

Abstract
The susceptibility gene for ataxia telangiectasia, ATM, is also an intermediate-risk breast-cancer-susceptibility gene. However, the spectrum and frequency distribution of ATM mutations that confer increased risk of breast cancer have been controversial. To assess the contribution of rare variants in this gene to risk of breast cancer, we pooled data from seven published ATM case-control mutation-screening studies, including a total of 1544 breast cancer cases and 1224 controls, with data from our own mutation screening of an additional 987 breast cancer cases and 1021 controls. Using an in silico missense-substitution analysis that provides a ranking of missense substitutions from evolutionarily most likely to least likely, we carried out analyses of protein-truncating variants, splice-junction variants, and rare missense variants. We found marginal evidence that the combination of ATM protein-truncating and splice-junction variants contribute to breast cancer risk. There was stronger evidence that a subset of rare, evolutionarily unlikely missense substitutions confer increased risk. On the basis of subset analyses, we hypothesize that rare missense substitutions falling in and around the FAT, kinase, and FATC domains of the protein may be disproportionately responsible for that risk and that a subset of these may confer higher risk than do protein-truncating variants. We conclude that a comparison between the graded distributions of missense substitutions in cases versus controls can complement analyses of truncating variants and help identify susceptibility genes and that this approach will aid interpretation of the data emerging from new sequencing technologies.
Introduction
The susceptibility gene for the autosomal-recessive disorder ataxia telangiectasia (A-T [MIM 208900]), ATM (MIM 607585), encodes a protein of 3056 amino acids that is activated in response to DNA damage and phosphorylates proteins involved in DNA repair and cell-cycle control.1–3 Before ATM was identified, investigation of the family histories of A-T patients revealed that heterozygous mutation carriers are at increased risk of cancer, particularly breast cancer.4 After the cloning of ATM, several investigators conducted mutation screening studies intended to clarify the role of ATM sequence variation in breast cancer risk. The results were controversial; some found evidence that truncating mutations in ATM were important, others found that missense substitutions were important, and others found little evidence of associated risk.5–13
Recently, Renwick et al. mutation-screened ATM in a series of familial breast cancer cases and ethnically similar controls and then compared the summed frequency of clearly pathogenic (for A-T) sequence variants in cases versus controls.14 Their results confirmed that ATM is an intermediate-risk breast cancer susceptibility gene: inheritance of variants that are clearly pathogenic for A-T confers increased risk of breast cancer with an odds ratio (OR) of 2 to 3, which is between the ORs conferred by high-risk variants in BRCA1 (MIM 113705) and BRCA2 (MIM 600185) and those due to common modest-risk SNPs in genes such as FGFR2 (MIM 176943) and TOX3 (alias TNRC9 [MIM 611416]).15,16 However, the combined bioinformatic and statistical analysis model employed by Renwick et al. was not sufficiently powerful to compare the relative contribution of protein-truncating variants and missense substitutions to the burden of breast cancer attributable to sequence variation in ATM.
To improve the power of case-control mutation-screening studies, we developed an analysis strategy to estimate risk attributable to rare missense substitutions in a known or candidate susceptibility gene.17 The analysis strategy involves two main steps. In the first step, evolutionarily unlikely missense substitutions are resolved from evolutionarily more likely missense substitutions along a graded trend. In the second step, the case and control distributions of graded missense substitutions are compared with a one degree of freedom (DF) test for log-linear trend. The strategy requires substantially complete mutation screening of the gene of interest in a suitably ascertained set of cases and controls and a protein multiple sequence alignment of sufficient phylogenetic depth to enable robust grading of the missense substitutions. Application of the strategy implies testing a null hypothesis that has three components: (1) the gene harbors missense substitutions that are pathogenic with respect to the disease of interest, (2) the probability that a missense substitution in the gene is pathogenic is directly associated with the probability that it is evolutionarily deleterious, and (3) the missense-substitution grading is directly associated with the probability that a missense substitution is evolutionarily deleterious. Should any of these three components be false, the data will show only random association between case-control status and missense-substitution grading: a significant p value therefore implies acceptance of all three components and rejection of the corresponding null hypothesis.
Here, we apply our analysis strategy to ATM mutation screening data pooled from seven published ATM case-control mutation-screening studies, including a total of 1544 breast cancer cases and 1224 controls, plus data from our own mutation screening of an additional 987 breast cancer cases and 1021 controls. We examine the results from two perspectives: the role of rare ATM sequence variants in risk of breast cancer and the contribution that analyses of rare missense substitutions can make to future, large-scale, case-control mutation-screening studies.
Subjects and Methods
Identification of Studies Included in the Meta-Analysis
To retrieve ATM mutation-screening data from the literature, we searched PubMed, Web of Science, and EMBASE databases, using the keywords [“ATM”], [“breast cancer” or “breast neoplasm” or “breast carcinoma”], and [“mutation” or ”polymorphism”] for reports up to January 2009. For our main analyses, we required that the studies reported substantially complete mutation screening of ATM in breast cancer cases and controls. Mutation-screening results from case-only or control-only studies were used as supplements to the main analyses. Several of the mutation-screening papers included in the meta-analysis supplemented their mutation screening with specific variant genotyping; we excluded these data. Papers were excluded for any of the following reasons: if patient ascertainment was on a phenotype other than breast cancer (i.e., Hodgkin disease before breast cancer,18 familial cancer in general,19 or breast plus breast-ovarian families in a format in which it was not possible to determine which variant was observed in which type of proband20); if patient selection was based on a specific tumor phenotype (i.e., breast cancer cases selected because their tumors showed LOH at 11q2321 or specific selection for early-stage breast tumors22); or if patients were specifically selected because of a radiotherapy complication or because of absence of a radiotherapy adverse reaction.23–25 There were several instances of overlap in breast cancer cases between mutation-screening studies. In these instances, the largest study (usually, the most recent one) was included in the meta-analysis. Consequently, several redundant studies26–29 were excluded. Discrepancies in nucleotide designation versus amino acid designation were checked with relevant authors, and their responses were used for correction of our data set.
ATM Sequences, Alignments, and Missense-Substitution Analysis
We constructed an ATM protein multiple sequence alignment that satisfied three criteria: (1) the individual sequences are full-length and encode proteins that appear to be structurally similar to human ATM from beginning to end, (2) the individual sequences are substantially free of cDNA (or gene model) structural errors, and (3) the alignment contains an average of at least three amino acid substitutions per position and meets the missense-substitution-analysis program Sorting Intolerant From Tolerant (SIFT) “median sequence conservation” criterion for confident prediction of substitutions that should “affect protein function.”30
The alignment contained full-length sequences from human (Homo sapiens), mouse (Mus musculus), pig (Sus scrofa), opossum (Monodelphis domestica), chicken (Gallus gallus), frog (Xenopus laevis), zebrafish (Danio rerio), lancelet (Branchiostoma floridae), and sea urchin (Strongylocentrotus purpuratus). Human (AAB65827.1), mouse (NP_031525.2), pig (AAT01608.2), chicken (XP_417160.2), frog (AAT72929.1), and partial zebrafish (BAD91491.1) ATM sequences were obtained from GenBank. To obtain ATM coding sequences from opossum, lancelet, and sea urchin, we used a combination of tBLASTn31 and splice-junction prediction to build initial gene models from the available genomic sequences.
In the case of the opossum sequence, two apparent anomalies in the genomic sequence interfered with assembly of a gene model matching the exonic structure of the other mammalian ATM sequences. In addressing these anomalies, cDNA was prepared from tissue samples of one gray short-tailed opossum (kindly provided by Paul B. Samollow), PCR amplified across the region of interest, and sequenced. After the resulting refinements were incorporated into the gene model, there remained four small differences between our opossum ATM predicted peptide sequence and that of Ensemble (ENSMODP00000018290), but these did not influence scoring of the human missense substitutions analyzed here.
Because the lancelet and sea urchin ATM sequences are much further diverged from mammalian ATM, there were many uncertain areas in our initial gene models. Accordingly, we PCR amplified their entire coding sequences from cDNA prepared from one lancelet and one sea urchin (kindly provided by Michael Schubert and R. Andrew Cameron, respectively) and sequenced them.
We then used the MCoffee alignment suite32 to build an initial protein multiple sequence alignment. The alignment was checked for anomalies, particularly near the splice junctions, that might be attributed to structural faults in the cDNA sequences rather than to sequence divergence. When such anomalies were found in one of the GenBank cDNA sequences, the corresponding genomic sequence was checked and, if gene prediction from the genomic sequence resulted in a better alignment than had been obtained with the original cDNA sequence, the cDNA sequence was repaired.
In the case of the chicken cDNA sequence, we used the genomic sequence to make two small edits to the GenBank gene model sequence, just after amino acids 1968 and 2327. In the case of the zebrafish cDNA sequence, we found that the amino terminus up to aa 327 (of the final complete sequence) was missing from GenBank entry BAD91491.1, the cDNA sequence appeared quite anomalous over a nine-amino-acid segment with respect to the other vertebrate sequences (aa 659–668 of the final complete sequence), and there were a number of additional ambiguity codes in the sequence. To obtain the missing amino end sequence, we used tBLASTn and splice-junction prediction on the Danio rerio build 7 genome sequence to create a gene model from the start codon into the ninth coding exon, PCR amplified it from cDNA prepared from one individual zebrafish (kindly provided by Laure Bernard), and sequenced it. We corrected the anomaly from 659–668 by reference to the Danio rerio genome sequence and corrected the remaining ambiguities by reference to the ENSEMBLE Danio rerio ATM gene model ENSDARP00000080608.
The sequences were then realigned, resulting in the alignment used for the analyses of missense substitutions described below. We counted substitutions per position in the alignment by using the Protpars routine in PHYLIP v 3.68 with the known underlying phylogeny, and we also used SIFT to confirm that the alignment met that program's “median sequence conservation” criterion for confident prediction of substitutions that should “affect protein function”30,33. Table 1 gives an idea of how much repair by gene prediction and repair by cDNA sequencing were applied to the sequences in the alignment. The complete alignment is available online as File S1, and the alignment (or updated versions thereof) is available for online use at the Align-Grantham Variation Grantham Deviation (Align-GVGD) web site (see Web Resources).
Table 1.
ATM and Ortholog Sequence Accession Numbers and Cross-Species Sequence Comparisons
| Organism | Accession Number | GenBank cDNA (%)a | Gene Model (%)b | Confirmation by Sequencing (%)c |
Pairwise Amino Acid Sequence Identity (%)d |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hs | Mm | Ss | Md | Gg | Xl | Dr | Bf | |||||
| Homo sapiens | AAB65827.1 | 100.0 | 0.0 | 0.0 | ||||||||
| Mus musculus | NP_031525.2 | 100.0 | 0.0 | 0.0 | 84.1 | |||||||
| Sus scrofa | AAT01608.1 | 100.0 | 0.0 | 0.0 | 88.4 | 82.6 | ||||||
| Monodelphis domestica | ACG68567.1, ACG68568.1 | 0.0 | 88.7e | 11.3 | 80.7 | 76.2 | 78.9 | |||||
| Gallus gallus | XP_417160.2 | 0.0 | 99.7 + 0.3f | 0.0 | 69.5 | 66.7 | 68.5 | 70.5 | ||||
| Xenopus laevis | AAT72929.1 | 100.0 | 0.0 | 64.5 | 62.7 | 63.6 | 65.9 | 64.6 | ||||
| Danio rerio | BAD91491.1 ACJ03990.1 | 89.4 | 0.3f | 10.6 | 54.2 | 53.2 | 53.4 | 53.9 | 53.4 | 54.3 | ||
| Branchiostoma floridae | ACG68443.1 | 0.0 | 0.0 | 100.0 | 36.9 | 36.3 | 36.6 | 36.5 | 37.2 | 37.7 | 36.6 | |
| Strongylocentrotus purpuratus | ABY60856.1 | 0.0 | 0.0 | 100.0 | 34.8 | 34.6 | 34.7 | 35.0 | 35.9 | 35.3 | 35.2 | 38.1 |
This is the percentage of the ATM amino acid sequence used in our alignment that was obtained directly from a GenBank cDNA entry.
This is the percentage of the ATM amino acid sequence used in our alignment that was obtained by gene prediction.
This is the percentage of the ATM amino acid sequence used in our alignment that we confirmed by RT-PCR and sequencing from model-organism cDNA.
The two-letter species-name abbreviations are as follows: Hs, Homo sapiens; Mm, Mus musculus; Ss, Sus scrofa; Md, Monodelphis domestica; Gg, Gallus gallus; Xl, Xenopus laevis; Dr, Danio rerio; Bf, Branchiostoma floridae. Note that the cross-comparison does not require a column for Strongylocentrotus purpuratus.
Gene model built at IARC, but similar to Ensemble prediction ENSMODP00000018290.
Corrections to apparent anomalies in a gene-model prediction obtained from GenBank, made by reference to the genomic sequence.
ATM missense substitutions reported in this study were scored by the use of this alignment with the missense analysis programs Align-GVGD and SIFT.17,30 With the use of Align-GVGD, the relevant output is the “C-score,” which provides seven discrete grades running from C0 (most likely neutral) to C65 (most likely deleterious). SIFT scores run from 1.00 (most likely neutral) to 0.00 (most likely deleterious) in steps of 0.01. Two specific variants required a more detailed treatment. For the di-amino acid substitution p.SV2855_2856RI, we scored both component substitutions (p.S2855R and p.V2856I). p.S2855R received the most severe possible scores, C65 and 0.00, respectively, from the two programs. Accordingly, we scored p.SV2855_2856RI as C65 and 0.00. The three-amino-acid in-frame deletion p.SRI2546_2548del3 (hereafter referred to as ΔSRI) was more difficult to score. The variant encodes a stable, essentially full-length protein;34 this makes it biologically more like a missense substitution than a protein-truncating variant, so we therefore gave it a score that would allow it to be included in the logistic regressions with the missense substitutions. To do so, we examined the scores of all possible missense substitutions to codons S2546, R2547, and I2548, as well as the degree of conservation of the surrounding sequences. We noted the following three points. (1) With SIFT, some individual missense substitutions at R2547 received a score of 0.00. (2) With Align-GVGD, the most severe possible substitutions at the three positions scored C0, C35, and C15, respectively. In the logistic regression trend tests, the x axis positions for C0, C35, and C15 were 1, 4, and 2, respectively. The sum of these x axis positions, 7, was the x axis position of C65. (3) This in-frame deletion is closely flanked by invariant residues, the spacing between which is also invariant in our alignment. Combining across these considerations, we chose to score ΔSRI as C65 and 0.00.
