

Abstract
Deficiency of cartilage-associated protein (CRTAP) or prolyl 3-hydroxylase 1(P3H1) has been reported in autosomal-recessive lethal or severe osteogenesis imperfecta (OI). CRTAP, P3H1, and cyclophilin B (CyPB) form an intracellular collagen-modifying complex that 3-hydroxylates proline at position 986 (P986) in the α1 chains of collagen type I. This 3-prolyl hydroxylation is decreased in patients with CRTAP and P3H1 deficiency. It was suspected that mutations in the PPIB gene encoding CyPB would also cause OI with decreased collagen 3-prolyl hydroxylation. To our knowledge we present the first two families with recessive OI caused by PPIB gene mutations. The clinical phenotype is compatible with OI Sillence type II-B/III as seen with COL1A1/2, CRTAP, and LEPRE1 mutations. The percentage of 3-hydroxylated P986 residues in patients with PPIB mutations is decreased in comparison to normal, but it is higher than in patients with CRTAP and LEPRE1 mutations. This result and the fact that CyPB is demonstrable independent of CRTAP and P3H1, along with reported decreased 3-prolyl hydroxylation due to deficiency of CRTAP lacking the catalytic hydroxylation domain and the known function of CyPB as a cis-trans isomerase, suggest that recessive OI is caused by a dysfunctional P3H1/CRTAP/CyPB complex rather than by the lack of 3-prolyl hydroxylation of a single proline residue in the α1 chains of collagen type I.
Main Text
Collagen type I is the major protein component of the extracellular matrix of bone and skin.1 It is a trimeric molecule in which each chain consists of repeating Gly-X-Y triplets in which proline and hydroxyproline (4Hyp) usually occupy the X and Y positions, respectively. Collagen type I is first synthesized as procollagen type I, consisting of two proα1(I) chains and one proα2(I) chain containing N- and C-terminal propeptides.2 After correct alignment of these three chains, propagation of triple helical folding in the C-to-N direction occurs in the rough endoplasmic reticulum (rER) in a zipper-like fashion3 during post-translational modification by specific rER proteins.2 In the literature, two important post-translational modification systems of collagen type I are known: (i) hydroxylation of multiple proline and lysine residues by, respectively, prolyl 4-hydroxylase, providing the triple helix with thermal stability, and lysyl-hydroxylase, providing attachment sites for carbohydrate units as well as stability of intermolecular cross links, and (ii) 3-hydroxylation of a single residue, proline at position 986 (P986), in the α1 chain by the rER protein complex consisting of P3H1(MIM 610339), CRTAP (MIM 605497), and CyPB (MIM 123841).1
Osteogenesis imperfecta (OI [MIM 166200, 166210, 259420, and 166220]) is an inherited connective-tissue disorder characterized clinically by bone fragility and increased susceptibility to fractures. Autosomal-dominant OI is usually caused by COL1A1 (MIM 120150) and COL1A2 (MIM 120160) mutations that disturb the primary structure of collagen type I and result in delayed triple helical folding and post-translational overmodification. CRTAP4 and LEPRE15 mutations, encoding CRTAP and P3H1, respectively, cause autosomal-recessive lethal or severe OI characterized by decreased 3-prolyl hydroxylation of P986 in the α1 chain of collagen type I and by post-translational overmodification.1 The function of 3-hydroxyproline in the α1 chains of collagen type I is not quite clear, but it has been hypothesized that 3-hydroxyproline takes part in protein-collagen interactions required for bone formation.6 In vitro studies have shown that P3H1 is responsible for prolyl 3-hydroxylase activity because the presence of CRTAP and CyPB is not required for full 3-prolyl hydroxylation activity.7 CRTAP and P3H1 share the same N terminal domain, but CRTAP lacks the catalytic-hydroxylation domain. Remarkably, CRTAP mutations appear to cause decreased 3-prolyl hydroxylation as well. Therefore, it was hypothesized that deficiency in collagen 3-Hyp might simply be a marker for the “dysfunctional” complex rather than an actual cause of lethal or severe OI.8 Also, the role of the third member CyPB in this 3-prolyl hydroxylation complex remained obscure. CyPB is a 21 kDA protein encoded by the PPIB gene9 and is known to belong to the cyclophilins (Cyps), a conserved class of intracellular and/or secreted proteins originally identified as cellular binding proteins for the immunosuppressive drug cyclosporin A (CsA).10 The Cyps are a family of peptidyl-prolyl cis-trans isomerases (PPIases), which catalyze the cis-trans isomerisation of peptide bonds. CyPB has a signal sequence directing it to the rER11, but it is also secreted extracellularly