mean of 66.9 ± 13.4 years. In total, 178 patients (44.4%) had a family history of glaucoma. All patients were also screened for myocilin (MYOC), optineurin (OPTN), and WD repeat domain 36 (WDR36)35 mutations, as determined by direct sequencing of all coding regions.
The 376 control subjects were all of German origin and recruited from the same geographic regions as those of the patients. In addition, control subjects were ophthalmologically examined and age and sex matched to the patients. Thus, at the time of examination and inclusion in this study, the age of control subjects ranged from 51 to 92 years, with a mean of 73.9 ± 6.4 years. They had IOP below 20 mmHg, no glaucomatous disc damage, and no family history of glaucoma. Visual acuity was at least 0.8, or 20/25, and the ocular media were clear for examination.
The first replication group consisted of 282 unrelated patients, 224 with NTG and 58 with POAG. The age of the patients ranged from 40 to 86 years, with a mean of 64.8 ± 11.9 years. The patients were clinically investigated at the University Eye Hospital in Würzburg and Tübingen. Clinical details have been reported elsewhere.36 Control DNA samples were obtained from 67 unrelated subjects of German descent, selected with the use of the same criteria described above.
The second replication group consisted of 211 POAG cases (age range 56 to 94 years, with a mean of 75.5 ± 7.4 years) from the Rotterdam Study. The rationale and study design of this study have been described elsewhere.37 Glaucoma diagnosis was based on Goldmann kinetic perimetry (Haag Streit, Bern, Switzerland) and on optical nerve head appearance. For details see Wolfs et al.38
The third replication cohort consisted of 69 POAG patients and 42 NTG patients who had been recruited in three local hospitals in the Netherlands as part of the Genetic Research in Isolated Populations (GRIP) program.39,40 Ages ranged from 52 to 90 years, with a mean age of 74.4 ± 9.0. The diagnosis of glaucoma was made by a glaucoma specialist and was based on a glaucomatous appearance of the optic disc (notching or thinning of the neuroretinal rim), combined with a matching glaucomatous visual field defect.
Mutation Screening
Genomic DNA was prepared from peripheral blood samples by a standard salting-out protocol and with the automated AutoGenFlex (Genelimited, UK) with the use of the Flexi-Gene Kit (QIAGEN, Germany). Complete coding regions of the NTF4 gene, including flanking intronic and UTR sequences, were amplified by polymerase chain reaction (PCR) with the use of appropriate amplification protocols. Primer sequences (depicted in Table S1, available online) were selected with the use of Primer3 software and were supplied from Invitrogen (Karlsruhe, Germany). Purified PCR fragments were sequenced with Big Dye Termination Chemistry v. 3.1 (Applied Biosystems, ABI, Weiterstadt, Germany) on an automated capillary sequencer (ABI 3730 Genetic Analyzer, Weiterstadt, Germany). Each variant was confirmed by a second independent analysis. GenBank accession number NM_ 006179, version NM_006179.3, was used as cDNA reference sequence and NT_ 011109 as genomic reference sequence. We used P34130 from the Swiss-Prot and Trembl database as reference preproprotein sequence. Evolutionary conservation of nonsynonymous variants was investigated with protein sequence alignment generated by ClustalW and compared with that presented by Ensembl Genome Browser.
Structural Analysis and Molecular Modeling
Modeling of all NTF4 mutations except R209G was performed on the basis of the crystal structure of the NTF4-TrkB complex (PDB code: 1HCF41). Because R209 is not resolved in the NTF4-TrkB crystal structure, the R209G mutation was modeled on the basis of the NTF4 heterodimer with brain-derived neutrophic factor (PDB code: 1B8M42). Mutations were inserted with the use of the Sybyl 7.2 program package (Tripos) and subsequently refined by 100 steps of energy minimization. Side chain interactions were analyzed with the use of LigPlot,43 with standard cutoff criteria used. The biophysical properties of the N-terminal pro-peptide, which contains the site of the C7Y mutation, was investigated with the ProtScale server, with standard settings used.44
Recombinant NTF4
An NTF4 expression clone with a CMV promoter was obtained from GeneCopoeia (Germantown, MD, USA). The NTF4 insert without a stop codon was then transferred into an additional expression vector containing a myc C-terminal fusion tag followed by a stop codon (delivered by GeneCopoeia).
Mutagenesis
R206W-myc NT-4 and wild-type (WT)-myc N-4 were obtained with the use of standard protocols of the QuickChange II Site-Directed Mutagenesis Kit (Stratagene, Amsterdam Zuidoost, The Netherlands). The following oligonucleotides were used for the mutagenesis: 5′-GCACACTCCTCAGCTGGACTGGCGAGCAGAAAC-3′and 5′-GTTTCTGCTCGCCAGTCCAGCTGAGGAGTGTGC-3′ (Invitrogen, Karlsruhe, Germany).
Production of Recombinant NT-4
COS-7 cells were transfected with the use of LipofectAmine2000 (Invitrogen, CA, USA) with 6 μg of empty vector or plasmids encoding WT NT-4, WT-myc NT-4, or mutant R206W-myc NT-4. After 2 days, conditioned media were collected and concentrated in Amicon 30,000 MWCO columns (Millipore, MA, USA). The concentrations of WT and WT-myc NT-4 were determined by immunoblotting with rabbit anti-NT-4 (1:500, sc-545, Santa Cruz Biotechnology, CA, USA) with known concentrations of recombinant human NT-4 as standard purified from CHO-conditioned medium (Genentech). The concentration of R206W-myc NT-4 was determined by immunoblotting with goat anti-c-myc (1:500, sc-789-G, Santa Cruz Biotechnology) and compared with WT-myc NT-4 as standard.
TrkB-Expressing nnr5-PC12 Cells
nnr-5 PC12 cells45 were stably transfected with a cDNA encoding full-length rat trkB. TrkB-expressing nnr5 PC12 cells (TrkB-nnr5 cells) were cultured on poly-D-lysine-coated plates and maintained in Dulbecco's modified Eagle's medium (GIBCO, Paisley, Scotland) containing 0.1% or 0.5% fetal calf serum (GIBCO) for TrkB activation assay or neurite outgrowth assay, respectively.