Selection of Cases and Controls for Additional Mutation Screening
Study 8
Breast cancer case individuals mutation screened at Regensburg were Australian women selected from the Kathleen Cuningham Foundation Consortium for Research into Familial Aspects of Breast Cancer (kConFab) pedigrees35 with the use of these criteria: no known pathogenic mutation in BRCA1, BRCA2, PTEN (MIM 601728), or TP53 (MIM 191170) (more than 95% of the cases have been screened for mutations in BRCA1 and BRCA2); ‘Manchester score’ for BRCA2 of > 5;36 and at least two blood samples available from female family members affected with breast cancer (to allow for future family genotyping and segregation analysis). The female who was affected with breast cancer at the youngest age and had available DNA was then selected for ATM screening. Female Australian control samples sequenced at Regensburg were recruited as controls for the Australian Cancer Study (ACS).37 None had a personal history of breast cancer at the time of recruitment. These cases and controls were recruited from all Australian states and territories during the last ten years. The self-reported ethnicity of the kConFab cases comprised 97% white, 1% other, and 2% unknown or not reported. The self-reported race and/or ethnicity of the ACS controls comprised 95% white, 2% Asian, and 3% other (including unknown and Torres Strait islander). This study had approval from the Queensland Institute for Medical Research (QIMR), the Regensburg University institutional review board (IRB), and all other participating centers' IRBs. The kConFab and ACS data are referred to as study 8.
Study 9
The case-control-series mutation screened at the International Agency for Research on Cancer (IARC) consisted of subjects subselected from five sources: kConFab35 (13 of these cases were also screened at Regensburg, thereby providing quality control data), the three population-based centers of the Breast Cancer Family Registries (BCFR) (Cancer Care Ontario, the Northern California Cancer Center, and the University of Melbourne),38 and the National Cancer Institute of Thailand.39 Subjects were recruited between 1995 and 2005, and the genetics studies included in this project had approval from the IARC IRB and the local IRBs of every center from which we received samples.
Selection of Cases
Selection criteria for cases were a combination of age at diagnosis, family history of breast cancer, and race and/or ethnicity, as follows:
Age at Diagnosis. Noting that in the US, Canada, and Australia, the 20th percentile age of diagnosis for breast cancer is approximately 51 (Age20) and the first percentile age at diagnosis is approximately 33 (Age1), we applied the following equation: (Age20 − Dx) × (20 / [Age20 − Age1]) points (scores can be negative).
This resulted in cases diagnosed at age1 receiving 20 age points, cases diagnosed at age20 receiving 0 age points, and older cases receiving negative age points.
Family History. The family history component of the score depended on whether or not the index case had bilateral breast cancer, the number of first-degree relatives with breast cancer, the number of second-degree relatives with breast cancer, and the number of third-degree relatives with breast cancer. The score was calculated as: index case with bilateral breast cancer: 9 points; each first-degree relative with breast cancer: 6 points; each second-degree relative with breast cancer: 3 points; and each third-degree relative with breast cancer: 1 point.
The total score was the sum of the age at diagnosis and family history components. For kConFab and the three Breast CFR centers, our minimum criterion was a score of 15 points. For the Thai samples, our minimum criterion was a score of 10 points. Thus, kConFab and CFR cases diagnosed at less than age 37 years (less than age 43 years for the Thai cases) qualified even if they had no family history. Progressively older cases required progressively stronger family histories in order to qualify. Finally, we also applied an absolute age at diagnosis cutoff at diagnosis of age 50 years.
Race and/or Ethnicity. Using the self-reported race and/or ethnicity and grandparent country-of-origin information available in the kConFab and BCFR databases, we selected cases of European or East Asian ancestry from the Cancer Care Ontario and University of Melbourne BCFR centers; we selected cases of East Asian ancestry from the Northern California BCFR center; and we limited our selection of kConFab cases to individuals of European ancestry. We assumed that cases from the National Cancer Institute of Thailand are of East Asian ancestry. Finally, our kConFab cases were selected very early in the project and were selected under the additional constraints of only one subject per pedigree and availability of a lymphoblastoid cell line (LCL) for that subject. These LCLs were used extensively for process development. The racial and/or ethnic composition of the resulting case series was 62.9% European and 37.1% East Asian.
Selection of Controls
Controls were obtained from the three population-based BCFR centers and the National Cancer Institute of Thailand. The selection criteria applied were that they were from the same racial and/or ethnic group as the cases selected from that center and that their age at ascertainment was not more than three years beyond the age range of the cases from the same center. The racial and/or ethnic composition of the resulting control series was 62.7% European and 37.3% East Asian.
The number and age distribution of the cases and controls screened in studies 8 and 9 is given in Table 2.
Table 2.
Distribution of Subjects from Studies 8 and 9 by Center and Age
| Study Designation (Subject Source) | Mutation-Screening Site | Cases | Average | (Range) | Controls | Average | (Range) |
|---|---|---|---|---|---|---|---|
| 8 (kConFab) | Regensburg | 364 | 44.3 | (21–71) | – | ||
| 8 (ACS) | Regensburg | – | 362 | 58.0 | (19–80) | ||
| 9a (kConFab)a | IARC | 21 | 40.0 | (28–48) | – | ||
| 9a (Melbourne CFR)a,b | IARC | 260 | 34.7 | (23–49) | 262 | 36.9 | (22–45) |
| 9a (Ontario CFR)a | IARC | 112 | 37.4 | (25–48) | 153 | 40.0 | (25–50) |
| 9b (No. Cal CFR)c | IARC | 90 | 35.6 | (23–49) | 42 | 43.9 | (31–52) |
| 9b (Thai NCI)c | IARC | 140 | 35.3 | (17–47) | 202 | 35.0 | (18–46) |
Except for three subjects noted immediately below (footnote b), all of the subjects in these studies were of recent European ancestry.
The Melbourne CFR sample series included one case and two controls of recent East Asian ancestry. In logistic regressions of the bona fide case-control studies, these were considered as part of study 9b.
All of the subjects in these studies were of recent East Asian ancestry.
Mutation Screening
Mutation screening of the ATM gene at Regensburg from 377 familial breast cancer cases and 362 controls (study 8) was performed by PCR from genomic DNA followed by dye-terminator sequencing. All 65 ATM exons including the promoter region were PCR amplified and bidirectionally sequenced with the use of 64 primer pairs. Sixty-two primer pairs were tailed with the M13 sequences 5′-TGTAAAACGACGGCCAGT-3′ and 5′-CAGGAAACAGCTATGACC-3′, which served as universal forward and reverse sequencing primers, respectively. Two fragments were amplified and sequenced with the use of primers without the M13 tails.
We set up 15 μL PCR reactions in 384-well plates, using the Liquidator96 multi-channel pipettor (Steinbrenner Laborsysteme GmbH). Each reaction contained 30 ng of DNA, 1 U AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA), 8% glycerol, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, and 2.4 pmol of each primer. The cycling conditions were as follows: 94°C for 5 min, 40 cycles of 94°C for 30 s, 60°C for 45 s, 72°C for 45 s, final extension at 72°C for 10 s. We purified 10 μL of the amplification product with the Ampure®Kit (Agencourt Bioscience), using a 96-channel pipetting robot (Biomek NX, Beckman Coulter). We performed the purification in accordance with the manufacturer's protocol. The PCR products were eluted from the magnetic beads and diluted 4- to 6-fold with 40 μl LC-MS water (Merck), depending on the amount of amplicon determined in agarose gels by ethidium bromide staining.
Cycle sequencing was performed in a final reaction volume of 10 μl that contained 0.25 μl BigDye Terminator v.3.1 (Applied Biosystems), 3.2 pmol primer, 1× reaction buffer, 5 μl HPLC water, and 2 μl of the diluted purified PCR product. Cycle sequencing conditions were as follows: 96°C for 1 min, 25 cycles of 96°C for 10 s, 50°C for 5 s, 60°C for 90 s. The sequencing products were purified with the CleanSeq Kit (Agencourt), in accordance with the manufacturer's protocol, with the use of a 96-channel pipetting robot (Biomek NX). The products were eluted from the magnetic beads with 20 μl 75% HiDi-Formamide (Applied Biosystems). We transferred 17 μL to the final plate and analyzed the DNA fragments with an AB-3730 48-Capillary Sequencer. Sequence traces were aligned and analyzed with SeqScape v.2.5 (Applied Biosystems). Sequences of the mutation-screening primers used are available from P.O.
Mutation screening of the ATM gene at IARC (study 9) was performed from whole-genome amplified (WGA) DNA with the use of a nested PCR strategy, followed by high-resolution melting curve analysis (HRM analysis),40,41 and then dye-terminator resequencing of samples that contained a melt curve aberration indicative of the presence of a sequence variant.
For ATM amplicons harboring SNPs with frequency ≥ 1% in the population, we applied a simultaneous mutation scanning and genotyping approach by using HRM analysis to improve the sensitivity and the efficiency of the mutation screening.42 This method combines both fluorescent LCGreen Plus dye and unlabeled oligonucleotide probes that target the common SNP in an asymmetric PCR, leading to simultaneous production of probe-target and whole-amplicon double-stranded DNA duplexes that can be analyzed from the same HRM run. It thereby allows stratification of the samples according to their probe-target melting, i.e to their genotype for the common SNP. Hence, the data analysis component of mutation scanning is performed separately on heterozygous and homozygous sample subsets.
Whole-genome amplifications were performed on genomic DNAs with the use of the GenomiPhi DNA Amplification Kit (GE Healthcare). Fifty nanograms of genomic DNA and 9 μl of a sample buffer containing random hexamer primers were heat denatured and cooled, allowing random priming of the hexamers, then 9 μl of reaction buffer and 1 μl of Phi29 DNA polymerase were added and incubated overnight at 30°C for linear DNA synthesis. Concentrations of WGA DNAs were measured by standard picogreen titration. WGA DNAs were normalized at 6 ng/μL, and 30 ng of WGA DNAs were plated and dried into 384-well plates before being stored at 4°C for further use.
Primary PCR (PCR1), usually set up as a three amplicon triplex, was performed in an 8 μl reaction volume containing 30 ng of template DNA that had been prealiquoted and dried into the sample well, 10% sucrose, 20 mM Tris base, 3.2 mM acetic acid, 10 mM Na citrate, 16 mM MgSO4, 0.01% Triton X-100, 200 nM dNTP, 200 nM forward and reverse primers for each amplicon, and 0.04 U/μL of Platinum Taq Polymerase (Invitrogen). The PCR consisted of 25 cycles of amplification with priming temperature and elongation time optimized for each amplicon multiplex.
For standard HRM mutation scanning, simplex secondary PCRs (PCR2) were then performed in 6 μl reaction volume containing 1.5 μl of 1:100 diluted PCR1 product, 1X Invitrogen PCR buffer (20 mM Tris-HCl pH 8.4, 50 mM KCl), 1.5 mM MgCl2, 500 nM dNTP, 400 nM forward and reverse primers, 0.5X LCGreen Plus (Idaho Technology), and 0.04 U/μL of Platinum Taq Polymerase. For the simultaneous mutation scanning and genotyping procedure, the same conditions were used, except that (1) a primer asymmetry ratio of 1:5 (100 nM limiting primer, 500 nM excess primer) was used to favor the production of the DNA strand targeted by the probe, and (2) the unlabelled 3′ end-capped probe was included at 500 nM. For an optimal efficiency of HRM, PCR2 amplicons were no longer than 350 bp and amplified with 40 cycles for standard mutation scanning and 55 cycles for simultaneous mutation scanning and genotyping.
Prior to HRM analysis, PCR2 products were heated to 94°C, then slowly cooled to 20°C to promote heteroduplex formation and detection. Melting was monitored from 65°C to 95°C for standard mutation scanning and 35°C to 95°C for simultaneous mutation scanning and genotyping on a LightScanner instrument (Idaho Technology). HRM analyses were carried out with the LightScanner software (Idaho Technology) with the “Scanning” mode used for standard mutation scanning and, in the case of common SNPs, the “Genotyping” mode used for the region of the probe melting followed by an analysis with the “Scanning” mode for the region of DNA melting.
PCR2 products with melting curves that differed from the reference group were rearrayed onto new 96-well plates and treated with exonuclease I and shrimp alkaline phosphatase for the removal of excess primers and nucleotide triphosphates (exo-SAP treatment). Dye-terminator sequencing reactions (BigDye Terminator, version1.1, Applied Biosystems) were inoculated with the exo-SAP-treated PCR products, thermocycled, then purified with Montage SEQ96 sequencing reaction cleanup kits (Millipore). Sequencing reaction products were then run on a 96-capillary Spectrumedix Sequencer (Transgenomics) in accordance with the manufacturer's recommendations.