and appears to play a role in inflammation, viral infection, and cancer.10 Previous studies showed that CyPB directly interacts with procollagen, which is necessary because procollagen contains many cis-conformers. These have to be converted to trans-conformers because only trans peptide bonds can be incorporated into the triple collagen helix.12 CyPB is also involved in procollagen export and secretion along with HSP47, a collagen-binding heat-shock protein.11 It was assumed that, because of coexistence in the complex with P3H1 and CRTAP, a CyPB deficiency would also result in decreased 3-prolyl hydroxylation, post-translational overmodification, and autosomal-recessive OI,8 suggesting PPIB as an important candidate gene for OI.13,14
Therefore, we performed sequence analysis of the PPIB gene in four affected individuals from two families in whom no COL1A1/2, CRTAP, or LEPRE1 mutations were detected. The procedures followed were in accordance with institutional and national standards on human experimentation. Appropriate informed consent was obtained from the families. Proband 1 from family 1 (P1-1) (Figure 1A) was delivered after termination of pregnancy at 22 weeks and 1 day of gestation. She was the second child of nonconsanguineous North European parents. During pregnancy, the diagnosis OI was suspected on the basis of advanced ultrasounds. Radiographs and an autopsy showed an absence of rib fractures and the presence of shortened, bowed, and fractured long bones without evident rhizomelia, consistent with a diagnosis of OI type II-B. Weight and head circumference were normal for the gestational age, and no other abnormalities were noted. Bone histology was indicative of OI. Overmodification of collagen type I in fibroblasts was evident on electrophoresis.
Figure 1.

Radiological Features of Affected Probands from Families 1 and 2
(A) Fetal anteroposterior radiographs of proband 1 from family 1 at 21 + 2 weeks of gestation show the following: normal skull mineralization for gestational age; slender ribs without fractures; incomplete ossification of T5 and T12; somewhat irregular proximal metaphyses of the humeri, radii, and ulnae; and bowing of the ulnae. Bowed femora with fractures and some loss of modeling and bowed tibiae and fibula, possibly with fractures, are apparent. The radiological features are compatible with Sillence OI type II-B/III.
(B and C) Radiographs of proband 2 from family 2 show normal mineralization of the skull, fractures of both humeri and radii and the left ulna with callus formation and some loss of modeling of the humeri. Discontinuously beaded ribs and a small, bell-shaped thorax are noted. Multiple fractures with callus formation of femora, tibiae, and fibula exist with moderate loss of modeling of the femora. The radiological features are compatible with Sillence OI type II-B/III.
Proband 2 from family 1 (P1-2) was delivered after termination of pregnancy at 16 weeks and 1 day of gestation (Figure S1A). During pregnancy the diagnosis OI was made on the basis of advanced ultrasounds and overmodification of collagen type I in chorionic villi cells. Radiographs and autopsy showed osteopenia, no apparent rib fractures, and short long bones with fractures but without evident rhizomelia. Weight and head circumference were normal for the gestational age, and no other abnormalities were noted. The histology was indicative of OI. A diagnosis of OI type II-B was made.
Proband 1 from family 2 (P2-1) (Figures S1B–S1H) was delivered at 38 weeks of gestation by Caesarian section to consanguineous Pakistani parents (first cousins). Birth weight was 2.89 kg (p25). Multiple long-bone fractures had been detected at 20 weeks gestation on an ultrasound scan and were evident at birth. A large head with large anterior fontanelle was noted in combination with gray colored sclera typical of severe OI; flexed and abducted hips; and short, bowed femurs with anterior bowing of the tibiae. Hypermobility of the joints, especially the hip and finger joints, was seen. Based on clinical features and X-ray findings, P2-1 was diagnosed with OI type III. Treatment with 0.5 mg/kg/day intravenous Pamidronate for 3 days every 6 weeks was started at age 2 weeks. Gross motor development was delayed; P2-1 achieved unsupported sitting at age 2.5 years and standing with support at age 4.5 years. No new fractures occurred between birth and the age of 3 years. There were no signs of dentinogenesis imperfecta. Kyphoscoliosis of the thoracic and lumbar spine was evident. P2-1 never walked, and at the age of 7 years P2-1 uses a wheelchair. Current height at the age of 8 years is 79.9 cm (SDS −8.4), which is at the 50th percentile for a 17-month-old child.