TrkB-Activation Assay
For quantification of TrkB activation by neurotrophins, immunoblotting was performed with the use of anti-phospho-TrkA (1:500, no. 9141, Cell Signaling Technology, MA, USA) or rabbit anti-pan Trk (1:500, sc-139, Santa Cruz Biotechnology). Image analysis software (ImageQuant, Molecular Dynamics, CA, USA) was used for determination of the relative expression levels of proteins, and the results are presented as the ratio of induced to basal levels.
Neurite-Outgrowth Assay
Neurite outgrowth was assessed by scoring the number of cells bearing neurites in four randomly chosen areas per well; each area contained approximately 80–100 cells. Cells bearing at least one neurite with a minimal length corresponding to the diameter of the cell body were positively scored. Four independent experiments were performed for confirmation of reproducibility.
Statistical Analysis
Statistical significance was evaluated with one-way analysis of variance followed by Newman-Keuls test for multiple groups, and probability values of less than 5% were considered significant.
Whole-Mount In Situ Hybridization and Microscopy
Retinal specimens were obtained from one human eye. One normal donor eye (age 70 years, male without history of ocular disease) was obtained at autopsy and processed within 6 hr after death. Informed consent for tissue donation was obtained from the patient, and the study protocol adhered to the tenets of the Declaration of Helsinki for experiments involving human tissues. The eyes were fixed in 4% PFA/PBS for 1.5 hr. RNA in situ hybridization was performed on free-floating pieces of whole-mount retina. For detection of NTF4, digoxigenin (DIG)-labeled RNA probes were prepared with the use of components of the riboprobe in vitro transcription systems (Promega, Mannheim, Germany) and DIG-labeled UTP (Roche, Mannheim, Germany). NTF4 cDNA fragment was obtained from human retina mRNA by reverse transcription coupled to PCR by the use of primers NTF4-SF (5′-CTGCAGCTGGCGGCAGTCC-3′) and NTF4-SR (5′-ATTACCCTCAAGTTGCTCCA-3′). The resulting fragment, 466 bp long and corresponding to nucleotide position 374-839 from the first ATG, was directly cloned into the pCR4-TOPO vector of the TOPO-PCR-Cloning Kit (Invitrogen, Karlsruhe, Germany), in accordance with the manufacturer's instructions. We sequenced isolated plasmids with the ABI Prism Big Dye Terminator cycle sequencing kit and analyzed them on an ABI Genetic Analyzer 3730 (Applied Biosystems, Foster City, CA, USA) to ensure that the constructs were correct. Two different clones containing the NTF4 cDNA fragment in opposite orientation were chosen for generation of the sense and antisense probes. Hybridization of labeled probe was visualized with the use of alkaline phosphatase conjugated anti-DIG (1:2.500), and the color reaction (5.5 hr, 30°C) was developed with NBT/BCIP (Roche). We processed treated and control retinas in parallel to ensure identical reaction conditions. Finally, the retina was sectioned vertically at 18 μm on a cryostat. For light microscopic analysis, sections were examined and photographed with a Zeiss Axio Imager.Z1 (Zeiss, Jena, Germany) fitted with a Canon A620 digital camera. Images were adjusted for contrast and brightness in Adobe Photoshop CS, and panels were arranged in Corel Draw 11.
Results
Extensive screening for mutations in the complete coding sequence of NTF4, including untranslated 5′, 3′, and intronic flanking regions, was performed in the discovery groups of patients and control subjects. This systematic mutation screen led to the identification of six heterozygous mutations leading to amino acid substitutions in NT-4; namely, C7Y, E84K, A88V, R90H, R206W, and R206Q (Figure 1, Table 1). Together, the amino acid changes account for over 2% of POAG in the discovery patient population (9/399). No variants were found in the complete NTF4 coding region in 376 matched, healthy control subjects (p < 0.004). All patients carrying an NT-4 mutation were sporadic cases, and no other family members were affected to date. The parents of all nine patients were deceased, and it is unknown whether they were also affected; thus, no familial segregation could be studied. All nine patients were negative for MYOC, OPTN, CYP1B1, and WDR3646 mutations. The mutation-positive patient group comprised individuals with both late juvenile and adult-onset POAG, with age at diagnosis varying from 36 to 80 years (Table 2). Seven had elevated (maximum) IOP ranging from 25 to 40 mmHg, whereas two had pressure measurements in the normal range: 16 and 20 mmHg. Apart from glaucoma, patients presented without neurological manifestations, and arterial hypertension was the only other abnormal feature, found in four of them.
Figure 1.
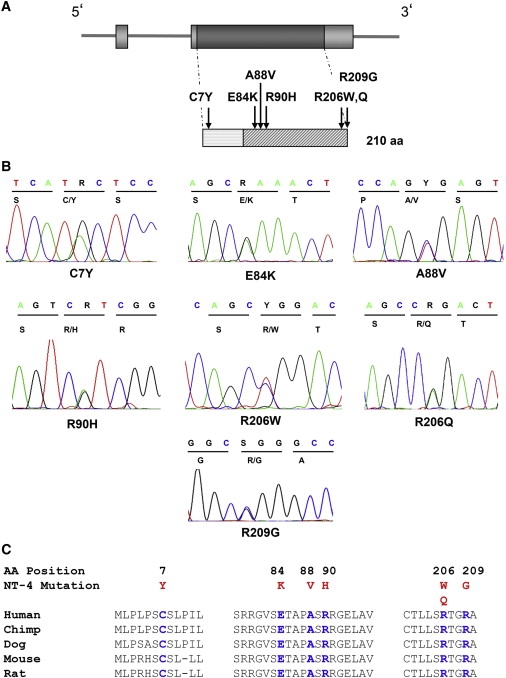
Location of NT-4 Amino Acid Changes on Protein Structure and Evolutionary Conservation
(A) Schematic representation of the intron-exon organization of the NTF4 gene and of the derived protein. NT-4 mature protein (starting from G81) is highlighted with gray transverse lines. All six mutations identified in this study are shown above the protein. Five variants are located in the NGF domain, and one is located in the N terminus of the preproprotein.
(B) DNA chromatograms of the mutation sites.