The resulting chromatograms were analyzed with the program Java SnpScreen. Very similar to the software used for research resequencing and BRACAnalysis at Myriad Genetics,43,44 the program starts with the canonical text sequence of each amplicon, aligns all of the forward chromatograms to the canonical sequence, reverse complements the reverse chromatograms and then aligns them to the canonical sequence, normalizes the signal strength from all of the chromatograms, then displays them as aligned forward-reverse chromatogram pairs. The software contains algorithms that spot potentially heterozygous positions on the basis of the joint data from the target sequence and each forward and reverse chromatogram pair. Alternatively, the user can scan the superimposed chromatogram sets visually. After the screening has been completed, the program creates an output report that contains an amplicon-specific genotype for each sample screened.
All samples found to carry a rare sequence variant were reamplified from genomic DNA for confirmation of the presence of the variant.
Every step of our automated laboratory process was tracked by a Laboratory Information Management System (LIMS) that had been internally developed.45 Sequences of the mutation-screening primers used are available from S.V.T., and the code for Java SnpScreen is available from A.T.
DNAs from 13 kConFab breast cancer cases were mutation screened at both Regensburg and IARC, as were those of 30 other individuals that are part of another study being conducted by these centers. The independently determined genotypes were identical for all 43 individuals. Results from the kConFab samples that were analyzed twice are included in the study 9 results.
Statistical Methods
To assess evidence of risk from the case-control frequency distribution of protein-truncating variants (T), known or very likely spliceogenic splice-junction variants (SJ), and rare missense substitutions (rMSs), we constructed a single table with one entry per subject, zero or one rare sequence variant per subject, and annotations for study, case-control status, probability of being of recent African ancestry, and the estimated efficiency of mutation-screening method used.
For mutation-screening data extracted from the seven published case-control studies and 17 published case-only or control-only studies, our assumption of no more than one rare variant per subject was necessary because the studies pooled did not systematically report co-occurrence between rare variants. Because the summed allele frequencies of the rare variants in these studies (excluding the four that used the protein-truncation test [PTT) only) was about 4.2%, we would expect that by chance, about 0.18% (∼six subjects in the entire pooled data set) might have been compound heterozygotes; unless the compound heterozygotes were spread very unevenly among the various grades of sequence variants, the slight implied counting error should have had minimal effect on our overall results. For subjects in our own mutation screening study who carried more than one rare variant, only the variant belonging to the most likely pathogenic grade was considered. We did not observe co-occurrence between any two rare variants of grade C35 or higher.
Because of variation in study parameters between study sites, including case and/or control selection criteria, ethnic groups sampled, and mutation-screening methodology, multivariable unconditional logistic regression analyses were performed. Analyses of the bona fide case-control studies were adjusted for study site. The European and East Asian components of study 9 were treated as two separate studies, 9a and 9b, for this purpose. However, adjustment for study site was not possible for expanded analyses that included the case-only and control-only studies. For these subsidiary expanded analyses, we adjusted for ethnicity and mutation-screening methodology as described below.
The frequency of rare variants in individuals of recent African ancestry is approximately twice as high as it is in individuals of European, Asian, or Latino/Hispanic ancestry.46 Accordingly, ethnicity was treated as a continuous variable reflecting the probability of a subject to be of recent African ancestry and was estimated from the case and control selection criteria described in each study.
Mutation detection is rarely 100% sensitive, and there are notable sensitivity differences between methods. Therefore, we treated mutation-screening-method sensitivity as a continuous variable equal to 1/s, with s corresponding to the sensitivity of the method. The values were based on a recent review of mutation-screening methods47 and were defined as follows: 0.95 for HRM, denaturing high-performance liquid chromatography (DHPLC) and sequencing, 0.90 for denaturing gradient gel electrophoresis (DGGE), 0.75 for single-strand conformation polymorphism (SSCP), 0.70 for fluorescent chemical cleavage of mismatch (FCCM), and conformation-sensitive gel electrophoresis (CSGE). We considered that the PTT had a sensitivity of 0.95 for detection of protein-truncating variants, and we considered that the mixed application of DHPLC and restriction endonuclease finger-printing analysis had a sensitivity of 0.60 for detection of missense substitutions. Finally, we estimated that the nonisotopic RNase cleavage-based assay (NIRCA) had a sensitivity of 0.50.
Logistic regression trend tests were formatted such that subjects who did not carry any rare variant and carriers of the seven grades of rMSs (C0, C15, C25, C35, C45, C55, and C65) defined by Align-GVGD17 were assigned the default row labels 0,1,2,3,4,5,6, and 7, respectively. These row labels were then used as a continuous variable in the logistic regressions. Regression coefficients and trend test p values (“Ptrend”) were estimated from the resulting ln(OR)s with the logit function of STATA. We used the Fisher's exact test (FET) to obtain the lower bound of the 95% confidence interval for single-category tests that had one or more cases but zero controls.
The reference noncarrier group (assigned logistic regression row label 0) comprised the subjects who were not reported to carry a rMS, an in-frame deletion, or a T+SJ variant anywhere in the gene. Thus, the same reference group of noncarriers was used for whole-gene analyses and domain-specific subanalyses.
Post hoc power calculations were performed by specifying a hypothetical OR and population prevalence for each class of variant, together with the total probability of breast cancer prior to age 70. The ORs that we specified for the individual grades of sequence variants, relative to C0 and the noncarriers, were as calculated from the whole-gene analysis for the grades for which there were reasonable numbers of observations: 1.13, 1.23, 1.20, 4.82, and 2.33 for C15, C25, C55, C65, and T+SJ, respectively. Because of the very low numbers of observations in C35 and C45, those ORs were set equal to C55 at 1.20. From these, we calculated expected values and variances of the test statistics for the types of test considered: Pearson's chi-square for the two-category tests, and the Wald statistic from a logistic regression for the trend test. We then calculated the probability of these statistics exceeding the thresholds corresponding to p < 0.05 in each case, using a normal approximation.
Results
Published Data Available for Meta-Analysis
Review of the literature revealed seven studies reporting nonredundant primary data from the mutation screening of ATM in breast cancer cases and controls,6,10,12,14,48–50 as well as 17 additional studies that reported case-only or control-only mutation screening with ascertainment criteria that met our inclusion requirements.5,7–9,11,13,51–61 These studies provided bona fide case-control data from a total of 1544 cases and 1224 controls plus case-only and control-only data from an additional 1581 cases and 154 controls (Table 3 and Table S1, available online). The set of sequence variants reported from these 4503 subjects included seven common missense substitutions (carrier frequency ≥ 1%), 121 rare missense substitutions (frequency < 1%; rMSs), 20 protein-truncating variants (T), and 10 variants thought or expected to cause severe splice-junction defects (SJ) (Table S2). We considered analysis of the seven common ATM missense substitutions to be outside of the scope of this work. Thus all results from this point on are based on analyses of rMS, T, and SJ variants.
Table 3.
Number of Cases and/or Controls by Study
| Study Designation | Study | Cases | Controls | Total |
|---|---|---|---|---|
| 1 | Fitzgerald et al. 199748 | 401 | 202 | 603 |
| 2 | Teraoka et al. 20016 | 142 | 81 | 223 |
| 3 | Sommer et al. 200310 | 90 | 90 | 180 |
| 4 | Thorstenson et al. 200312a | 270 | 52 | 322 |
| 5 | Renwick et al. 200614a | 443 | 521 | 964 |
| 6 | Hirsch et al. 200849a | 37 | 95 | 132 |
| 7 | Soukupova et al. 200850a | 161 | 183 | 344 |
| 8 | This study, kConFab/Regensburga | 364 | 362 | 726 |
| 9a | This study, IARC- European | 392 | 414 | 806 |
| 9b | This study, IARC- East Asian | 231 | 245 | 476 |
| Bona Fide Case-Control Subtotal | 2531 | 2245 | 4776 | |
| 10 | Vorechovsky et al. 199651 | 38 | 0 | 38 |
| 11 | Chen et al. 199852a | 100 | 0 | 100 |
| 12 | Bebb et al. 199953 | 47 | 0 | 47 |
| 13 | Izatt et al. 19995a | 100 | 0 | 100 |
| 14 | Dörk et al. 20017 | 192 | 0 | 192 |
| 15 | Drumea et al. 200054 | 37 | 0 | 37 |
| 16 | Atencio et al. 20018 | 52 | 0 | 52 |
| 17 | Maillet et al. 20029 | 94 | 0 | 94 |
| 18 | Angele et al. 200311 | 51 | 0 | 51 |
| 19 | Buchholz et al. 200413 | 91 | 0 | 91 |
| 20 | Ho et al. 200757 | 131 | 0 | 131 |
| 21 | Broeks et al. 200858 | 437 | 0 | 437 |
| 22 | Brunet et al. 200859 | 43 | 0 | 43 |
| 23 | Tapia et al. 200860a | 42 | 0 | 42 |
| 24 | Gonzalez-Hormazabal et al. 200861a | 126 | 0 | 126 |
| 25 | Thorstenson et al. 200155b | 0 | 64 | 64 |
| 26 | NIEHS56 | 0 | 90 | 90 |
| All Studies Total | 4112 | 2399 | 6511 | |
Studies in which more than 50% of the cases had a family history of breast cancer.
We have used only 64 of the 93 controls described in Thorstenson et al (2001).55 The remaining 29 controls were of Middle Eastern, South Asian, or Oceanian descent, and there were essentially no breast cancer cases from these groups in the published studies.
Additional Mutation Screening
To increase the power of our analyses, we mutation screened the coding exons and adjacent proximal introns of ATM in 987 cases and 1021 controls: 364 cases and 362 controls were screened by direct sequencing (study 8), and 623 cases and 659 controls were screened by HRM, followed by sequencing of the individual samples that yielded an HRM aberration (study 9). The mutation screening revealed 76 rMSs, one in-frame deletion of three amino acids (ΔSRI) that we treated as a missense substitution, 12 protein-truncating variants, and one variant expected to destroy a splice acceptor. Only 28 of the 77 rMSs and two of the 13 T+SJ variants were present in the published mutation-screening data (Table S2).
Analysis of Truncating and Splice-Junction Variants
In analyses of known or candidate susceptibility genes in which simple loss of function is expected to be pathogenic, it is now becoming customary to pool data from rare truncating variants with data from rare splice-junction variants that are known to (or thought highly likely to) destroy a splice junction with the ultimate result of nonsense-mediated decay and a protein truncation because their effects on disease risk are often similar.14,62–65 Before we pooled the ATM T+SJ data, we reviewed the sequence context of all of the SJ variants that had been treated as likely pathogenic in previous studies. We found ten that appear to be correctly classified, but we also found two, c.1066-6T>G and c.3993+5G>T, that ought not be included in the T+SJ pool in the absence of further functional assay results. The variant c.1066-6T>G is no longer thought to be pathogenic for A-T because the homozygous A-T patient previously described7 has recently been found to harbor second-site mutations that are sufficient to explain the A-T phenotype on their own (Richard Gatti, personal communication). In addition, in silico analyses of the variant with splice site prediction by neural network (NNsplice) and maximum entropy modeling of short sequence motifs (MaxEntScan)66,67 are not indicative of a severe effect on the fitness of the intron 10 splice acceptor; both programs give scores for this sequence variant that are above the mean for the pool of all wild-type splice acceptors in ATM+BRCA1+BRCA2. Similarly, despite the argument that Dörk et al. made in favor of the idea that c.3993+5G>T should interfere with splicing,7 both NNsplice and MaxEntScan score this variant above the mean for the pool of all wild-type splice donors in ATM+BRCA1+BRCA2.
Excluding these two sequence variants, a total of 41 distinct T+SJ variants were present in the combination of the published ATM breast cancer case and control mutation-screening literature plus our own mutation-screening data. One, c.3802delG, has been reported four times, two have been reported twice each, and the remaining 38 were reported once each (Table S2). With a focus on the bona fide case control studies, there were a total of 26 T+SJ variants observed among 2531 cases and ten among 2245 controls (OR = 2.32, p = 0.024) (Table 4). Expansion for inclusion of the 15 case-only and two control-only data sets had little effect on these results (OR = 2.08, p = 0.042).
Table 4.
Analysis of Truncating and Spliceogenic Splice-Junction Variants
| Cases | Controls | Crude OR [95% CI] | Adjusted OR [95% CI]a | |
|---|---|---|---|---|
| Bona Fide Case-Control Studiesb | ||||
| Noncarrier | 2505 | 2235 | ref | ref |
| T+SJ | 26 | 10 | 2.33 [1.12–4.84] | 2.32 [1.12–4.83] |
| All Studies | ||||
| Noncarrier | 4076 | 2389 | ref | ref |
| T+SJ | 36 | 10 | 2.10 [1.04–4.24] | 2.08 [1.03–4.21] |
Abbreviations are as follows: OR, odds ratio; CI, confidence interval;ref, reference category (OR = 1.0).
The OR from the analysis of the bona fide case-control studies was adjusted for study. The OR from the analysis of all studies was adjusted for ethnicity and sensitivity of the mutation-screening method employed.
The bona fide case-control studies included both mutation-screened cases and mutation-screened controls that met our ascertainment criteria.
Analysis of Rare Missense Substitutions
There is as yet no community consensus on how to handle rMSs. With 170 distinct rMSs in the present ATM data set, 117 of which were observed only once, it is clear that any analysis of individual rMSs will be overwhelmed by either the number of degrees of freedom inherent in the analysis or the adjustment of significance thresholds required to take account of multiple testing, depending on the format of the test. However, when all of the rMSs reported in the bona fide case-control studies were pooled, there was no notable difference in their pooled frequency in cases versus controls (OR = 1.14, p = 0.29) (Table 5). Recently, Li and Leal suggested using frequency to collapse rare variants into a limited set of n pools, followed by an n-1 degree of freedom test for heterogeneity over the pools.68 When we collapsed the rMS case-control distribution into a series of four pools based on apparent frequency, we again found no obvious difference between cases and controls (p = 0.39) (Table 5).