Proband 2 from family 2 (P2-2) (Figure 1B) was born by elective Caesarian section at 37 weeks of gestation. On an antenatal ultrasound scan at 20 weeks of gestation, multiple fractures indicative of OI were observed. Birth weight was 2.32 kg (p10), and a skeletal survey confirmed multiple fractures of ribs and limbs. A large head with a large anterior fontanelle was observed in combination with some occipital flattening. There was no chest deformity. The legs were abducted at the hips in a frog-leg position. Bowing of the femora and anterior bowing of the tibae were noted. At the age of 6 months, her length is 47.4 cm (SDS −8.8). Treatment with 0.5 mg/kg/day intravenous Pamidronate on 3 consecutive days every 8 weeks commenced shortly after birth.
In Family 1, sequencing of the five exons of PPIB and adjacent intronic sequences with M13 tailed primers (Table S1) yielded a homozygous mutation c.556–559 delAAGA in exon 5; this mutation resulted in p.Lys186GlnfsX8 (Figure 2A) in both affected siblings (P1-1 and P1-2). The mutation leads to a frameshift that replaces 31 C-terminal amino acids that are highly conserved in evolution, and it is therefore expected to affect protein function. The parents proved to be heterozygous carriers. In family 2, we detected a homozygous mutation c.451 C > T in exon 4 of the PPIB gene; this mutation resulted in p.Gln151X (Figure 2B) in both affected siblings (P2-1 and P2-2). The parents were heterozygous carriers. This mutation removes the last 65 amino acids at the C-terminal. Therefore, the mutation can lead either to diminished protein function or to nonsense-mediated decay (NMD). These PPIB mutations were not found in 192 alleles from clinically normal controls. The homozygous mutation c.556–559 delAAGA in exon 5 leads to a premature stop in the last exon, and therefore NMD is unlikely to occur. Quantitative RT-PCR (Lightcycler480, Roche, Nutley, NJ, USA) (Table S2) showed equal expression of both wild-type and mutant PPIB mRNA in the parents of P1-1 and P1-2, indicating that no NMD occurs, as was expected. The total mRNA level of mutant and wild-type PPIB in fibroblasts form the parents as well as the level of mutant mRNA in P1-1 were comparable to the level of wild-type PPIB mRNA in healthy control fibroblasts. Interestingly, intracellular CyPB was detectable in fibroblasts from the control cell line and from the father of P1-1 and P1-2 but was undetectable in fibroblasts from P1-1 (Figure 3), as measured by immunoblot with a monoclonal CyPB antibody raised against the C-terminal tail (HPA012720, Atlas Antibodies, Stockholm, Sweden) and two different polyclonal antibodies raised against the whole protein (11607-1-AP, Proteintech, Manchester, United Kingdom and AF5410, R&D systems, Minneapolis, MN, USA). The absence of truncated CyPB in the proband P1-1 might be explained in one of the following ways: (i) truncated protein is generated, but the amount is insufficient to be detected by immunoblot, or (ii) truncated protein is rapidly degraded. To investigate the function of CyPB in the complex and the interactions of CyPB with P3H1 and CRTAP in complex assembly, we performed immunohistochemistry as reported elsewhere15 with CRTAP antibody (sc-100920, Santa Cruz, Santa Cruz, CA, USA), P3H1 antibody (H00064175-B01P, Abnova, Taipei, Taiwan) and the CyPB antibodies raised against the C-terminal tail (HPA012720, Atlas Antibodies, Stockholm, Sweden) and the whole CyPB protein (11607-1-AP, Proteintech, Manchester, United Kingdom) on bone tissue from P1-1 and P1-2 and from two probands with autosomal-recessive OI due to CRTAP and LEPRE1 mutations (Figures 4A–4O). Interestingly, in bone tissue of patients with CRTAP or LEPRE1 mutations, antibody staining was observed for neither CRTAP nor P3H1 (4G-M), whereas in P1-2 with PPIB mutations, no signal of CyPB was noted, and a positive signal of CRTAP and P3H1 was identified (4M-O). Apparently, the presence of both CRTAP and P3H1 is needed for a stable protein complex, whereas CyPB is demonstrably independent of the presence of either CRTAP or P3H1. This has been demonstrated previously (W. Chang et al., 2008, American Society for