(C) Multiple amino acid sequence alignments show evolutionary conservation of mutated residues (shown in blue) among mamalian species. Amino acid changes are highlighted in red.
Table 1.
NTF4 Sequence Variants Found in Patients and Control Individuals
| Nucleotide Alteration | Amino Acid Change | Patients | Controls |
|---|---|---|---|
| Discovery group | |||
| n = 399 | n = 376 | ||
| c.20G>A | C7Y | 1 | 0 |
| c.250C>T | E84K | 1 | 0 |
| c.263C>T | A88V | 1 | 0 |
| c.269G>A | R90H | 1 | 0 |
| c.616C>T | R206W | 4 | 0 |
| c.617G>A | R206Q | 1 | 0 |
| Subtotala | 9 | 0 | |
| Replication group I | |||
| n = 282 | n = 67 | ||
| c.263C>T | A88V | 2 | 0 |
| c.616C>T | R206W | 1 | 0 |
| c.625C>G | R209G | 1 | 0 |
| Replication group II | |||
| n = 211 | n = 452 | ||
| c.263C>T | A88V | 2 | 0 |
| c.620C>G | T207S | 0 | 1 |
| Totalb | |||
| n = 892 | n = 895 | ||
| 15 | 1 | ||
p < 0.004.
p < 0.0002 (two-tailed Fischer's exact test).
Table 2.
Phenotypic Characteristics of Patients with NT-4 Mutations
| Patient ID | Glaucoma Type | NT-4 Mutation | Age at Diagnosis (Yrs) | Sex | Eye | Optic Disc (Jonas) | Disc Area (mm2) | Visual Field (md/clv) | Max IOP (mmHg) | Chamber Angle (Shaffer) | Additional Disease |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery group | |||||||||||
| 99149 | JOAG | C7T | 36 | M | L | III | 2.8 | 19.9/56.0 | 30 | Open, III | Arterial. hypertension |
| 20532 | POAG | E84K | 80 | M | R | IV | 2.4 | 23.4/- | 22 | Open, III | Arterial. hypertension |
| 99406 | POAG | A88V | 48 | M | L | IV | 2.5 | 11.8/89.1 | 35 | Open, III, mal differentiated | Otherwise healthy |
| 18780 | POAG | R90H | 54 | F | L | III-IV | 3.9 | 11.0/48.8 | 40 | Open, III, uv. tissue | Type II Diabetes mellitus |
| 20096 | POAG | R206Q | 43 | M | L | III | 2.9 | 11.2/88.0 | 28 | Open, III, uv. tissue | Poliomyelitis vaccination damage, otherwise healthy |
| 7290 | POAG | R206W | 53 | F | R | III | 2.5 | 8.2/61.7 | 25 | Open, III | History of Tubercolosis |
| 11299 | POAG | R206W | 51 | M | L | II | 2.6 | 3.7/ 9.6 | 34 | Open, III | Arterial. hypertension, goit |
| 99394 | POAG | R206W | 41 | M | L | II | 2.1 | 6.7/58.5 | 32 | Open, III | Otherwise healthy |
| 6551 | NTG | R206W | 51 | M | R | III | 3.0 | 5.9/116.9 | 16 | Open III | Arterial. hypertension |
| Replication group I | |||||||||||
| 179 | NTG | A88V | 42 | F | L | I | 2.8 | 2.0/2.6 | 12 | Open III | Arterial hypotonia |
| 225 | NTG | A88V | 49 | F | R | I | 2.4 | 0.2/4.1 | 19 | Open III | Arterial hypotonia |
| 78 | NTG | R206W | 78 | F | L | II | 2.1 | 11.7/48.1 | 15 | Open III | Arterial hypertension |
| 65 | NTG | R209G | 66 | M | R | IV | 2.1 | 22.3/55.6 | 20 | Open III | Arterial hypotonia |
| Replication group II | |||||||||||
| 4134001 | NTG | A88V | 79 | F | R | II | 3.6 | Goldmann | 21 | Open | Hypertension |
| 4904001 | NTG | A88V | 87 | M | L | I | NA | Goldmann | 13 | Open | Hypertension |
Eye measurements refer to the worst affected eye (r, right; l, left), grade of optic disc atrophy according to Jonas, disc area (mm2), automated visual field (md dB/clv dB2; in replication group II, Goldmann kinetic perimetry was used), maximum intraocular pressure (IOP mmHg), width of chamber angle (Shaffer's scale; in replication group II according to Von Herik).
To replicate the observed data, we screened the complete NTF4 coding region in an additional German cohort of 282 glaucoma patients and 67 control subjects. Two amino acid changes, A88V and R206W, previously identified in the discovery group, as well as an additional one, R209G, were detected in four unrelated patients (Table 1). Two patients suffered from progressed NTG and two from beginning NTG (Table 2). It is important to note that no variants leading to amino acid changes were detected in all control individuals apart from a single one, a variant leading to the same amino acid, D196D, found in one person of the control replication cohort. In addition, one known polymorphism in the 3′ untranslated region (rs11669977) was identified in patients as well in control individuals of either discovery or replication groups. This is not surprising, given that NTF4 is evolutionarily conserved.47 In the second replication group, originating from The Netherlands, two patients carried one NTF4 mutation (A88V). The control group was population based and had not been ophthalmologically examined, unlike the other two German groups. In this group, variant T207S, not previously found in the patient groups, was identified in one individual.
Altogether, we identified a total of 15 glaucoma patients (1.7%) carrying an NTF4 mutation but only one person carryinng the mutation in the three control groups (p < 0.0002; Table 1). The identified mutations are located in the mature domain of the NT-4 protein, with the exception of mutation C7Y, which is located in the N-terminal part of the pro-part of the NT-4 protein (Figure 1A). All affected amino acid positions show strong evolutionary conservation among orthologs in chimpanzee, dog, mouse, and rat (Figure 1C).
Molecular modeling based on the known homodimeric NT-4 crystal structure in complex with its receptor TrkB41,42 predicts that the identified mutations affect residues of the TrkB binding site (E84, A88, R90) or residues relevant for the stabilization of the tertiary structure of NT-4 (R206, R209) (Figure 2 and Figure 3). Thus, mutated residues are likely to disrupt several interactions that appear crucial for the formation and stabilization of this complex (Figure 3).