Table 5.
Whole-Gene Analysis of Rare Missense Substitutions, Unstratified or Stratified by Frequency
|
Bona Fide Case-Control Studies |
All Studies |
|||||||
|---|---|---|---|---|---|---|---|---|
|
Test of Significance: OR [95% CI], p Value, or Regression Coefficient [95% CI] |
Test of Significance: OR [95% CI], p Value, or regression coefficient [95% CI] |
|||||||
| Cases | Controls | Crude | Adjusteda | Cases | Controls | Crude | Adjustedb | |
| Noncarrierc | 1788 | 1717 | ref | ref | 3125 | 1850 | ref | ref |
| Any rMSd | 160 | 135 | 1.14 [0.90–1.44] | 1.14 [0.90–1.44] | 248 | 156 | 0.94 [0.76–1.16] | 1.06 [0.86–1.31] |
| Stratification by Frequency | ||||||||
| rMSs observed 1×–3× | 69 | 63 | 1.05 [0.74–1.49] | 1.05 [0.74–1.49] | 113 | 79 | 0.86 [0.64–1.15] | 0.89 [0.66–1.20] |
| rMSs observed 4×–10× | 55 | 43 | 1.23 [0.82–1.84] | 1.23 [0.82–1.84] | 74 | 46 | 0.95 [0.66–1.38] | 1.01 [0.69–1.47] |
| rMSs observed 11×–30× | 20 | 21 | 0.91 [0.49–1.69] | 0.91 [0.49–1.69] | 37 | 23 | 0.95 [0.56–1.61] | 0.96 [0.57–1.62] |
| rMSs observed > 30× | 23 | 12 | 1.84 [0.91–3.71] | 1.84 [0.91–3.71] | 33 | 12 | 1.63 [0.84–3.16] | 1.59 [0.82–3.10] |
| Test of heterogeneity | p = 0.39 | p = 0.39 | p = 0.49 | p = 0.62 | ||||
Abbreviations are as follows: OR, odds ratio; CI, confidence interval; ref, reference category (OR = 1.0).
Use of unconditional logistic regression with an adjustment for study.
Use of unconditional logistic regression with adjustments for ethnicity and sensitivity of mutation-screening method employed.
Carriers of T+SJ variants are excluded.
Individuals in studies 8 or 9 who carried two (10) or three (1) rare variants are coded according to the highest grade of rare variant that they carried. The co-occurrences are detailed in the footnotes to Table S2.
Previously, we suggested collapsing rMSs into a graded series of pools ordered by the probability that missense substitutions in each pool are evolutionarily deleterious and then conducting a test for trend over the ordered pools.17 A number of missense-substitution-analysis programs, including Align-GVGD, MAPP, and SIFT, output a variable that can be used to order missense substitutions with respect to the probability that they are evolutionarily deleterious.69–71 A common thread is that these programs require a protein multiple sequence alignment of the gene of interest, and their performance is sensitive to the quality of the alignment used.72 To enable grading of ATM rMSs, we constructed and carefully curated a protein multiple sequence alignment from seven full-length vertebrate plus two additional deuterostomate ATM ortholog sequences that were determined in the course of this project. The alignment is similar in phylogenetic depth to those that we have found useful for analyzing missense substitutions in BRCA1, BRCA2, and CHEK2.17,73 A maximum parsimony count revealed that the alignment contains an average of 3.08 amino acid substitutions per position, and SIFT reported “median sequence conservation” of 3.07, meeting that program's criterion for confident prediction of which substitutions should “affect protein function.” Thus, the alignment meets externally defined criteria of sufficient informativeness to support grading of missense substitutions.71,74 Sequence accession numbers and pairwise percentage sequence identities are reported in Table 1.
The missense substitutions were then assessed in silico with the use of Align-GVGD with our sequence alignment, and the raw scores were converted into an ordered series of seven grades: C0, C15, C25, C35, C45, C55, and C65.17 These grades provide a ranking of missense substitutions from evolutionarily most likely to least likely. The pooled rMS observational data are summarized in Table 6, the complete set of sequence variants is described in Table S2, and their distribution and frequency are displayed graphically in Figure 1. After excluding T+SJ carriers from the data set, we performed a log-linear trend test across noncarriers (grade 0) and carriers of the seven grades of missense substitutions. Applied to the bona fide case-control studies, the trend test, which is against the null hypothesis of no change in OR with increasing grade of missense substitution, yielded a ln(OR) increase of 0.13 per grade (Ptrend = 0.0035). Expansion for inclusion of the case-only and control-only data sets had little effect on these results (ln(OR) increase of 0.11 per grade and Ptrend = 0.0073).
Table 6.
Analyses of Rare Missense Substitutions, Stratified by Align-GVGD Grade
|
Bona Fide Case-Control Studies |
All Studies |
|||||||
|---|---|---|---|---|---|---|---|---|
|
Test of Significance: ln(OR) [95%CI] or Regression Coefficient [95%CI] |
Test of Significance: ln(OR) [95%CI] or Regression Coefficient [95%CI] |
|||||||
| Cases | Controls | Crude | Adjusteda | Cases | Controls | Crude | Adjustedb | |
| Whole-Gene Analysis; Stratification by Align-GVGD Grade | ||||||||
| Noncarrierc | 1788 | 1717 | ref | ref | 3125 | 1850 | ref | ref |
| C0d | 86 | 89 | −0.07 [−0.38–0.23] | −0.08 [−0.38–0.23] | 140 | 107 | −0.26 [−0.51–0.00] | −0.09 [−0.36–0.18] |
| C15d | 34 | 29 | 0.12 [−0.38–0.62] | 0.12 [−0.38–0.62] | 46 | 30 | −0.10 [−0.56–0.37] | −0.07 [−0.54–0.39] |
| C25d | 9 | 7 | 0.21 [−0.78–1.20] | 0.21 [−0.78–1.20] | 14 | 8 | 0.04 [−0.84–0.91] | 0.011 [−0.76–0.98] |
| C35 | 0 | 1 | - | - | 0 | 1 | - | - |
| C45 | 1 | 0 | - | - | 1 | 0 | - | - |
| C55 | 5 | 4 | 0.18 [−1.13–1.50] | 0.18 [-1.13–1.50] | 10 | 5 | 0.17 [−0.91–1.24] | 0.19 [−0.89–1.27] |
| C65 | 25 | 5 | 1.57 [0.61–2.53] | 1.57 [0.61–2.53] | 37 | 5 | 1.48 [0.54–2.41] | 1.51 [0.58–2.45] |
| ln(OR) regression coefficients [95% CI]e | 0.13 [0.044–0.22] | 0.13 [0.044–0.22] | 0.085 [0.0077–0.16] | 0.11 [0.026–0.18] | ||||
| Analysis from Position Ile1960 to the End of the Protein; Stratification by Align-GVGD Grade | ||||||||
| Noncarrierc | 1788 | 1717 | ref | ref | 3125 | 1850 | ref | ref |
| C0 | 22 | 21 | 0.01 [−0.60–0.61] | 0.01 [−0.59–0.61] | 35 | 25 | −0.19 [−0.70–0.33] | −0.08 [−0.60–0.44] |
| C15 | 3 | 1 | 1.06 [−1.21–3.32] | 1.06 [−1.21–3.32] | 4 | 1 | 0.86 [−1.33–3.05] | 0.91 [−1.28–3.10] |
| C25 | 2 | 2 | −0.04 [−2.00–1.92] | −0.04 [−2.00–1.92] | 3 | 2 | −0.12 [−1.91–1.67] | −0.07 [−1.86–1.72] |
| C35 | 0 | 0 | - | - | 0 | 0 | - | - |
| C45 | 1 | 0 | - | - | 1 | 0 | - | - |
| C55 | 4 | 1 | 1.35 [−0.85–3.54] | 1.34 [−0.85–3.54] | 7 | 2 | 0.73 [−0.84–2.30] | 0.76 [−0.82–2.33] |
| C65 | 18 | 1 | 2.85 [0.84–4.86] | 2.85 [0.83–4.86] | 24 | 1 | 2.65 [0.65–4.66] | 2.65 [0.65–4.65] |
| ln(OR) regression coefficients [95% CI]e | 0.31 [0.14–0.48] | 0.31 [0.14–0.48] | 0.23 [0.083–0.37] | 0.24 [0.091-0.39] | ||||
| Analysis Limited to the Restrictively Defined FAT, Kinase, and FATC Domains; Stratification by Align-GVGD Grade | ||||||||
| Noncarrierc | 1788 | 1717 | ref | ref | 3125 | 1850 | ref | ref |
| C0 | 11 | 10 | 0.05 [−0.80–0.91] | 0.06 [−0.80–0.91] | 20 | 12 | −0.01 [−0.73–0.70] | 0.09 [−0.64–0.81] |
| C15 | 0 | 0 | - | - | 0 | 0 | - | - |
| C25 | 0 | 0 | - | - | 0 | 0 | - | - |
| C35 | 0 | 0 | - | - | 0 | 0 | - | - |
| C45 | 0 | 0 | - | - | 0 | 0 | - | - |
| C55 | 3 | 1 | 1.06 [−1.21–3.32] | 1.05 [−1.21–3.32] | 6 | 1 | 1.27 [−0.85–3.39] | 1.28 [−0.84–3.40] |
| C65 | 17 | 0 | Infinite [1.45f–∞] | g | 22 | 0 | Infinite [3.39e –∞] | g |
| ln(OR) regression coefficients [95% CI]e | 0.41 [0.15–0.68] | 0.41 [0.15–0.68] | 0.38 [0.13–0.63] | 0.40 [0.13–0.64] | ||||
Bold font is used to indicate point estimates or trend coefficients with p < 0.05. Abbreviations are as follows: OR, odds ratio; CI, confidence interval; ref, reference category (OR = 1.0).
Using unconditional logistic regression with an adjustment for study.
Using unconditional logistic regression with adjustments for ethnicity and sensitivity of mutation-screening method employed.
Carriers of T+SJ variants are excluded. Carriers of rMSs that fall outside of the specified region (and no rMS occurring in the region) are excluded.
Individuals in studies 8 or 9 who carried two (8) or three (1) rare variants are coded according to the highest grade of rare variant that they carried. Categories that lose a subject(s) are marked “d.” The co-occurrences are detailed in the footnotes to Table S2.
From a standard logistic regression of form ln(OR) = a + b(x) in which a = 0, b is the logistic regression OR trend coefficient, and x is, in this case, missense-substitution grade. Note that the regression coefficient is significant if its 95% CI excludes 0.00.
Lower boundary of this 95% CI was obtained from Fisher's exact test.
Could not be calculated with the use of the adjusted model.
Figure 1.
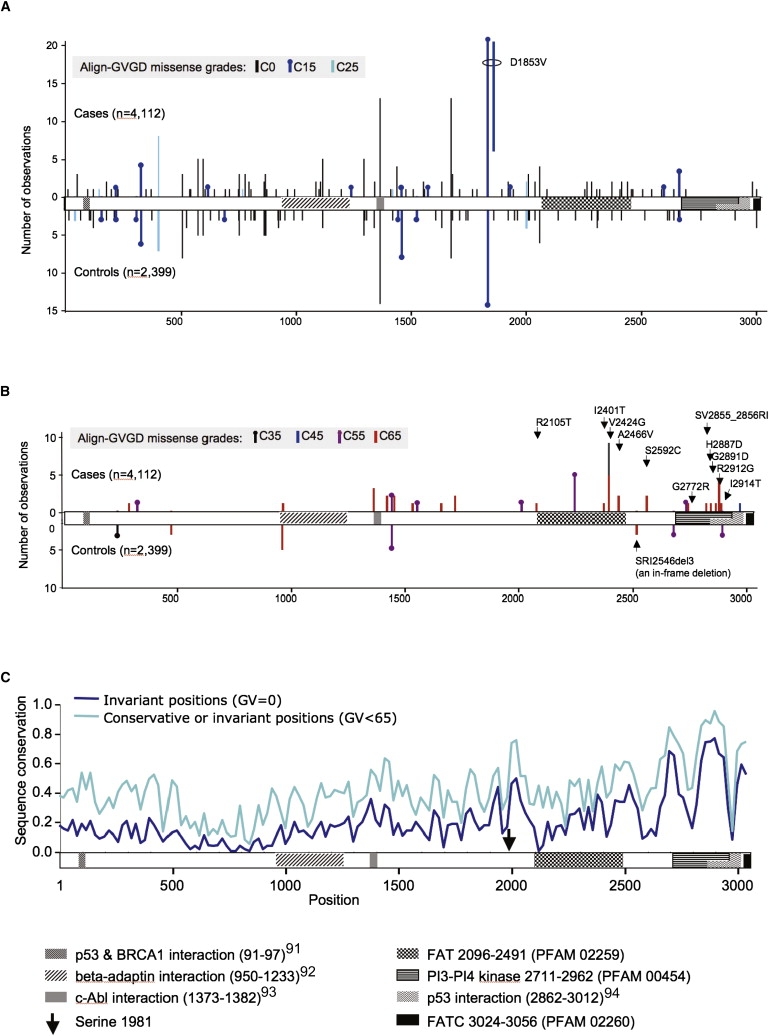
Domain Organization of ATM and Case-Control Distribution of Missense Substitutions by Align-GVGD Grade
(A) Distribution of rare C0, C15, and C25 missense substitutions superimposed on the domain organization of ATM. Note that if two distinct substitutions are located very close to each other, we shifted one by a few amino acids so that the presence of both is visible.
(B) Distribution of rare C35, C45, C55, and C65 missense substitutions. We labeled the C65 missense substitutions falling from Ile1960 until the end of the protein.