Bone and Mineral Research, Abstract). Recently, it was reported that the P3H1/CRTAP/CyPB complex has three distinct activities: it is a prolyl 3-hydroxylase, a PPIase, and a molecular chaperone. Consequently, it was hypothesized that autosomal-recessive OI due to LEPRE1 and CRTAP mutations resulted not only from a lack of 3-Hyp residues in the collagen type I chains but also from a disturbance in the additional function of the P3H1/CRTAP/CyPB complex as a chaperone and PPIase.16 To further investigate this hypothesis, we performed tandem mass spectrometry of collagen tryptic peptides17 in individuals with OI due to LEPRE1, CRTAP, COL1A1, and PPIB mutations (technical details can be found in Figure S3). In P1-2, homozygous for a PPIB mutation, the P986 3-hydroxylation level was lower (33%) than control levels (93%–100%) but still higher than that observed in patients with CRTAP (16%) and LEPRE1 (22%) mutations (Figure 5). This result and the fact that CyPB itself is stable and does not require the presence of CRTAP or P3H1, as well as both reported decreased 3-prolyl hydroxylation due to deficiency of CRTAP4 lacking the catalytic hydroxylation domain and the known function of CyPB, suggest that OI is caused by a dysfunctional CRTAP/P3H1/CyPB complex rather than by a lack of 3-hydroxylation of a single proline residue in the α1 chains of collagen type I. Given the above, PPIB mutations might also disrupt the folding of the type I collagen helix as a result of interference with PPIase activity and/or chaperone function, two other recently reported functions of the P3H1/CRTAP/CyPB complex. Further investigations will need to shed more light on this. It is already known that inhibition of CyPB in cultured dermal fibroblasts with cyclosporine A increases modification of the type I collagen chains, which indicates that overmodification of collagen type I in autosomal-recessive OI due to PPIB mutations results from a decreased rate of cis-trans isomerization by CyPB.18 Collagen type I electrophoresis performed as reported elsewhere19 showed overmodification of collagen type I chains comparable to collagen type I overmodification observed in patients with CRTAP and LEPRE1 mutations and in patients with COL1A1 mutations causing lethal or severe OI (Figure 6). In conclusion, we describe mutations in the PPIB gene encoding the third member, CyPB, of the collagen-modifying complex. PPIB mutations result in autosomal-recessive OI with a clinical picture compatible with OI type II-B/III, as is the case with CRTAP4,13,20 and LEPRE15,13 mutations. Our findings, in combination with recent literature, support the hypothesis that OI due to PPIB mutations results from a disruption of the function of the P3H1/CRTAP/CyPB complex as a prolyl 3-hydroxylase, a PPIase, and a molecular chaperone.16
Figure 2.

PPIB Mutations in Severe OI
(A) A homozygous c.556–559 delAAGA in exon 5 resulting in p.Lys186GlnfsX8 is shown in probands of family 1.
(B) A homozygous c.451 C > T mutation in exon 4 resulting in p.Gln151X is shown in probands of family 2.
Figure 3.

Immunoblot in Family 1
An immunoblot is shown of fibroblast CyPB, which is lacking in the affected infant P1-1 and present in the parent and the control (polyclonal antibodies raised against the whole protein were used in this immunoblot, 11607-1-AP, Proteintech, Manchester, UK). CRTAP is present in P1-1, the parent, and the control.
Figure 4.

Results of Immunohistochemistry with Antibodies against CRTAP, P3H1, and CyPB
Immunohistochemistry performed on bone tissue with CyPB-CRTAP and P3H1 antibodies shows the following:
(A–C) a positive signal of CyPB, CRTAP, and P3H1 in a control without OI;
(D–F) a positive signal of CyPB, CRTAP, and P3H1 in a patient with a COL1A1 mutation (c.1238G > A [p.Gly413Asp]);
(G–I) no signal of either CRTAP or P3H1 but a signal of CyPB in a patient with a homozygous c.404 delG CRTAP mutation;
(J–L) no signal of either P3H1 or CRTAP and a signal of CyPB in a patient with a homozygous c.401_402 delAA LEPRE1 mutation; and
(M–O) no signal of CyPB but signals of both CRTAP and P3H1 antibodies in P1-2 with a homozygous PPIB mutation.
AB: antibody.
Figure 5.