Figure 2.
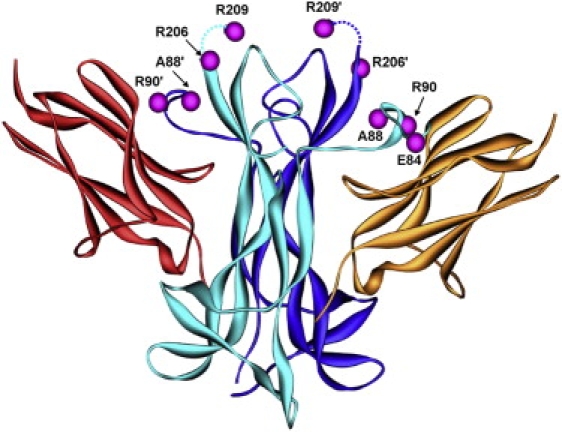
Location of the Mutations in the Crystal Structure of the NT-4-TrkB Complex
The two chains of the dimeric NT-4 are colored in cyan and blue, respectively. The sites of the mutations are highlighted by magenta balls (prime symbols denote residues of the second subunit), indicating that these residues are located within or close to the TrkB binding interface. TrkB domains bound by NT-4 are colored in red and orange. Residue R209, which is C-terminally adjacent to R206, is not resolved in this crystal structure. Therefore, the approximate position of this residue is indicated in accordance with another NT-4 structure,42 in which the respective region is better resolved. The latter structure also provided the basis for the detailed analysis of the R209G mutation shown in Figure 3E.
Figure 3.
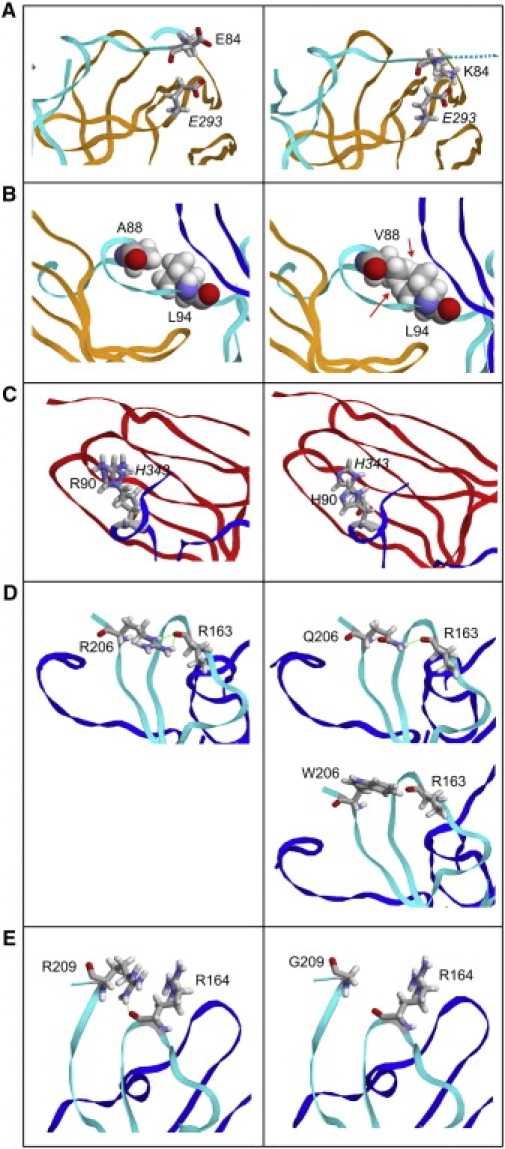
Predicted Effects of NT-4 Amino Acid Changes on NT-4 Structure and Interaction with TrkB
Contacts formed by the WT residues (left) and effects of the mutations (right). Residues of TrkB are denoted in italics.
(A) Effect of the E84K mutation: The side chain of E84 is oriented toward the solvent. K84 is expected to adopt a different orientation as a result of electrostatic attraction by E293 of TrkB (green dotted line). As a consequence, the contacts from NTF4 residues 81–84, representing the N terminus of mature NT-4 to TrkB, will be disrupted (indicated by the broken line for the protein backbone).
(B) Effect of the A88V mutation. The larger size of the V88 side chain results in steric clashes with L94 (red arrows), most probably causing structural rearrangement of the binding region and thus a poorer fit to the receptor.
(C) Effect of the R90H mutation: R90 stacks on the side chain of H343 from TrkB. The shorter side chain of H90 results in poorer stacking in the mutant, suggesting a weaker binding.
(D) Effect of the R206W and R206Q mutations: R206 forms hydrogen bonds (green dotted lines) to the backbone carbonyl of R163, thus stabilizing the three-dimensional structure of NT-4 in a region close to the receptor binding site. In the R206Q mutant, only one hydrogen bond can be formed, whereas no hydrogen bonds can be formed in the R206W mutant because of the length and chemical properties of the W side chain.
(E) Effect of the R209G mutation: R209 forms a hydrogen bond (green dotted line) to the backbone carbonyl of R164, thereby stabilizing the three-dimensional structure of NT-4 in a fashion similar to that of R206 (see panel D).
For the mutation C7Y, located in the N-terminal part of the unprocessed protein, no molecular modeling was possible because no three-dimensional structural information is available for this region. Analysis of the biophysical properties of the pro-peptide, based on the common hydrophathy scale,48 revealed that the C7Y mutation causes a significant decrease in the hydrophobicity of the pro-peptide. The hydrophobic region of signal peptides was shown to be a determinant for signal recognition particle and protein translocation across the endoplasmatic reticulum membrane.49 Thus, we postulate that the C7Y mutation, which significantly reduces the hydrophobicity of the pro-region, could impair the cleavage and/or transport process, leading to a decreased concentration or stability of NT-4.
The possibility that the most frequent mutation, R206W, may reduce TrkB activation was tested by comparing the activity of recombinant WT versus mutant NT-4 (Figures 4A–4C). These experiments revealed that the R206W mutation has a reduced ability to activate TrkB, as measured by tyrosine phosphorylation of the receptor (Figure 4C). The functional consequences in terms of neurotrophic activity were assessed with the use of a PC12-based assay with cells stably expressing the TrkB receptor. At concentrations identical to WT NT-4, R206W was significantly less active, as measured by the fiber-outgrowth response (Figure 4D) that can readily be quantified with this assay system (Figure 4E).