(C) Sequence-conservation profile across ATM. The fraction of invariant positions (GV = 0) across the ATM protein multiple sequence alignment was measured in a 20-residue sliding window. Results were smoothed by inclusion of (1/e × sequence invariance) in the ten residues preceding and trailing each window, then normalized. The analysis was repeated with the use of a conservation criterion of only conservative substitution or invariance (GV < 65) across species.
Citations correspond to Fernandes et al.,91 Lim et al.,92 Shafman et al.,93 and Khanna et al.94
Combining mutation-screening data from a population sampling with ATM sequence variation between primates, Oefner and co-workers argued that there is stronger selection against missense substitutions falling in the carboxy-third of ATM than in the rest of the gene.55 Accordingly, we analyzed separately the missense substitutions located in this region of the protein. Using the relatively relaxed Prosite definition of residue Ile1960 as the start of the FAT domain (Prosite entry PS51189, last updated February 2009) to provide a domain-based definition of the carboxy-third of ATM, we reran the same set of missense trend tests described above (Table 6). Applied to the bona fide case-control studies, the rMS trend test over the carboxy-third of the protein yielded a ln(OR) increase of 0.31 per grade (Ptrend = 0.00048). In contrast, the trend test applied to the segment 1-1959 returned a ln(OR) increase of 0.0095 per grade (Ptrend = 0.87). Expanded to include all of the studies, the ln(OR) increase for the carboxy-third was 0.24 per grade (Ptrend = 0.0016). That the whole-gene, amino two-thirds, and carboxy-third analyses produce different ln(OR) coefficients for overlapping sets of rMSs highlights the point that none of these are perfect models of reality. No model will ever be exactly correct, so we preplanned a relatively simple analysis strategy17 that potentially sacrifices OR accuracy to avoid hidden multiple testing that would erode the validity of the p values obtained.
Using the bona-fide case-control data, we performed two additional analyses of the carboxy-third of the protein. First, in order to test for a difference between OR trend estimates for the amino two-thirds versus carboxy-third of the protein, we performed a likelihood-ratio test to compare two models. In one model, we used an indicator variable to specify whether the rMSs fall in the carboxy-third of the protein or not; in the other model, all rMSs were treated similarly. The result from this likelihood ratio test was significant (p = 0.0021), indicating that risk conferred by rMSs falling before and after Ile1960 are not equivalent. Second, we were concerned that the evidence for risk conferred by rMSs falling in the carboxy-third of the protein might be entirely due to p.V2424G. This was the most common of the clearly pathogenic (for A-T) variants in our data set, observed nine times in the cases and zero times in the controls. After exclusion of this variant, a trend test over the carboxy-third of the protein still returned substantial evidence for risk attributable to rMSs (ln(OR) increase of 0.25 per grade and Ptrend = 0.0088).
For ATM, the specific domains in which missense substitutions have been most closely tied to A-T are the FAT, kinase, and FATC domains.75,76 Therefore, there is also a rationale for focusing our analysis of missense substitutions very tightly on these three domains. Using the relatively restrictive PFAM FAT (PFAM PF02259, 2096–2489), PI3_PI4_kinase (PFAM PF00454, 2711–2962), and FATC (PFAM PF02260, 3024–3056) domain definitions, we reiterated our set of rMS trend tests. In this iteration, Align-GVGD produced an essentially binary classification; the missense substitutions were either C0 (21 distinct substitutions in all studies) or C55–C65 (13 distinct substitutions in all studies) (Table 6 and Table S2). When the missense-substitution trend test was applied to the FAT+kinase+FATC rMSs observed in the bona fide case-control series, we found a ln(OR) increase of 0.41/grade (Ptrend = 0.0022). Expanded to include all of the studies, the ln(OR) increase was 0.40/grade (Ptrend = 0.0030).
Noting the estimated OR for T+SJ variants (2.32, 95% confidence interval [CI] 1.12–4.83) and the OR predicted at C65 from the fitted trend of the FAT+kinase+FATC analysis (18.0, 95% CI 2.82–117) (Table 4 and calculation from Table 6), we asked whether the risk conferred by inheritance of FAT+kinase+FATC C65 missense substitutions is higher than that for T+SJ variants. A Fisher's exact test revealed that the proportion of cases among FAT+kinase+FATC C65 missense-substitution carriers (17/17 when confined to case-control studies, 22/22 for all studies) was different from the proportion of cases among T+SJ carriers (26/36 when confined to case-control studies, 36/46 for all studies) (PFET = 0.021 and 0.024 for the two comparisons, respectively). When we excluded from the FAT+kinase+FATC rMS versus T+SJ comparison the four studies (1, 7, 11, and 12) that used only the protein-truncation test for their mutation screen, the differences remained significant (PFET = 0.019 for case-control only and PFET = 0.022 for all studies). Thus, results from the two-sided Fisher's exact tests support the interpretation, derived from the logistic regression OR point estimates, that FAT+kinase+FATC C65 rMSs confer on average greater risk than do T+SJ variants.
Comparison between Align-GVGD and SIFT
The ability to detect statistical evidence of risk attributable to rMSs in ATM was not unique to Align-GVGD. For example, we used SIFT to set up a binary comparison between noncarriers and carriers of rMSs with SIFT score ≤ 0.05, which is the standard binary classification cutoff with this algorithm. In the whole-gene missense analysis of the bona fide case-control data, the SIFT analysis returned OR = 1.58 (p = 0.014), a result that would clearly contribute toward evidence that ATM is a breast cancer susceptibility gene (data not shown). Confined to rMSs in the carboxy-third of the protein, this SIFT analysis returned OR = 3.60 (p = 0.0014), reiterating the strength of this subset analysis. Finally, for the restrictive FAT+kinase+FATC analysis, we obtained OR = 5.27 (p = 0.0023). However, analysis with SIFT did not provide any evidence that a subset of rMSs might confer greater risk than do T+SJ variants. For example, a Fisher's exact test did not indicate any difference in the proportion of cases among FAT+kinase+FATC SIFT ≤ 0.05 missense-substitution carriers (22/26 when confined to case-control studies) and the proportion of cases among T+SJ carriers (PFET = 0.36, or p = 0.34 after exclusion of studies 1 and 7). The most severe grade of missense substitutions that SIFT can define is SIFT score = 0.00. Even upon restriction of the rMS analysis to the proportion of cases among FAT+kinase+FATC SIFT = 0.00 missense-substitution carriers (19/21 when confined to case-control studies), the difference with the proportion of cases who carry T+SJ variants remained null (PFET = 0.18, or p = 0.16 after exclusion of studies 1 and 7).
We suspected that an analysis using Align-GVGD detected a difference between the most severe grade of FAT+kinase+FATC missense substitutions versus T+SJ variants whereas an analysis using SIFT did not because Align-GVGD C65 provides, on average, a slightly higher standard for missense-substitution severity than does SIFT score = 0.00. For example, across the whole gene and with the inclusion of all of the studies reporting rMS data, 19/21 rMSs that scored C65 also had SIFT score = 0.00 (the remaining two, p.I2401T and p.I2914T, had SIFT score = 0.01) (ΔSRI was excluded from this and the following comparisons because analysis of in-frame deletions is very awkward). In contrast, 15/34 rMSs with a SIFT score = 0.00 had Align-GVGD grades that are distributed from C0 to C55. When the rMSs with SIFT scores = 0.00 were stratified into those that were also C65 versus those that were not, the group with SIFT scores = 0.00 and C65 appeared to be associated with a higher OR than those that were SIFT score = 0.00 but not C65 (ORs of 5.22 [1.86–20.24] and 0.93 [0.37–2.44], respectively, with PFET for the difference = 0.011). The difference between these scoring criteria is made apparent in Figure 2. Substitutions scored as C65 fell at positions that either are invariant or have cross-species variation that is limited to Ile-Leu-Met, and the substitutions were clearly nonconservative with respect to the position at which they fell. Substitutions that were SIFT = 0.00 but not C65 were either relatively conservative substitutions that fell at invariant positions (specifically, the standard Grantham difference is < 65) or nonconservative substitutions that fell at positions having slightly greater cross-species variation than the extremely conservative Ile-Leu-Met set, as judged by their Grantham variations.
Figure 2.
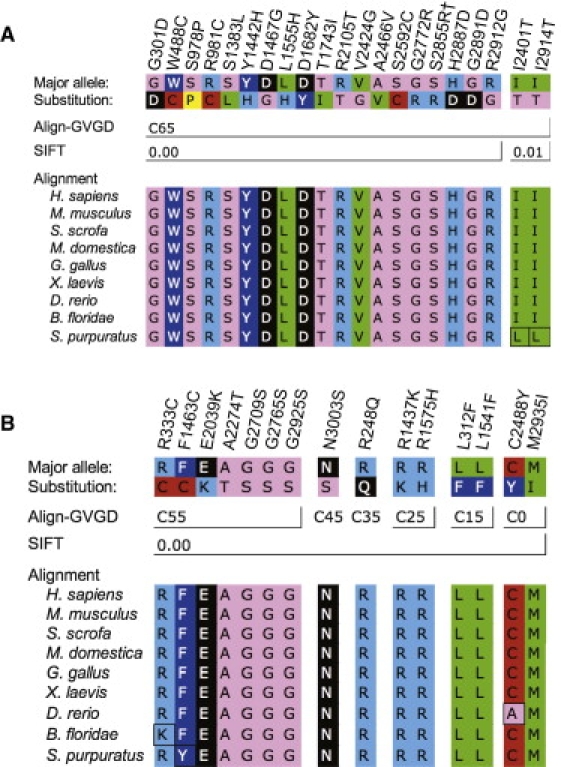
ATM Missense Substitutions Graded C65 by Align-GVGD and/or Scored 0.00 by SIFT
Substitution designations are given over their respective positions in the ATM alignment. Amino acid symbols are colored to represent standard Dayhoff groupings.
(A) Substitutions graded as C65; although most of these were scored 0.00 by SIFT, note that the last two fall at slightly variable positions and were scored as 0.01 by SIFT. “†” indicates that p.S2855R is the first substitution of the two-amino-acid substitution p.SV2855_2856RI.
(B) Substitutions scored as 0.00 by SIFT but as C55 or lower by Align-GVGD.
Sensitivity
To explore whether any of the individual studies affected the significance or magnitude of our summary OR estimates, we conducted leave-one-out tests of sensitivity (Table 7) in which each of the ten bona fide case-control studies was removed in turn (for this analysis, studies 9a and 9b were considered as separate studies; note also that there were no significant effects attributable to inclusion or exclusion of single case-only or control-only studies [data not shown]). Of our four main tests—T+SJ variants, the whole-gene rMS trend test, the carboxy-third rMS trend test, and the FAT+kinase+FATC rMS trend test—the analysis of T+SJ variants proved to be the most sensitive. For this test, 7/10 leave-one-out tests rejected the null with p < 0.05; two of the remaining had 0.05 ≤ p < 0.10, and one (exclusion of study 5) resulted in p = 0.178. The three missense-substitution trend tests were more robust, with 23/24 leave-one-out tests rejecting the null with p < 0.05 and the remaining test returning p = 0.06. Because of loss of power, removing a relatively large study could render the pooled result from the remaining studies nonsignificant even if there was little or no change in the OR point estimate. For the T+SJ tests, the leave-one-out OR point estimates were all between 0.77× and 1.18× of the overall OR point estimate. Exponentiating the ln(OR) regression coefficients obtained from analyses of the rMSs to convert them to OR space, we found that the resulting exponentiated coefficients were all between 0.95× and 1.45× of their respective complete data analyses. Finally, analysis of the FAT+kinase+FATC C65 rMS versus T+SJ comparison revealed that each of the four largest studies was required in order to obtain p < 0.05. Even for this analysis, the ratio of the OR estimated for these C65 rMSs from the logistic-regression trend coefficients to the OR estimated for T+SJ variants stayed above 0.75× of its value for all of the case-control studies combined.
Table 7.