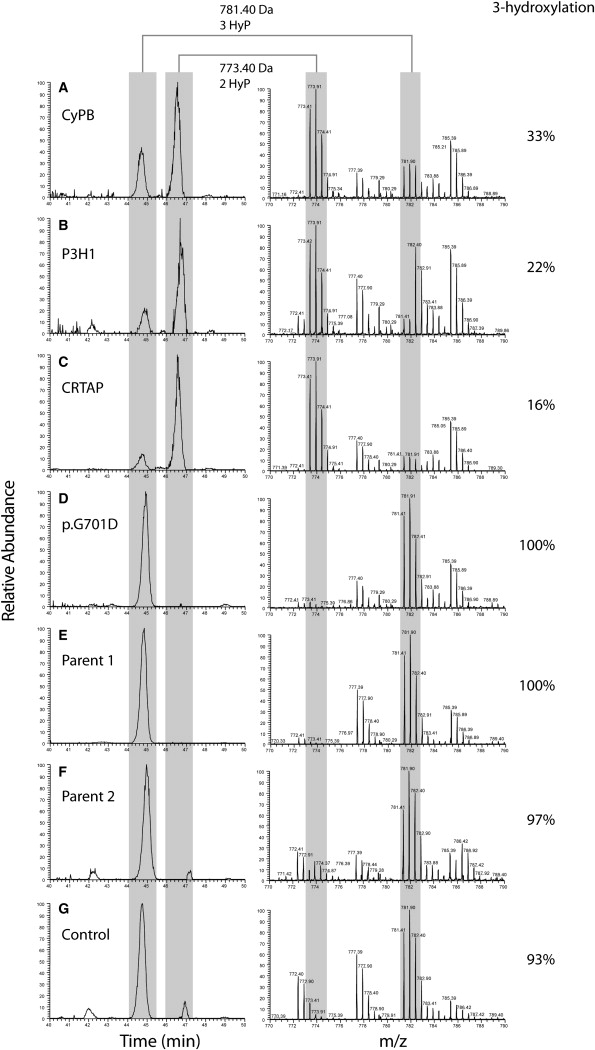
Collagen I α-1 P986 3-Hydroxylation Level in Patients with OI Due to Different Molecular Genetic Causes
Figure 5 shows mass spectrometry analysis of the C1A1 P986-containing tryptic peptide, modified with either two or three hydroxyl groups (as indicated by an asterisk below the modified proline residue in Figure S2) in patients with homozygous PPIB (A), LEPRE1 (B), or CRTAP (C) mutations or a heterozygous COL1A1 mutation (D), in the parents of the patient with PPIB mutations (E and F), and in a control (G). The left panel shows extracted ion chromatograms (XIC), generated at m/z 773.4 and 781.4 ± 0.05, and the right panel shows the corresponding summed mass spectra (40–50 min.). XIC peak areas were determined for doubly and triply hydroxylated peptides. The C1A1 P986 3-hydroxylation level (%) was calculated as triple-hydroxylation area (triple-hydroxylation area + double-hydroxylation area). Collagen I α-1 peptides that show normal P986 3-hydroxylation have an m/z ratio of 781.4, whereas peptides lacking 3-hydroxylation have an m/z ratio of 773.4. Controls (D–F) show normal 3-hydroxylation (>90%). P1-1 with homozygous PPIB mutations (A) shows decreased 3-hydroxylation (33%). Patients with homozygous LEPRE1 (p.Glu134fs) or CRTAP (p.Ala8fs) mutations (B and C, respectively), however, show even more decreased 3-hydroxylation levels (22% and 16%, respectively).
Figure 6.

Electrophoresis of Collagen Type I Chains in Fibroblasts and Chorionic Villi of Patients with a Homozygous CRTAP, LEPRE1, or PPIB Mutation or a Heterozygous COL1A1 Mutation
Backstreaking and/or increased band width is visible in cells (c) and media (m), indicating overmodification of collagen type I chains in chorionic villi cells and fibroblasts of patients with lethal or severe OI due to homozygous CRTAP (p.Ala8fs), LEPRE1 (p.Glu134fs), or PPIB (p.Lys186GlnfsX8) mutations or a heterozygous COL1A1 (p.G965S in chorionic villi cells and p.G662V in fibroblasts) mutation. The slight apparent overmodification of other collagens appears to be an effect typically evident in chorionic villi.
Acknowledgments
We thank the families of the affected individuals for their kind cooperation. Y. Moutaouakil established fibroblast cell lines of affected individuals and parents, E. van den Akker conducted prior collagen electrophoretic studies on affected individuals from family 1, and S. Elshof and K. van der Ven performed immunohistochemistry. We thank B. Zandiehdoulabi for his help and advice, L. Scheffer for her technical support, and J. Hewitt for providing additional data on family 2.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov
Accession Numbers
The GenBank accession number of the PPIB reference sequence, used for identification of the mutations reported in this paper, is NM_000942.4.
Note Added in Proof
This version of this paper differs from that originally published online in that an Accession Numbers section has been added to reflect a newly available GenBank number.