Figure 4.
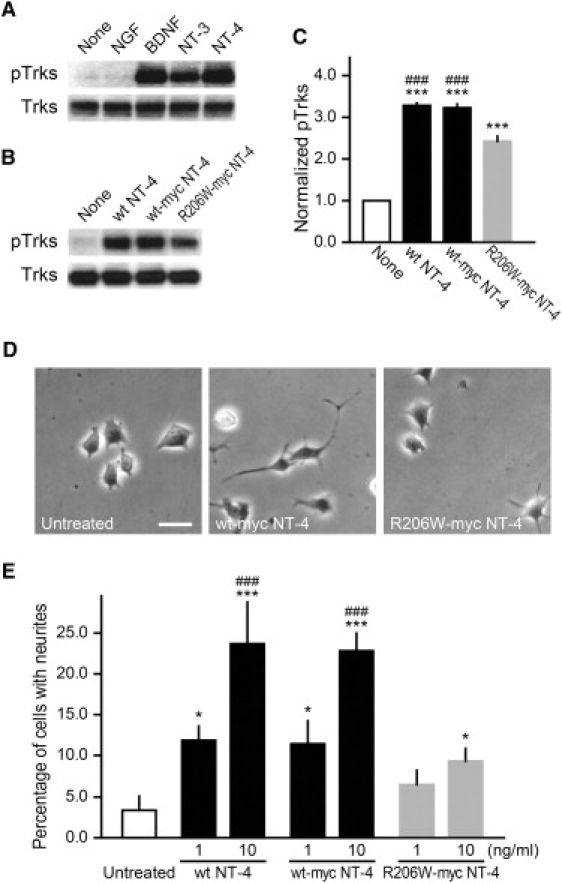
R206W-myc NT-4 Shows Decreased TrkB Activation and Neurite Outgrowth in Comparison to WT-myc NT-4
(A) Treatment of TrkB-nnr5 cells with BDNF and NT-4 for 10 min similarly induced TrkB phosphorylation, whereas NT-3 was less active and NGF was inactive (100 ng/ml, respectively).
(B) Treatment with R206W-myc NT-4 for 10 min induced TrkB activation significantly less than WT and WT-myc NT-4 (10 ng/ml, respectively).
(C) Densitometry analysis of “phospho-TrkB” was performed after immunoblotting in (B). Data represent mean ± SEM (n = 4). ∗∗∗ indicates p < 0.001 versus untreated, and ### indicates p < 0.001 versus R206W-myc NT-4.
(D) Representative phase contrast images of TrkB-nnr5 cells treated with WT-myc or R206W-myc NT-4 (10 ng/ml) for 2 days. Scale bar represents 10 mm.
(E) Graph shows the percentage of cells with neurites. TrkB-nnr5 cells were treated with WT, WT-myc, or R206W-myc NT-4 at 1 or 10 ng/ml for 2 days. Data represent mean ± SEM (n = 3). ∗ indicates p < 0.05, ∗∗∗ indicates p < 0.001 versus untreated, and ### indicates p < 0.001 versus R206W-myc NT-4 (10 ng/ml).
Finally, we tested for the presence and localization of NTF4 in the adult retina by in situ hybridization (Figure 5). Given the fact that glaucoma typically develops in the elderly, NTF4 would be expected to be expressed in the adult retina if it is to play a direct role in the pathogenesis of glaucoma. We obtained a human retina a few hours after the death of a 70-year-old patient with no previous history of eye disease. In situ hybridization detected a specific NTF4 signal that could clearly be localized to the ganglion cell layer (Figure 5A), confirming recent findings.50
Figure 5.
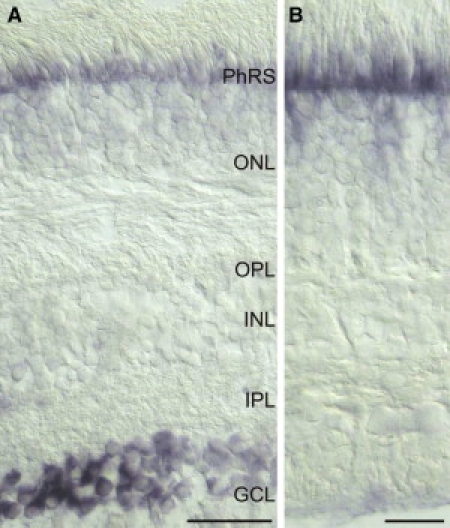
NTF4 mRNA Is Expressed in Human Retinal Ganglion Cells
(A) Ganglion cells in the ganglion cell layer (GCL) label for NTF4 transcript.
(B) In control sections (sense probe), no specific signal is detectable.
Scale bars represent 50 mm (A) and 20 mm (B). Abbreviations are as follows: PhRs, photoreceptor segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer.
Discussion
We hypothesized that retinal ganglion cell loss in POAG is not mainly due to elevated intraocular pressure but that factors compromising survival of such cells may also be involved. We therefore screened NTF4 for mutations in POAG on the basis of its function as a neurotrophic factor protecting neurons. We sequenced the entire coding sequence in a large cohort of POAG patients because we expected to find only rare variants in few patients, in line with previous findings in other glaucoma genes. We were surprised to find different missense variants in NTF4 in almost 2% of cases, because to our knowledge, no such variants had ever been described before. When we attempted replication of our findings in a second cohort of German POAG patients, we likewise found missense variants at a similar frequency, thus confirming our hypothesis of a link between NT-4 and neuroprotection. We also investigated two ophthalmologically examined cohorts of control individuals but did not find a single missense variant. To further validate our findings, we screened a third POAG cohort from The Netherlands. Among 211 POAG cases from the Rotterdam Study, we found one previously discovered variant in two patients (0.9%). In this instance, a population-based cohort was used as control, although these individuals were not investigated for glaucoma. Thus, on the basis of the incidence of the disease, we can expect at least five individuals from this group to develop glaucoma during their lifetime. Not unexpectedly, we found one person (0.2%) carrying an additional missense variant, likely to have similar effect as the previously identified ones. Overall, 15 patients (1.7%) but only one control individual (0.1%) carried a missense variant (p = 0.0002), thus giving strong genetic evidence that NTF4 variants are associated with POAG.