Tests of Sensitivity
|
Test Scenario A | ||
|---|---|---|
| OR [CI] | p-Logistic | |
| All case-control studies | 2.32 [1.12–4.83] | 0.024 |
| Excluding study 1 (Fitzgerald et al.48)a | 2.93 [1.31–6.55] | 0.009 |
| Excluding study 2 (Teraoka et al.6) | 2.41 [1.16–5.02] | 0.019 |
| Excluding study 3 (Sommer et al.10) | 2.31 [1.11–4.80] | 0.025 |
| Excluding study 4 (Thorstenson et al.) | 2.14 [1.01–4.53] | 0.047 |
| Excluding study 5 (Renwick et al.14) | 1.78 [0.77–4.15] | 0.178 |
| Excluding study 6 (Hirsch et al.49) | 2.53 [1.18–5.40] | 0.017 |
| Excluding study 7 (Soukupova et al.50)a | 2.01 [0.95–4.24] | 0.066 |
| Excluding study 8 (kConFab/Regensburg) | 2.04 [0.93–4.46] | 0.076 |
| Excluding study 9a (IARC, European) | 2.61 [1.17–5.82] | 0.019 |
| Excluding study 9b (IARC, East Asian) | 2.74 [1.23–6.10] | 0.041 |
| Test Scenario B | ||
| Coefficientb | p-Trend | |
| All case-control studies | 0.1318 | 0.00350 |
| Excluding study 1 (Fitzgerald et al.48)a | NA | NA |
| Excluding study 2 (Teraoka et al.6) | 0.1237 | 0.00720 |
| Excluding study 3 (Sommer et al.10) | 0.1288 | 0.00490 |
| Excluding study 4 (Thorstenson et al.) | 0.1129 | 0.01630 |
| Excluding study 5 (Renwick et al.14) | 0.0896 | 0.05950 |
| Excluding study 6 (Hirsch et al.49) | 0.1416 | 0.00220 |
| Excluding study 7 (Soukupova et al.50)a | NA | NA |
| Excluding study 8 (kConFab/Regensburg) | 0.1151 | 0.03120 |
| Excluding study 9a (IARC, European) | 0.1966 | 0.00051 |
| Excluding study 9b (IARC, East Asian) | 0.1478 | 0.00180 |
| Test Scenario C | ||
| Coefficientb | p-Trend | |
| All case-control studies | 0.3082 | 0.00048 |
| Excluding study 1 (Fitzgerald et al.48)a | NA | NA |
| Excluding study 2 (Teraoka et al.6) | 0.2913 | 0.00082 |
| Excluding study 3 (Sommer et al.10) | 0.2953 | 0.00064 |
| Excluding study 4 (Thorstenson et al.) | 0.2694 | 0.00190 |
| Excluding study 5 (Renwick et al.14) | 0.2664 | 0.00370 |
| Excluding study 6 (Hirsch et al.49) | 0.3206 | 0.00050 |
| Excluding study 7 (Soukupova et al.50)a | NA | NA |
| Excluding study 8 (kConFab/Regensburg) | 0.2745 | 0.00410 |
| Excluding study 9a (IARC, European) | 0.5168 | 0.00170 |
| Excluding study 9b (IARC, East Asian) | 0.3202 | 0.00057 |
| Test Scenario D | ||
| Coefficientb | p-Trend | |
| All case-control studies | 0.4129 | 0.00220 |
| Excluding study 1 (Fitzgerald et al.48)a | NA | NA |
| Excluding study 2 (Teraoka et al.6) | 0.3978 | 0.00250 |
| Excluding study 3 (Sommer et al.10) | 0.3977 | 0.00230 |
| Excluding study 4 (Thorstenson et al.) | 0.3605 | 0.00480 |
| Excluding study 5 (Renwick et al.14) | 0.3673 | 0.00850 |
| Excluding study 6 (Hirsch et al.49) | 0.4305 | 0.00280 |
| Excluding study 7 (Soukupova et al.50)a | NA | NA |
| Excluding study 8 (kConFab/Regensburg) | 0.3758 | 0.00710 |
| Excluding study 9a (IARC, European) | 0.7865 | 0.04762 |
| Excluding study 9b (IARC, East Asian) | 0.4207 | 0.00250 |
| Test Scenario E | ||
| p-FETc,d | p-FETc,e | |
| All case-control studies | 0.0210 | 0.0187 |
| Excluding study 1 (Fitzgerald et al.48)a | 0.0384 | NA |
| Excluding study 2 (Teraoka et al.6) | 0.0218 | 0.0366 |
| Excluding study 3 (Sommer et al.10) | 0.0210 | 0.0187 |
| Excluding study 4 (Thorstenson et al.) | 0.0204 | 0.0337 |
| Excluding study 5 (Renwick et al.14) | 0.0357 | 0.0568 |
| Excluding study 6 (Hirsch et al.49) | 0.0226 | 0.0342 |
| Excluding study 7 (Soukupova et al.50)a | 0.0103 | NA |
| Excluding study 8 (kConFab/Regensburg) | 0.0413 | 0.0695 |
| Excluding study 9a (IARC, European) | 0.0845 | 0.0705 |
| Excluding study 9b (IARC, East Asian) | 0.0393 | 0.0662 |
Bold font is used to indicate leave-one-out analyses resulting in point estimates or trend coefficients with p > 0.05. Abbreviations are as follows: OR, odds ratio; CI, confidence interval; NA, not applicable.
Test scenario A: logistic regression ORs and p value for T+SJ.
Test scenario B: trend test on missense substitutions across the whole gene (excluding carriers of T+SJ variants and adjusting for study).
Test scenario C: trend test on missense substitutions after residue Ile1960 (excluding carriers of T+SJ variants and adjusting for study).
Test scenario D: trend test on missense substitutions in the FAT+Kinase+FATC domains (excluding carriers of T+SJ variants and adjusting for study).
Test scenario E: comparison between C65 rMSs in the FAT+Kinase+FATC domains versus T+SJ variants.
Study included in tests of T+SJ variants only.
ln(OR) regression coefficient.
Fisher's exact test.
All of the case-control studies were used.
After exclusion of studies 1 and 7, which used the PTT test and consequently had zero sensitivity for detection.
Discussion
Our meta-analysis of T+SJ variants in ATM is consistent with an OR for breast cancer of slightly above 2.0 and a frequency in controls of around 0.5%. Combined with a recent study of the “Mennonite” ATM founder mutation p.E1978X,77 there can be little doubt but that this class of ATM sequence variants confer increased risk of breast cancer. Our point estimate lies within the 95% confidence intervals of all of the bona fide case-control studies (data not shown). Thus, the perceived differences between studies that have led to controversy over the breast cancer risk associated with truncating variants in ATM can easily be attributed to stochastic sampling variation. However, because case individuals were typically young or had family history of breast cancer, even our summary ORs may be inflated in comparison to effects in the general population.
To our knowledge, our meta-analysis of rMSs in ATM is unique in the biomedical literature. The whole-gene rMS trend test across noncarriers and the seven grades of missense substitutions amounts to a test of a null hypothesis with three underlying components: rare missense substitutions in ATM have no role in breast cancer, the probability that such ATM missense substitutions are pathogenic is unrelated to the probability that they are evolutionarily deleterious, or the Align-GVGD grading of ATM missense substitutions does not predict evolutionary fitness. Rejection of this hypothesis with p = 0.0035 implies the alternative: rare missense substitutions in ATM are associated with breast cancer, the probability that such substitutions are pathogenic is related to the probability that they are deleterious, and the Align-GVGD grading predicts evolutionary fitness. Therefore, the p value obtained for the overall missense test for trend ought to be considered a fair measure of the strength of evidence that at least a subset of rare missense substitutions in ATM confer increased risk of breast cancer. This being the case, we note that, were ATM a candidate gene, evidence extracted from the case-control distribution of rMSs would complement evidence extracted from the case-control distribution of T+SJ variants to help establish the gene's status as a susceptibility gene.
In the whole-gene analysis across the seven grades of missense substitutions defined by Align-GVGD, there appears to be only a modest trend from C0 to C55 followed by a step function to much higher risk at C65. In the subanalysis of the carboxy-third of the protein, the data from C0 to C55 are more consistent with a trend toward increasing risk, but there again appears to be a step at C65. The degree to which the series of ORs resemble a step function rather than a log-linear trend does not weigh against the validity of the p value obtained from the test for trend. Moreover, although it might be tempting to report a p value for C65 versus noncarriers as a main result, such a p value would be invalid because it involves post hoc optimization over the observed data.78 If future analyses of rare missense-substitution case-control data from this or other susceptibility genes consistently show that ORs for the grade C65 are disproportionately high in comparison to the trend across the other grades, then we can modify the parameters of the test to better fit the previously observed data. Within the paradigm of the test for log-trend, such a change could be incorporated by assigning to the grades C0 to C65 row values that have been determined from regressions against already published data.
The two rMS positional analyses that we have conducted, e.g., over the carboxy-third of the protein and the more restrictive PFAM-defined FAT+kinase+FATC concatenation, are both subset analyses analogous to those routinely reported in more conventional molecular epidemiology studies. Thus, the risk estimates and p values obtained need to be treated with caution because of the effects of case and control ascertainment criteria, post hoc analysis, and hidden multiple testing. Still, the results obtained lead us to propose two hypotheses: (1) that rMSs conferring increased risk of breast cancer are more concentrated in the last third of the protein than elsewhere and, more tentatively (2) that a subset of these rMSs actually confer higher risk of breast cancer than do T+SJ variants on average. This second hypothesis resembles that proposed by Gatti et al., who argued that there should be a class of common dominant-negative missense substitutions in ATM that confer markedly increased risk of breast cancer but a less severe A-T phenotype.79 We hypothesize that the relatively high-risk missense substitutions that we have tentatively identified, typified by C65 missense substitutions falling in the FAT, kinase, and perhaps FATC domains, are very rare in the general population, whereas Gatti et al. proposed that they would be more common. We also note that recent results from the WECARE study virtually eliminate the possibility that any of the relatively common ATM missense substitutions individually confer more than very modestly increased risk of breast cancer.80
On the basis of our tests of sensitivity, the hypothesis that specific missense substitutions falling in the last one-third of ATM may confer greater risk of breast cancer than do T+SJ variants was the least robust of our principal findings. Nonetheless, this hypothesis enjoys two lines of experimental support. First, there is functional assay evidence that some missense substitutions and in-frame deletions falling in the FAT and kinase domains are biochemically dominant negative;81–85 this observation is a prerequisite for the hypothesis. Second, Spring et al. constructed mice that carry the three-amino-acid in-frame deletion p.SRI2556-2558del3, which corresponds to the pathogenic human allele ΔSRI. The allele encodes a moderately stable protein with biochemically dominant-negative features34,82 and is therefore more like a pathogenic missense substitution than like a pathogenic protein-truncating variant. The ATM+/− mice had little increase in tumor incidence, whereas the ΔSRI heterozygote mice had a notable increase in incidence (relative risk = 3.4, p = 0.004).82 Thus, one could argue that our result is a human-genetics confirmation of a published mouse-genetics result.
If the relatively high-risk for FAT, kinase, and perhaps FATC domain C65 missense substitutions is replicated in large, population-based studies, the results would pose an interesting clinical cancer genetics dilemma. One can immediately recognize that most truncating variants, and many variants at canonical GT-AG splice-junction dinucleotides, damage function and will be pathogenic. But, in contrast to BRCA1 and BRCA2, such variants in ATM do not by themselves confer enough risk to achieve clinical relevance.86 Nonetheless, our statistical inference is that C65 missense substitutions in these three domains may confer, on average, greater risk than do T+SJ variants and may, therefore, have greater clinical relevance to heterozygous carriers. However, in the absence of further characterization, missense substitutions are almost always considered unclassified variants. Hence, under current clinical guidelines, carriers of such substitutions would be counseled only on the basis of their family history, without modification with respect to their ATM genotype.87 If our hypothesis is confirmed, then it will become important to complement the bioinformatic and statistical inferences used here with pedigree-based genetic analysis and validated functional assays to reclassify a subset of these missense substitutions as likely or clearly pathogenic.87–89 In doing so, we should keep two points in mind. First, some evolutionarily conserved residues outside of the restrictively defined FAT, kinase, and FATC domains may also harbor clinically relevant missense substitutions. Second, we should expect heterogeneity of effect among missense substitutions that fall into specific Align-GVGD or SIFT score categories. Aside from the fact that these programs do not have perfect specificity, a simple reason that this should be so is that missense substitutions falling in this region of ATM can result in proteins that are quite stable, of intermediate stability, or very unstable.90 Evolutionarily deleterious missense substitutions that result in very unstable proteins would not be expected to have dominant-negative effects, whereas those that result in stable but functionally compromised proteins are more likely to have dominant-negative effects.
Several limitations should be considered for this study. Foremost among them is heterogeneity across the studies, including design (case-control, case-only, control and/or population sampling only), case-ascertainment criteria, and sensitivity of the mutation-screening technique employed. We handled the problem of study design by basing our primary analyses on the bona fide case-control studies and then adding in data from the case-only and control-only studies to show that their addition did not result in any substantial changes. For case-ascertainment criteria, we excluded studies that restricted breast cancer cases by treatment response or specific tumor characteristics in an effort to exclude selection criteria that might have biased toward (or away from) any specific genetic predisposition. An additional source of heterogeneity was the race and/or ethnicity distribution in the individual studies. For many of the studies, we know the fraction of subjects who were members of one or another ethnic group, but the published data were not usually detailed enough to allow us to ascribe individual sequence variants to subjects of specific ethnicity. Consequently, it was not possible to do a stratified analysis. The largest non-Northwest European groups were the African American cases and controls screened by Hirsch et al.49 and the East Asian cases and controls screened in the IARC study 9b. For the logistic regressions, each of these comprised a single study; consequently, the logistic-regression adjustment for study acted as a proxy for ethnicity. Finally, the effect of leaving these studies out is summarized in the tests of sensitivity presented in Table 7.
The analyses reported here have implications for any disease in which rare variants, especially missense substitutions of unknown function, are likely to play a role in susceptibility. These implications will be magnified as mutation screening of whole transcriptomes becomes economically feasible. To use the breast cancer analogy, recognition of high-risk genes, such as BRCA1 and BRCA2, would be accomplished easily by mutation screening of a limited number of cases. But recognition of the intermediate-risk genes, such as ATM, CHEK2, and PALB2, may be much more challenging. Because controls carry pathogenic sequence variants in these genes at substantial frequency, results from case-only mutation screening would be quite misleading. Moreover, because about one-half of the observations of rare ATM C25–C65 and T+SJ variants are of variants that occur only once in > 5000 individuals, mutation screening of a limited series of subjects followed by genotyping of cases and controls could miss a substantial fraction of variants of interest—hence the importance of case-control mutation screening as a method of addressing the problem of rare variants. Even so, at the ORs and frequencies that we have reported (Tables 4 and 6), 1350 of cases and of controls are required for the ability to detect evidence of risk with 80% power with the use of a model that combines assessment of rare missense substitutions with T+SJ variants, 2200 of each are required for detecting evidence of risk in a rare missense-substitution-only model, and 3800 of each are required for a T+SJ-only model. Analysis of rare missense substitutions along the lines of the strategy described here provides a gain in power relative to either analyses focused on T+SJ variants alone or analyses that include rare missense substitutions via stratification on frequency followed by a test of heterogeneity. However, the gain in power offered by bioinformatic grading of missense substitutions followed by a test for trend over the ordered grades will carry the price of creating properly curated sequence alignments of appropriate phylogenetic depth. Moreover, with multiple testing taken into account, the number of subjects needing to be screened will be daunting, even if only all of the genes in a particular biochemical pathway are evaluated.