References
- 1.Marini J.C., Cabral W.A., Barnes A.M., Chang W. Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle. 2007;6:1675–1681. doi: 10.4161/cc.6.14.4474. [DOI] [PubMed] [Google Scholar]
- 2.Canty E.G., Kadler K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 3.Engel J., Prockop D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991;20:137–152. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 4.Barnes A.M., Chang W., Morello R., Cabral W.A., Weis M., Eyre D.R., Leikin S., Makareeva E., Kuznetsova N., Uveges T.E. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuno K., Peyton D.H., Hayashi T., Engel J., Bächinger H.P. Effect of the -Gly-3(S)-hydroxyprolyl-4(R)-hydroxyprolyl- tripeptide unit on the stability of collagen model peptides. FEBS J. 2008;275:5830–5840. doi: 10.1111/j.1742-4658.2008.06704.x. [DOI] [PubMed] [Google Scholar]
- 7.Vranka J.A., Sakai L.Y., Bächinger H.P. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004;279:23615–23621. doi: 10.1074/jbc.M312807200. [DOI] [PubMed] [Google Scholar]
- 8.Krane S.M. The importance of proline residues in the structure, stability and susceptibility to proteolytic degradation of collagens. Amino Acids. 2008;35:703–710. doi: 10.1007/s00726-008-0073-2. [DOI] [PubMed] [Google Scholar]
- 9.Price E.R., Zydowsky L.D., Jin M.J., Baker C.H., McKeon F.D., Walsh C.T. Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc. Natl. Acad. Sci. USA. 1991;88:1903–1907. doi: 10.1073/pnas.88.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Q., Li M., Yang H., Chai H., Fisher W., Chen C. Roles of cyclophilins in cancers and other organ systems. World J. Surg. 2005;29:276–280. doi: 10.1007/s00268-004-7812-7. [DOI] [PubMed] [Google Scholar]
- 11.Smith T., Ferreira L.R., Hebert C., Norris K., Sauk J.J. Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 1995;270:18323–18328. doi: 10.1074/jbc.270.31.18323. [DOI] [PubMed] [Google Scholar]
- 12.Zeng B., MacDonald J.R., Bann J.G., Beck K., Gambee J.E., Boswell B.A., Bächinger H.P. Chicken FK506-binding protein, FKBP65, a member of the FKBP family of peptidylprolyl cis-trans isomerases, is only partially inhibited by FK506. Biochem. J. 1998;330:109–114. doi: 10.1042/bj3300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T.K., Pace J.M., Pepin M.G., Weis M., Eyre D.R., Walsh J. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008;29:1435–1442. doi: 10.1002/humu.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willaert A., Malfait F., Symoens S., Gevaert K., Kayserili H., Megarbane A., Mortier G., Leroy J.G., Coucke P.J., De Paepe A. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: Clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 2009;46:233–241. doi: 10.1136/jmg.2008.062729. [DOI] [PubMed] [Google Scholar]
- 15.Groenendaal F., Vles J., Lammers H., De V.J., Smit D., Nikkels P.G. Nitrotyrosine in human neonatal spinal cord after perinatal asphyxia. Neonatology. 2008;93:1–6. doi: 10.1159/000106432. [DOI] [PubMed] [Google Scholar]
- 16.Ishikawa Y., Wirz J., Vranka J.A., Nagata K., Bächinger H.P. Biochemical characterization of the prolyl 3-hydroxylase 1/CRTAP/cyclophilin B complex. J. Biol. Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shevchenko A., Wilm M., Vorm O., Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 18.Steinmann B., Bruckner P., Superti-Furga A. Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 1991;266:1299–1303. [PubMed] [Google Scholar]
- 19.Korkko J., la-Kokko L., De P.A., Nuytinck L., Earley J., Prockop D.J. Analysis of the COL1A1 and COL1A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1A1 mutations in 15 patients with osteogenesis imperfecta type I: Identification of common sequences of null-allele mutations. Am. J. Hum. Genet. 1998;62:98–110. doi: 10.1086/301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Dijk, F.S., Nesbitt, I.M., Nikkels, P.G., Dalton, A., Bongers, E.M., van de Kamp, J.M., Hilhorst-Hofstee, Y., Den Hollander, N.S., Lachmeijer, A.M., Marcelis, C.L., et al. (2009) CRTAP mutations in lethal and severe osteogenesis imperfecta: the importance of combining biochemical and molecular genetic analysis. Eur. J. Hum. Genet. Published online June 24, 2009. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.