On the basis of molecular modeling, all NT-4 variants were predicted to affect either dimer stability of NT-4 or interaction between the NT-4 dimer and its receptor, TrkB, suggesting that these are bona fide mutations. For the most frequent variant, R206W, we demonstrated with in vitro experiments that, indeed, both ligand-mediated TrkB signaling and neurite outgrowth are impaired. This suggests that these variants have a subtle but significant effect on neuronal survival. The effect is probably small and might be compensated for during many years. Age and/or other risk factors might be required for manifestation of the disease, in line with POAG being a late-onset disease.
We are not aware of previous reports on mutations in the neurotrophin-4 gene (NTF4), and our results suggest an essential role of the neurotrophin signaling system in preventing neuronal degeneration in humans. Taken together, these results suggest a relevant pathway in the pathophysiology of glaucoma. They complement the previous findings in rodents that a chronic reduction of TrkB levels leads to a progressive loss of RGCs that becomes apparent only in the adult.51 Likewise, a chronic, suboptimal activation of TrkB resulting from the secretion of a mutated form of NT-4 may lead to the progressive loss of retinal ganglion cells, a finding that may open the possibility of treating glaucoma by using agents activating TrkB.
In addition, these and previous data support the hypothesis that POAG is characterized by a high locus and allelic heterogeneity with different rare variants in numerous genes. In fact, rare mutations with low frequencies have also been reported for the other known glaucoma genes, such as MYOC52 and WDR36.46 Jointly, these observations suggest that POAG belongs to the same category of traits under the “frequent disease-rare variant” hypothesis, such as epilepsy53 and macular degeneration,54 with rare, highly penetrant variants in numerous genes, some familial but most occurring sporadically.55,56 This could explain the so-far-elusive quest for identification of more glaucoma genes and would have important consequences for the design of future studies aimed at unraveling the molecular basis of this blinding disease.
Supplemental Data
Supplemental Data include one table containing the list of primers used in this work and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW, http://www.ebi.ac.uk/clustalw/
Ensembl genome browser, http://www.ensembl.org/index.html
Expert Protein Analysis System (ExPasy) proteomic server, http://www.expasy.org
Geneseeker, http://www.cmbi.kun.nl/GeneSeeker/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer 3, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi/
Protein Data Bank (PDB), http://www.rcsb.org
University of California Santa Cruz (UCSC) genome browser, http://genome.ucsc.edu/cgi-bin/hgTracks
Acknowledgments
We thank all of the patients and control individuals for their participation, Claudia Preller and Freya Boggasch for technical assistance, and Juliane Niedzella for patient recruitment support. This work was supported by grants from the SFB539 to A.R., C.Y.M., B.R., F.E.K., and J.H.B. and by an individual grant to B.H.F.W., both funded by the Deutsche Forschungsgemeinschaft (DFG).
References
- 1.Tuck M.W., Crick R.P. The projected increase in glaucoma due to an ageing population. Ophthalmic Physiol. Opt. 2003;23:175–179. doi: 10.1046/j.1475-1313.2003.00104.x. [DOI] [PubMed] [Google Scholar]
- 2.Nickells R.W. Retinal ganglion cell death in glaucoma: the how, the why, and the maybe. J. Glaucoma. 1996;5:345–356. [PubMed] [Google Scholar]
- 3.Quigley H.A. Neuronal death in glaucoma. Prog. Retin. Eye Res. 1999;18:39–57. doi: 10.1016/s1350-9462(98)00014-7. [DOI] [PubMed] [Google Scholar]
- 4.Soto I., Oglesby E., Buckingham B.P., Son J.L., Roberson E.D., Steele M.R., Inman D.M., Vetter M.L., Horner P.J., Marsh-Armstrong N. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J. Neurosci. 2008;28:548–561. doi: 10.1523/JNEUROSCI.3714-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutjen-Drecoll E., Kruse F.E. Ophthalmologe. 2007;104:167–178. doi: 10.1007/s00347-007-1481-5. [DOI] [PubMed] [Google Scholar]
- 6.Medeiros F.A., Weinreb R.N. Medical backgrounders: glaucoma. Drugs Today (Barc) 2002;38:563–570. doi: 10.1358/dot.2002.38.8.704676. [DOI] [PubMed] [Google Scholar]
- 7.Leydhecker W. Springer Verlag; Berlin, Göttingen, Heidelberg: 1960. Glaukom ein Handbuch. [Google Scholar]
- 8.Wiggs J.L. Genetic etiologies of glaucoma. Arch. Ophthalmol. 2007;125:30–37. doi: 10.1001/archopht.125.1.30. [DOI] [PubMed] [Google Scholar]
- 9.Fan B.J., Wang D.Y., Lam D.S., Pang C.P. Gene mapping for primary open angle glaucoma. Clin. Biochem. 2006;39:249–258. doi: 10.1016/j.clinbiochem.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Stone E.M., Fingert J.H., Alward W.L., Nguyen T.D., Polansky J.R., Sunden S.L., Nishimura D., Clark A.F., Nystuen A., Nichols B.E. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 11.Rezaie T., Child A., Hitchings R., Brice G., Miller L., Coca-Prados M., Heon E., Krupin T., Ritch R., Kreutzer D. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–1079. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 12.Monemi S., Spaeth G., DaSilva A., Popinchalk S., Ilitchev E., Liebmann J., Ritch R., Heon E., Crick R.P., Child A. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum. Mol. Genet. 2005;14:725–733. doi: 10.1093/hmg/ddi068. [DOI] [PubMed] [Google Scholar]