Supplemental Data
Supplemental Data include two tables and an ATM protein multiple sequence alignment and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Align-GVGD algorithm, http://agvgd.iarc.fr/agvgd_input.php
Align-GVGD alignments library, http://agvgd.iarc.fr/alignments.php
ATM domain definitions, http://www.ebi.ac.uk/interpro/ISpy?mode=single&ac=Q13315
EMBASE, http://www.embase.com/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
MaxEntScan, http://genes.mit.edu/burgelab/maxent/
MCoffee, http://www.igs.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi
National Institute of Environmental Health Sciences (NIEHS) SNP database, http://egp.gs.washington.edu/data/ATM/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Web of Knowledge, http://www.isiwebofknowledge.com/
Acknowledgments
We would like to thank David Goldgar, Douglas Easton, and KumKum Khanna for helpful comments on the manuscript; Paul Pharoah for ancestry-informative marker analysis of the National Institute of Environmental Health Sciences (NIEHS) data; Paul B. Samollow, Laure Bernard, Michael Schubert, and R. Andrew Cameron for model-organism tissue samples; Annegien Broeks, Ariel Hirsch, Yvonne Thorstenson, and Pilar Carvallo for clarifying data from their ATM mutation-screening work; Heather Thorne, Eveline Niedermayr, the Kathleen Cuningham Foundation Consortium for Research into Familial Aspects of Breast Cancer (kConFab) research nurses and staff, and the heads and staff of the Family Cancer Clinics and the Clinical Follow Up Study (CFUS) for their contributions to kConFab; and the families who have contributed to kConFab and the Breast Cancer Family Registries (BCFR). D.B. received an International Agency for Research on Cancer (IARC) Special Training Award. G.C.T., D.W., and P.W. are supported by National Health and Medical Research Council (NHMRC) research fellowships. This work was supported by National Institutes of Health (NIH) grants RO1-CA121245 and RO1-CA100352. The BCFR was funded under RFA-CA-06-503 and through cooperative agreements with BCFR members, including Cancer Care Ontario (U01 CA69467), the Northern California Cancer Center (U01 CA69417), and the University of Melbourne (U01 CA69638). kConFab and the CFUS are supported by grants from the National Breast Cancer Foundation, the NHMRC (including grants 145684, 288704, and 454508), and multiple state-based cancer foundations. The American Cancer Study (ACS) was funded by NHMRC grant 199600. This work was also funded by BayGene. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute (NCI) or any of the collaborating centers in the BCFR, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government or the BCFR.
References
- 1.Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 2.Uziel T., Savitsky K., Platzer M., Ziv Y., Helbitz T., Nehls M., Boehm T., Rosenthal A., Shiloh Y., Rotman G. Genomic Organization of the ATM gene. Genomics. 1996;33:317–320. doi: 10.1006/geno.1996.0201. [DOI] [PubMed] [Google Scholar]
- 3.Lee J.H., Paull T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- 4.Swift M., Reitnauer P.J., Morrell D., Chase C.L. Breast and other cancers in families with ataxia-telangiectasia. N. Engl. J. Med. 1987;316:1289–1294. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- 5.Izatt L., Greenman J., Hodgson S., Ellis D., Watts S., Scott G., Jacobs C., Liebmann R., Zvelebil M.J., Mathew C. Identification of germline missense mutations and rare allelic variants in the ATM gene in early-onset breast cancer. Genes Chromosomes Cancer. 1999;26:286–294. [PubMed] [Google Scholar]
- 6.Teraoka S.N., Malone K.E., Doody D.R., Suter N.M., Ostrander E.A., Daling J.R., Concannon P. Increased frequency of ATM mutations in breast carcinoma patients with early onset disease and positive family history. Cancer. 2001;92:479–487. doi: 10.1002/1097-0142(20010801)92:3<479::aid-cncr1346>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 7.Dork T., Bendix R., Bremer M., Rades D., Klopper K., Nicke M., Skawran B., Hector A., Yamini P., Steinmann D. Spectrum of ATM gene mutations in a hospital-based series of unselected breast cancer patients. Cancer Res. 2001;61:7608–7615. [PubMed] [Google Scholar]
- 8.Atencio D.P., Iannuzzi C.M., Green S., Stock R.G., Bernstein J.L., Rosenstein B.S. Screening breast cancer patients for ATM mutations and polymorphisms by using denaturing high-performance liquid chromatography. Environ. Mol. Mutagen. 2001;38:200–208. doi: 10.1002/em.1072. [DOI] [PubMed] [Google Scholar]
- 9.Maillet P., Bonnefoi H., Vaudan-Vutskits G., Pajk B., Cufer T., Foulkes W.D., Chappuis P.O., Sappino A.P. Constitutional alterations of the ATM gene in early onset sporadic breast cancer. J. Med. Genet. 2002;39:751–753. doi: 10.1136/jmg.39.10.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sommer S.S., Jiang Z., Feng J., Buzin C.H., Zheng J., Longmate J., Jung M., Moulds J., Dritschilo A. ATM missense mutations are frequent in patients with breast cancer. Cancer Genet. Cytogenet. 2003;145:115–120. doi: 10.1016/s0165-4608(03)00119-5. [DOI] [PubMed] [Google Scholar]
- 11.Angele S., Romestaing P., Moullan N., Vuillaume M., Chapot B., Friesen M., Jongmans W., Cox D.G., Pisani P., Gerard J.P. ATM haplotypes and cellular response to DNA damage: association with breast cancer risk and clinical radiosensitivity. Cancer Res. 2003;63:8717–8725. [PubMed] [Google Scholar]
- 12.Thorstenson Y.R., Roxas A., Kroiss R., Jenkins M.A., Yu K.M., Bachrich T., Muhr D., Wayne T.L., Chu G., Davis R.W. Contributions of ATM mutations to familial breast and ovarian cancer. Cancer Res. 2003;63:3325–3333. [PubMed] [Google Scholar]
- 13.Buchholz T.A., Weil M.M., Ashorn C.L., Strom E.A., Sigurdson A., Bondy M., Chakraborty R., Cox J.D., McNeese M.D., Story M.D. A Ser49Cys variant in the ataxia telangiectasia, mutated, gene that is more common in patients with breast carcinoma compared with population controls. Cancer. 2004;100:1345–1351. doi: 10.1002/cncr.20133. [DOI] [PubMed] [Google Scholar]
- 14.Renwick A., Thompson D., Seal S., Kelly P., Chagtai T., Ahmed M., North B., Jayatilake H., Barfoot R., Spanova K. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 15.Thompson D., Easton D. The genetic epidemiology of breast cancer genes. J. Mammary Gland Biol. Neoplasia. 2004;9:221–236. doi: 10.1023/B:JOMG.0000048770.90334.3b. [DOI] [PubMed] [Google Scholar]
- 16.Easton D.F., Pooley K.A., Dunning A.M., Pharoah P.D., Thompson D., Ballinger D.G., Struewing J.P., Morrison J., Field H., Luben R. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavtigian S.V., Byrnes G.B., Goldgar D.E., Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum. Mutat. 2008;29:1342–1354. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Offit K., Gilad S., Paglin S., Kolachana P., Roisman L.C., Nafa K., Yeugelewitz V., Gonzales M., Robson M., McDermott D. Rare variants of ATM and risk for Hodgkin's disease and radiation-associated breast cancers. Clin. Cancer Res. 2002;8:3813–3819. [PubMed] [Google Scholar]
- 19.Vorechovsky I., Luo L., Lindblom A., Negrini M., Webster A.D., Croce C.M., Hammarstrom L. ATM mutations in cancer families. Cancer Res. 1996;56:4130–4133. [PubMed] [Google Scholar]
- 20.Heikkinen K., Rapakko K., Karppinen S.M., Erkko H., Nieminen P., Winqvist R. Association of common ATM polymorphism with bilateral breast cancer. Int. J. Cancer. 2005;116:69–72. doi: 10.1002/ijc.20996. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez C., Valles H., Causse A., Johannsdottir V., Eliaou J.F., Theillet C. Involvement of ATM missense variants and mutations in a series of unselected breast cancer cases. Genes Chromosomes Cancer. 2002;33:141–149. doi: 10.1002/gcc.1222. [DOI] [PubMed] [Google Scholar]
- 22.Shafman T.D., Levitz S., Nixon A.J., Gibans L.A., Nichols K.E., Bell D.W., Ishioka C., Isselbacher K.J., Gelman R., Garber J. Prevalence of germline truncating mutations in ATM in women with a second breast cancer after radiation therapy for a contralateral tumor. Genes Chromosomes Cancer. 2000;27:124–129. [PubMed] [Google Scholar]
- 23.Shayeghi M., Seal S., Regan J., Collins N., Barfoot R., Rahman N., Ashton A., Moohan M., Wooster R., Owen R. Heterozygosity for mutations in the ataxia telangiectasia gene is not a major cause of radiotherapy complications in breast cancer patients. Br. J. Cancer. 1998;78:922–927. doi: 10.1038/bjc.1998.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramsay J., Birrell G., Lavin M. Testing for mutations of the ataxia telangiectasia gene in radiosensitive breast cancer patients. Radiother. Oncol. 1998;47:125–128. doi: 10.1016/s0167-8140(98)00014-0. [DOI] [PubMed] [Google Scholar]
- 25.Oppitz U., Bernthaler U., Schindler D., Sobeck A., Hoehn H., Platzer M., Rosenthal A., Flentje M. Sequence analysis of the ATM gene in 20 patients with RTOG grade 3 or 4 acute and/or late tissue radiation side effects. Int. J. Radiat. Oncol. Biol. Phys. 1999;44:981–988. doi: 10.1016/s0360-3016(99)00108-x. [DOI] [PubMed] [Google Scholar]
- 26.Broeks A., Urbanus J.H., Floore A.N., Dahler E.C., Klijn J.G., Rutgers E.J., Devilee P., Russell N.S., van Leeuwen F.E., van 't Veer L.J. ATM-heterozygous germline mutations contribute to breast cancer-susceptibility. Am. J. Hum. Genet. 2000;66:494–500. doi: 10.1086/302746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iannuzzi C.M., Atencio D.P., Green S., Stock R.G., Rosenstein B.S. ATM mutations in female breast cancer patients predict for an increase in radiation-induced late effects. Int. J. Radiat. Oncol. Biol. Phys. 2002;52:606–613. doi: 10.1016/s0360-3016(01)02684-0. [DOI] [PubMed] [Google Scholar]
- 28.Bernstein J.L., Teraoka S., Haile R.W., Borresen-Dale A.L., Rosenstein B.S., Gatti R.A., Diep A.T., Jansen L., Atencio D.P., Olsen J.H. Designing and implementing quality control for multi-center screening of mutations in the ATM gene among women with breast cancer. Hum. Mutat. 2003;21:542–550. doi: 10.1002/humu.10206. [DOI] [PubMed] [Google Scholar]
- 29.Broeks A., Braaf L.M., Huseinovic A., Nooijen A., Urbanus J., Hogervorst F.B., Schmidt M.K., Klijn J.G., Russell N.S., Van Leeuwen F.E. Identification of women with an increased risk of developing radiation-induced breast cancer: a case only study. Breast Cancer Res. 2007;9:R26. doi: 10.1186/bcr1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng P.C., Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Wallace I.M., O'Sullivan O., Higgins D.G., Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34:1692–1699. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 34.Spring K., Cross S., Li C., Watters D., Ben-Senior L., Waring P., Ahangari F., Lu S.L., Chen P., Misko I. Atm knock-in mice harboring an in-frame deletion corresponding to the human ATM 7636del9 common mutation exhibit a variant phenotype. Cancer Res. 2001;61:4561–4568. [PubMed] [Google Scholar]
- 35.Mann G.J., Thorne H., Balleine R.L., Butow P.N., Clarke C.L., Edkins E., Evans G.M., Fereday S., Haan E., Gattas M. Analysis of cancer risk and BRCA1 and BRCA2 mutation prevalence in the kConFab familial breast cancer resource. Breast Cancer Res. 2006;8:R12. doi: 10.1186/bcr1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans D.G., Eccles D.M., Rahman N., Young K., Bulman M., Amir E., Shenton A., Howell A., Lalloo F. A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J. Med. Genet. 2004;41:474–480. doi: 10.1136/jmg.2003.017996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whiteman D.C., Sadeghi S., Pandeya N., Smithers B.M., Gotley D.C., Bain C.J., Webb P.M., Green A.C. Combined effects of obesity, acid reflux and smoking on the risk of adenocarcinomas of the oesophagus. Gut. 2008;57:173–180. doi: 10.1136/gut.2007.131375. [DOI] [PubMed] [Google Scholar]
- 38.John E.M., Hopper J.L., Beck J.C., Knight J.A., Neuhausen S.L., Senie R.T., Ziogas A., Andrulis I.L., Anton-Culver H., Boyd N. The Breast Cancer Family Registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6:R375–R389. doi: 10.1186/bcr801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sangrajrang S., Schmezer P., Burkholder I., Boffetta P., Brennan P., Woelfelschneider A., Bartsch H., Wiangnon S., Cheisilpa A., Popanda O. The XRCC3 Thr241Met polymorphism and breast cancer risk: a case-control study in a Thai population. Biomarkers. 2007;12:523–532. doi: 10.1080/13547500701395602. [DOI] [PubMed] [Google Scholar]
- 40.Reed G.H., Wittwer C.T. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin. Chem. 2004;50:1748–1754. doi: 10.1373/clinchem.2003.029751. [DOI] [PubMed] [Google Scholar]