- 13.Acharya M., Mookherjee S., Bhattacharjee A., Bandyopadhyay A.K., Daulat Thakur S.K., Bhaduri G., Sen A., Ray K. Primary role of CYP1B1 in Indian juvenile-onset POAG patients. Mol. Vis. 2006;12:399–404. [PubMed] [Google Scholar]
- 14.Chakrabarti S., Devi K.R., Komatireddy S., Kaur K., Parikh R.S., Mandal A.K., Chandrasekhar G., Thomas R. Glaucoma-associated CYP1B1 mutations share similar haplotype backgrounds in POAG and PACG phenotypes. Invest. Ophthalmol. Vis. Sci. 2007;48:5439–5444. doi: 10.1167/iovs.07-0629. [DOI] [PubMed] [Google Scholar]
- 15.Melki R., Colomb E., Lefort N., Brezin A.P., Garchon H.J. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J. Med. Genet. 2004;41:647–651. doi: 10.1136/jmg.2004.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasutto F., Chavarria-Soley G., Mardin C.Y., Michels-Rautenstrauss K., Ingelman-Sundberg M., Fernandez-Martinez L., Weber B.H., Rautenstrauss B., Reis A. Heterozygous loss of function variants in CYP1B1 predispose to primary open angle glaucoma. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3880. Published online July 30, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Murphy J.A., Clarke D.B. Target-derived neurotrophins may influence the survival of adult retinal ganglion cells when local neurotrophic support is disrupted: Implications for glaucoma. Med. Hypotheses. 2006;67:1208–1212. doi: 10.1016/j.mehy.2006.04.049. [DOI] [PubMed] [Google Scholar]
- 18.Blesch A. Neurotrophic factors in neurodegeneration. Brain Pathol. 2006;16:295–303. doi: 10.1111/j.1750-3639.2006.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawai H., Clarke D.B., Kittlerova P., Bray G.M., Aguayo A.J. Brain-derived neurotrophic factor and neurotrophin-4/5 stimulate growth of axonal branches from regenerating retinal ganglion cells. J. Neurosci. 1996;16:3887–3894. doi: 10.1523/JNEUROSCI.16-12-03887.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudzinski M., Wong T.P., Saragovi H.U. Changes in retinal expression of neurotrophins and neurotrophin receptors induced by ocular hypertension. J. Neurobiol. 2004;58:341–354. doi: 10.1002/neu.10293. [DOI] [PubMed] [Google Scholar]
- 21.Taylor S., Srinivasan B., Wordinger R.J., Roque R.S. Glutamate stimulates neurotrophin expression in cultured Muller cells. Brain Res. Mol. Brain Res. 2003;111:189–197. doi: 10.1016/s0169-328x(03)00030-5. [DOI] [PubMed] [Google Scholar]
- 22.Takahata K., Katsuki H., Kume T., Nakata D., Ito K., Muraoka S., Yoneda F., Kashii S., Honda Y., Akaike A. Retinal neuronal death induced by intraocular administration of a nitric oxide donor and its rescue by neurotrophic factors in rats. Invest. Ophthalmol. Vis. Sci. 2003;44:1760–1766. doi: 10.1167/iovs.02-0471. [DOI] [PubMed] [Google Scholar]
- 23.Bibel M., Barde Y.A. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- 24.Ip N.Y., Ibanez C.F., Nye S.H., McClain J., Jones P.F., Gies D.R., Belluscio L., Le Beau M.M., Espinosa R., Squinto S.P. Mammalian neurotrophin-4: structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. USA. 1992;89:3060–3064. doi: 10.1073/pnas.89.7.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng L., Sapieha P., Kittlerova P., Hauswirth W.W., Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J. Neurosci. 2002;22:3977–3986. doi: 10.1523/JNEUROSCI.22-10-03977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clarke D.B., Bray G.M., Aguayo A.J. Prolonged administration of NT-4/5 fails to rescue most axotomized retinal ganglion cells in adult rats. Vision Res. 1998;38:1517–1524. doi: 10.1016/s0042-6989(97)00341-6. [DOI] [PubMed] [Google Scholar]
- 27.Cohen A., Bray G.M., Aguayo A.J. Neurotrophin-4/5 (NT-4/5) increases adult rat retinal ganglion cell survival and neurite outgrowth in vitro. J. Neurobiol. 1994;25:953–959. doi: 10.1002/neu.480250805. [DOI] [PubMed] [Google Scholar]
- 28.Johnson J.E., Barde Y.A., Schwab M., Thoenen H. Brain-derived neurotrophic factor supports the survival of cultured rat retinal ganglion cells. J. Neurosci. 1986;6:3031–3038. doi: 10.1523/JNEUROSCI.06-10-03031.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peinado-Ramon P., Salvador M., Villegas-Perez M.P., Vidal-Sanz M. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Invest. Ophthalmol. Vis. Sci. 1996;37:489–500. [PubMed] [Google Scholar]
- 30.Harada C., Harada T., Quah H.M., Namekata K., Yoshida K., Ohno S., Tanaka K., Parada L.F. Role of neurotrophin-4/5 in neural cell death during retinal development and ischemic retinal injury in vivo. Invest. Ophthalmol. Vis. Sci. 2005;46:669–673. doi: 10.1167/iovs.04-0826. [DOI] [PubMed] [Google Scholar]
- 31.Wiggs J.L., Allingham R.R., Hossain A., Kern J., Auguste J., DelBono E.A., Broomer B., Graham F.L., Hauser M., Pericak-Vance M. Genome-wide scan for adult onset primary open angle glaucoma. Hum. Mol. Genet. 2000;9:1109–1117. doi: 10.1093/hmg/9.7.1109. [DOI] [PubMed] [Google Scholar]
- 32.Berkemeier L.R., Winslow J.W., Kaplan D.R., Nikolics K., Goeddel D.V., Rosenthal A. Neurotrophin-5: a novel neurotrophic factor that activates trk and trkB. Neuron. 1991;7:857–866. doi: 10.1016/0896-6273(91)90287-a. [DOI] [PubMed] [Google Scholar]
- 33.Jonas J.B., Gusek G.C., Naumann G.O. Optic disc morphometry in chronic primary open-angle glaucoma. I. Morphometric intrapapillary characteristics. Graefes Arch. Clin. Exp. Ophthalmol. 1988;226:522–530. doi: 10.1007/BF02169199. [DOI] [PubMed] [Google Scholar]
- 34.Jonas J.B., Papastathopoulos K. Ophthalmoscopic measurement of the optic disc. Ophthalmology. 1995;102:1102–1106. doi: 10.1016/s0161-6420(95)30905-0. [DOI] [PubMed] [Google Scholar]
- 35.Schlotzer-Schrehardt U., Pasutto F., Sommer P., Hornstra I., Kruse F.E., Naumann G.O., Reis A., Zenkel M. Genotype-correlated expression of lysyl oxidase-like 1 in ocular tissues of patients with pseudoexfoliation syndrome/glaucoma and normal patients. Am J Pathol. 2008;173:1724–1735. doi: 10.2353/ajpath.2008.080535. Published online October 30, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weisschuh N., Wolf C., Wissinger B., Gramer E. Variations in the WDR36 gene in German patients with normal tension glaucoma. Mol. Vis. 2007;13:724–729. [PMC free article] [PubMed] [Google Scholar]
- 37.Hofman A., Breteler M.M., van Duijn C.M., Krestin G.P., Pols H.A., Stricker B.H., Tiemeier H., Uitterlinden A.G., Vingerling J.R., Witteman J.C. The Rotterdam Study: objectives and design update. Eur. J. Epidemiol. 2007;22:819–829. doi: 10.1007/s10654-007-9199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolfs R.C., Borger P.H., Ramrattan R.S., Klaver C.C., Hulsman C.A., Hofman A., Vingerling J.R., Hitchings R.A., de Jong P.T. Changing views on open-angle glaucoma: definitions and prevalences–The Rotterdam Study. Invest. Ophthalmol. Vis. Sci. 2000;41:3309–3321. [PubMed] [Google Scholar]
- 39.Aulchenko Y.S., Heutink P., Mackay I., Bertoli-Avella A.M., Pullen J., Vaessen N., Rademaker T.A., Sandkuijl L.A., Cardon L., Oostra B. Linkage disequilibrium in young genetically isolated Dutch population. Eur. J. Hum. Genet. 2004;12:527–534. doi: 10.1038/sj.ejhg.5201188. [DOI] [PubMed] [Google Scholar]
- 40.Pardo L.M., MacKay I., Oostra B., van Duijn C.M., Aulchenko Y.S. The effect of genetic drift in a young genetically isolated population. Ann. Hum. Genet. 2005;69:288–295. doi: 10.1046/J.1469-1809.2005.00162.x. [DOI] [PubMed] [Google Scholar]
- 41.Banfield M.J., Naylor R.L., Robertson A.G., Allen S.J., Dawbarn D., Brady R.L. Specificity in Trk receptor:neurotrophin interactions: the crystal structure of TrkB-d5 in complex with neurotrophin-4/5. Structure. 2001;9:1191–1199. doi: 10.1016/s0969-2126(01)00681-5. [DOI] [PubMed] [Google Scholar]
- 42.Robinson R.C., Radziejewski C., Spraggon G., Greenwald J., Kostura M.R., Burtnick L.D., Stuart D.I., Choe S., Jones E.Y. The structures of the neurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4 heterodimer reveal a common Trk-binding site. Protein Sci. 1999;8:2589–2597. doi: 10.1110/ps.8.12.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 44.Wilkins M.R., Gasteiger E., Bairoch A., Sanchez J.C., Williams K.L., Appel R.D., Hochstrasser D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 45.Green S.H., Rydel R.E., Connolly J.L., Greene L.A. PC12 cell mutants that possess low- but not high-affinity nerve growth factor receptors neither respond to nor internalize nerve growth factor. J. Cell Biol. 1986;102:830–843. doi: 10.1083/jcb.102.3.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pasutto F., Mardin C.Y., Michels-Rautenstrauss K., Weber B.H., Sticht H., Chavarria-Soley G., Rautenstrauss B., Kruse F., Reis A. Profiling of WDR36 Missense Variants in German Patients with Glaucoma. Invest. Ophthalmol. Vis. Sci. 2008;49:270–274. doi: 10.1167/iovs.07-0500. [DOI] [PubMed] [Google Scholar]
- 47.Gotz R., Schartl M. The conservation of neurotrophic factors during vertebrate evolution. Comp. Biochem. Physiol. Pharmacol. Toxicol. Endocrinol. 1994;108:1–10. doi: 10.1016/1367-8280(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 48.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 49.Hatsuzawa K., Tagaya M., Mizushima S. The hydrophobic region of signal peptides is a determinant for SRP recognition and protein translocation across the ER membrane. J. Biochem. (Tokyo) 1997;121:270–277. doi: 10.1093/oxfordjournals.jbchem.a021583. [DOI] [PubMed] [Google Scholar]
- 50.Ghazi-Nouri S.M., Ellis J.S., Moss S., Limb G.A., Charteris D.G. Expression and localisation of BDNF, NT4 and TrkB in proliferative vitreoretinopathy. Exp. Eye Res. 2008;86:819–827. doi: 10.1016/j.exer.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Rohrer B., LaVail M.M., Jones K.R., Reichardt L.F. Neurotrophin receptor TrkB activation is not required for the postnatal survival of retinal ganglion cells in vivo. Exp. Neurol. 2001;172:81–91. doi: 10.1006/exnr.2001.7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gong G., Kosoko-Lasaki O., Haynatzki G.R., Wilson M.R. Genetic dissection of myocilin glaucoma. Hum Mol Genet. 2004;13 Spec No 1:R91–R102. doi: 10.1093/hmg/ddh074. [DOI] [PubMed] [Google Scholar]
- 53.Weber Y.G., Lerche H. Genetic mechanisms in idiopathic epilepsies. Dev. Med. Child Neurol. 2008;50:648–654. doi: 10.1111/j.1469-8749.2008.03058.x. [DOI] [PubMed] [Google Scholar]
- 54.Swaroop A., Branham K.E., Chen W., Abecasis G. Genetic susceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits. Hum Mol Genet. 2007;16 Spec No. 2:R174–R182. doi: 10.1093/hmg/ddm212. [DOI] [PubMed] [Google Scholar]
- 55.Iyengar S.K., Elston R.C. The genetic basis of complex traits: rare variants or “common gene, common disease”? Methods Mol. Biol. 2007;376:71–84. doi: 10.1007/978-1-59745-389-9_6. [DOI] [PubMed] [Google Scholar]
- 56.Pritchard J.K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.