- 41.Takano E.A., Mitchell G., Fox S.B., Dobrovic A. Rapid detection of carriers with BRCA1 and BRCA2 mutations using high resolution melting analysis. BMC Cancer. 2008;8:59. doi: 10.1186/1471-2407-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen-Dumont T., Calvez-Kelm F.L., Forey N., McKay-Chopin S., Garritano S., Gioia-Patricola L., De Silva D., Weigel R., Sangrajrang S., Lesueur F. Description and validation of high-throughput simultaneous genotyping and mutation scanning by high-resolution melting curve analysis. Hum. Mutat. 2009;30:884–890. doi: 10.1002/humu.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steck P.A., Pershouse M.A., Jasser S.A., Yung W.K., Lin H., Ligon A.H., Langford L.A., Baumgard M.L., Hattier T., Davis T. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 44.Tavtigian S.V., Oliphant A., Shattuck-Eidens D., Bartel P.L., Thomas A., Frank T.S., Pruss D., Skolnick M.H. Genomic organization, functional analysis, and mutation screening of BRCA1 and BRCA2. In: Fortner J.G., Sharp P.A., editors. Accomplishments in Cancer Research 1996. Lippincott-Raven; New York, USA: 1997. pp. 189–204. [Google Scholar]
- 45.Voegele C., Tavtigian S.V., de Silva D., Cuber S., Thomas A., Le Calvez-Kelm F. A Laboratory Information Management System (LIMS) for a high throughput genetic platform aimed at candidate gene mutation screening. Bioinformatics. 2007;23:2504–2506. doi: 10.1093/bioinformatics/btm365. [DOI] [PubMed] [Google Scholar]
- 46.Guthery S.L., Salisbury B.A., Pungliya M.S., Stephens J.C., Bamshad M. The structure of common genetic variation in United States populations. Am. J. Hum. Genet. 2007;81:1221–1231. doi: 10.1086/522239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tavtigian S.V., Le Calvez-Kelm F. Molecular Diagnostics: Methods and Limitations. In: Isaacs C., Rebbeck T.R., editors. Hereditary Breast Cancer. Informa Healthcare; New York, USA: 2007. pp. 179–206. [Google Scholar]
- 48.FitzGerald M.G., Bean J.M., Hegde S.R., Unsal H., MacDonald D.J., Harkin D.P., Finkelstein D.M., Isselbacher K.J., Haber D.A. Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat. Genet. 1997;15:307–310. doi: 10.1038/ng0397-307. [DOI] [PubMed] [Google Scholar]
- 49.Hirsch A.E., Atencio D.P., Rosenstein B.S. Screening for ATM sequence alterations in African-American women diagnosed with breast cancer. Breast Cancer Res. Treat. 2008;107:139–144. doi: 10.1007/s10549-007-9531-x. [DOI] [PubMed] [Google Scholar]
- 50.Soukupova J., Dundr P., Kleibl Z., Pohlreich P. Contribution of mutations in ATM to breast cancer development in the Czech population. Oncol. Rep. 2008;19:1505–1510. [PubMed] [Google Scholar]
- 51.Vorechovsky I., Rasio D., Luo L., Monaco C., Hammarstrom L., Webster A.D., Zaloudik J., Barbanti-Brodani G., James M., Russo G. The ATM gene and susceptibility to breast cancer: analysis of 38 breast tumors reveals no evidence for mutation. Cancer Res. 1996;56:2726–2732. [PubMed] [Google Scholar]
- 52.Chen J., Birkholtz G.G., Lindblom P., Rubio C., Lindblom A. The role of ataxia-telangiectasia heterozygotes in familial breast cancer. Cancer Res. 1998;58:1376–1379. [PubMed] [Google Scholar]
- 53.Bebb D.G., Yu Z., Chen J., Telatar M., Gelmon K., Phillips N., Gatti R.A., Glickman B.W. Absence of mutations in the ATM gene in forty-seven cases of sporadic breast cancer. Br. J. Cancer. 1999;80:1979–1981. doi: 10.1038/sj.bjc.6690630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drumea K.C., Levine E., Bernstein J., Shank B., Green S., Kaplan E., Mandell L., Cropley J., Obropta J., Braccia I. ATM heterozygosity and breast cancer: screening of 37 breast cancer patients for ATM mutations using a non-isotopic RNase cleavage-based assay. Breast Cancer Res. Treat. 2000;61:79–85. doi: 10.1023/a:1006463730337. [DOI] [PubMed] [Google Scholar]
- 55.Thorstenson Y.R., Shen P., Tusher V.G., Wayne T.L., Davis R.W., Chu G., Oefner P.J. Global analysis of ATM polymorphism reveals significant functional constraint. Am. J. Hum. Genet. 2001;69:396–412. doi: 10.1086/321296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Livingston R.J., Von Niederhausern A., Jegga A.G., Crawford D.C., Carlson C.S., Rieder M.J., Gowrisankar S., Aronow B.J., Weiss R.B., Nickerson D.A. Pattern of sequence variation across 213 environmental response genes. Genome Res. 2004;14:1821–1831. doi: 10.1101/gr.2730004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho A.Y., Fan G., Atencio D.P., Green S., Formenti S.C., Haffty B.G., Iyengar P., Bernstein J.L., Stock R.G., Cesaretti J.A. Possession of ATM sequence variants as predictor for late normal tissue responses in breast cancer patients treated with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2007;69:677–684. doi: 10.1016/j.ijrobp.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 58.Broeks A., Braaf L.M., Huseinovic A., Schmidt M.K., Russell N.S., van Leeuwen F.E., Hogervorst F.B., Van 't Veer L.J. The spectrum of ATM missense variants and their contribution to contralateral breast cancer. Breast Cancer Res. Treat. 2008;107:243–248. doi: 10.1007/s10549-007-9543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brunet J., Gutierrez-Enriquez S., Torres A., Berez V., Sanjose S., Galceran J., Izquierdo A., Menendez J.A., Guma J., Borras J. ATM germline mutations in Spanish early-onset breast cancer patients negative for BRCA1/BRCA2 mutations. Clin. Genet. 2008;73:465–473. doi: 10.1111/j.1399-0004.2008.00987.x. [DOI] [PubMed] [Google Scholar]
- 60.Tapia T., Sanchez A., Vallejos M., Alvarez C., Moraga M., Smalley S., Camus M., Alvarez M., Carvallo P. ATM allelic variants associated to hereditary breast cancer in 94 Chilean women: susceptibility or ethnic influences? Breast Cancer Res. Treat. 2008;107:281–288. doi: 10.1007/s10549-007-9544-5. [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez-Hormazabal P., Bravo T., Blanco R., Valenzuela C.Y., Gomez F., Waugh E., Peralta O., Ortuzar W., Reyes J.M., Jara L. Association of common ATM variants with familial breast cancer in a South American population. BMC Cancer. 2008;8:117. doi: 10.1186/1471-2407-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seal S., Thompson D., Renwick A., Elliott A., Kelly P., Barfoot R., Chagtai T., Jayatilake H., Ahmed M., Spanova K. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 63.Rahman N., Seal S., Thompson D., Kelly P., Renwick A., Elliott A., Reid S., Spanova K., Barfoot R., Chagtai T. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2006;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sandilands A., Terron-Kwiatkowski A., Hull P.R., O'Regan G.M., Clayton T.H., Watson R.M., Carrick T., Evans A.T., Liao H., Zhao Y. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat. Genet. 2007;39:650–654. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 65.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 67.Lin C.Y., Strom A., Vega V.B., Kong S.L., Yeo A.L., Thomsen J.S., Chan W.C., Doray B., Bangarusamy D.K., Ramasamy A. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome Biol. 2004;5:R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tavtigian S.V., Deffenbaugh A.M., Yin L., Judkins T., Scholl T., Samollow P.B., de Silva D., Zharkikh A., Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stone E.A., Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tavtigian S.V., Greenblatt M.S., Lesueur F., Byrnes G.B. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sodha N., Mantoni T.S., Tavtigian S.V., Eeles R., Garrett M.D. Rare germ line CHEK2 variants identified in breast cancer families encode proteins that show impaired activation. Cancer Res. 2006;66:8966–8970. doi: 10.1158/0008-5472.CAN-06-1990. [DOI] [PubMed] [Google Scholar]
- 74.Greenblatt M.S., Beaudet J.G., Gump J.R., Godin K.S., Trombley L., Koh J., Bond J.P. Detailed computational study of p53 and p16: using evolutionary sequence analysis and disease-associated mutations to predict the functional consequences of allelic variants. Oncogene. 2003;22:1150–1163. doi: 10.1038/sj.onc.1206101. [DOI] [PubMed] [Google Scholar]
- 75.Becker-Catania S.G., Chen G., Hwang M.J., Wang Z., Sun X., Sanal O., Bernatowska-Matuszkiewicz E., Chessa L., Lee E.Y., Gatti R.A. Ataxia-telangiectasia: phenotype/genotype studies of ATM protein expression, mutations, and radiosensitivity. Mol. Genet. Metab. 2000;70:122–133. doi: 10.1006/mgme.2000.2998. [DOI] [PubMed] [Google Scholar]
- 76.Jiang X., Sun Y., Chen S., Roy K., Price B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006;281:15741–15746. doi: 10.1074/jbc.M513172200. [DOI] [PubMed] [Google Scholar]
- 77.Bogdanova N., Cybulski C., Bermisheva M., Datsyuk I., Yamini P., Hillemanns P., Antonenkova N.N., Khusnutdinova E., Lubinski J., Dork T. A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer. Breast Cancer Res. Treat. 2009 doi: 10.1007/s10549-008-0189-9. in press. [DOI] [PubMed] [Google Scholar]
- 78.Altman D.G. Categorising continuous variables. Br. J. Cancer. 1991;64:975. doi: 10.1038/bjc.1991.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gatti R.A., Tward A., Concannon P. Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol. Genet. Metab. 1999;68:419–423. doi: 10.1006/mgme.1999.2942. [DOI] [PubMed] [Google Scholar]
- 80.Concannon P., Haile R.W., Borresen-Dale A.L., Rosenstein B.S., Gatti R.A., Teraoka S.N., Diep T.A., Jansen L., Atencio D.P., Langholz B. Variants in the ATM gene associated with a reduced risk of contralateral breast cancer. Cancer Res. 2008;68:6486–6491. doi: 10.1158/0008-5472.CAN-08-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scott S.P., Bendix R., Chen P., Clark R., Dork T., Lavin M.F. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc. Natl. Acad. Sci. USA. 2002;99:925–930. doi: 10.1073/pnas.012329699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spring K., Ahangari F., Scott S.P., Waring P., Purdie D.M., Chen P.C., Hourigan K., Ramsay J., McKinnon P.J., Swift M. Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nat. Genet. 2002;32:185–190. doi: 10.1038/ng958. [DOI] [PubMed] [Google Scholar]
- 83.Chenevix-Trench G., Spurdle A.B., Gatei M., Kelly H., Marsh A., Chen X., Donn K., Cummings M., Nyholt D., Jenkins M.A. Dominant negative ATM mutations in breast cancer families. J. Natl. Cancer Inst. 2002;94:205–215. doi: 10.1093/jnci/94.3.205. [DOI] [PubMed] [Google Scholar]
- 84.Waddell N., Jonnalagadda J., Marsh A., Grist S., Jenkins M., Hobson K., Taylor M., Lindeman G.J., Tavtigian S.V., Suthers G. Characterization of the breast cancer associated ATM 7271T>G (V2424G) mutation by gene expression profiling. Genes Chromosomes Cancer. 2006;45:1169–1181. doi: 10.1002/gcc.20381. [DOI] [PubMed] [Google Scholar]
- 85.Pylkas K., Tommiska J., Syrjakoski K., Kere J., Gatei M., Waddell N., Allinen M., Karppinen S.M., Rapakko K., Kaariainen H. Evaluation of the role of Finnish ataxia-telangiectasia mutations in hereditary predisposition to breast cancer. Carcinogenesis. 2007;28:1040–1045. doi: 10.1093/carcin/bgl237. [DOI] [PubMed] [Google Scholar]
- 86.Stratton M.R., Rahman N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 87.Plon S.E., Eccles D.M., Easton D., Foulkes W.D., Genuardi M., Greenblatt M.S., Hogervorst F.B., Hoogerbrugge N., Spurdle A.B., Tavtigian S.V. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goldgar D.E., Easton D.F., Byrnes G.B., Spurdle A.B., Iversen E.S., Greenblatt M.S. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008;29:1265–1272. doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Couch F.J., Rasmussen L.J., Hofstra R., Monteiro A.N., Greenblatt M.S., de Wind N. Assessment of functional effects of unclassified genetic variants. Hum. Mutat. 2008;29:1314–1326. doi: 10.1002/humu.20899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mitui M., Nahas S.A., Du L.T., Yang Z., Lai C.H., Nakamura K., Arroyo S., Scott S., Purayidom A., Concannon P. Functional and computational assessment of missense variants in the ataxia-telangiectasia mutated (ATM) gene: mutations with increased cancer risk. Hum. Mutat. 2009;30:12–21. doi: 10.1002/humu.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fernandes N., Sun Y., Chen S., Paul P., Shaw R.J., Cantley L.C., Price B.D. DNA damage-induced association of ATM with its target proteins requires a protein interaction domain in the N terminus of ATM. J. Biol. Chem. 2005;280:15158–15164. doi: 10.1074/jbc.M412065200. [DOI] [PubMed] [Google Scholar]
- 92.Lim D.S., Kirsch D.G., Canman C.E., Ahn J.H., Ziv Y., Newman L.S., Darnell R.B., Shiloh Y., Kastan M.B. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc. Natl. Acad. Sci. USA. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shafman T., Khanna K.K., Kedar P., Spring K., Kozlov S., Yen T., Hobson K., Gatei M., Zhang N., Watters D. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 94.Khanna K.K., Keating K.E., Kozlov S., Scott S., Gatei M., Hobson K., Taya Y., Gabrielli B., Chan D., Lees-Miller S.P. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat. Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.