

After several decades of intense research and various attempts of definition and classification, cardiomyopathies still remain disorders of remarkable and intriguing complexity. Once more, this aspect is elicited by the recent discovery that mutations in the cardiac ankyrin repeat protein or CARP, a protein functionally part of the sarcomere, can cause different types of cardiomyopathies, as reported in this issue (1,2), as well as a congenital heart disease (3).
ANKRD1 in normal heart and disease
CARP is a 36 kD protein encoded by the cardiac ankyrin repeat domain 1 gene ANKRD1, which maps on chromosome 10. ANKRD1 is a member of a conserved gene family, coding for muscle ankyrin repeat proteins (MARPs), involved in muscle stress response such as stretch, injury and hypertrophy (4). CARP is a nuclear transcription co-factor, a signaling molecule predominantly expressed in the heart. CARP is found in the sarcomere, where it co-localize with the N2A domain of titin and myopalladin in the I-band of the Z disk (Figure 1), and in the nucleus (4). The expression of CARP is controlled, at list in part, by the titin-based mechano-transduction signaling pathway, and it is increased in heart development and conditions of injury and stress. In heart development, CARP acts as transcriptional repressor of myocyte contractile elements. In heart failure, CARP is overexpressed, suggesting a role in the “fetal gene program” characteristic of the molecular remodeling of the failing heart (5).
Figure. Model for the titin-N2A signaling complex.
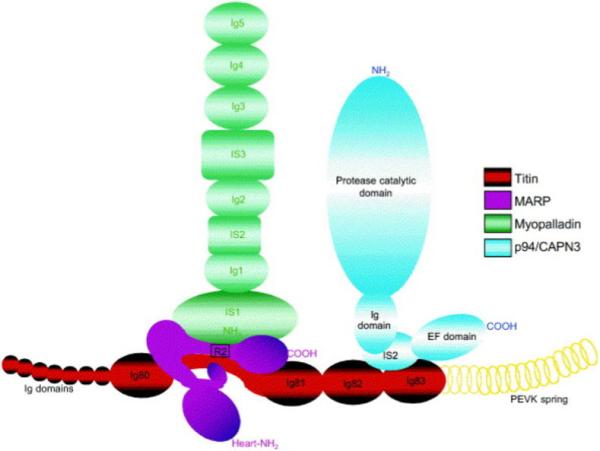
N2A titin’s sequence interacts with MARPs (CARP, ankrd2 or DARP). Myopalladin associates with MARP/N2A complex by interacting with the N-terminal domains of MARPs. Reprinted from Miller et al.(4) with permission from Elsevier.
Since titin was previously found to be associated to both HCM and DCM (6-8), also CARP, as part of the titin complex, was hypothesized to play a role in cardiomyopathies. In this issue, two reports confirm this hypothesis and show that in fact ANKRD1 mutations can cause both DCM and HCM (1,2).
In the first article, Arimura et al. (2) report the results of the ANKRD1 mutation screening in a large HCM population collected in Japan and in USA. In 384 index patients, they found 3 missense mutations (ANKRD1 Pro52Ala, Thr123Met and Ile280Val), accounting for ∼1% of HCM cases. Interestingly, they also investigated the N2A CARP-biding domain of titin, and found two additional mutations (TTN Arg8500 and Arg8604Gln) in their HCM cohort. In the second report, Moulik et al (1) investigated a series of 208 DCM index patients of Japanese and USA origin, and found 3 missense mutations (ANKRD1 Pro105Ser, which was recurrent in 2 families, Val107Leu, and Met1841Ile) accounting for in 2% of DCM cases, further supporting a role of the titin mechano-transduction complex in the pathogenesis of cardiomyopathies. But, how to explain two different cardiomyopathies with opposite patho-physiology caused by the same gene?
Phenotypic heterogeneity in cardiomyopathies
Phenotypic heterogeneity (also called “allelic variants” in OMIM (9)) is a well known and common phenomenon in genetics, referring to the occurrence of more than one phenotype caused by allelic mutations at a single locus (10): examples familiar to cardiologists are Duchenne and Becher muscular dystrophies caused by the same dystrophin gene, laminopathies ranging from progeria to lipodystrophy due to lamin A/C gene, LQT syndrome and congenital conduction defect caused by the cardiac sodium channel gene SCN5A.
The reason for the clinical variability in allelic disorders lies in the different function of the mutant proteins. In the case of sarcomeric genes, it appears that a “gain” of function usually results in increased energy demand, inefficient ATP utilization and hypertrophy, whereas a “loss” of function in decreased contractility (Table). The two studies published in this issue seem to follow the rule. Arimura et al. (2) show that ANKRD1 mutations in HCM increase binding of CARP to titin and myopalladin, and that titin mutations at the CARP-binding site have the same effect. On the other hand, Moulik et al. (1) show that ANKRD1 mutations in DCM cause a loss of CARP binding to talin 1, potentially leading to loss of stretch-sensing, disruption of the link between titin complex and cytoskeletal network, and transcriptional deregulation of genes involved in cell cycle and other pathways.
Table 1. Phenotypic heterogeneity of sarcomeric genes and differential changes of mutant protein function (8,9,12-16).
| Gene | Protein | DCM | Function | HCM | Function | Other allelic disorders |
|---|---|---|---|---|---|---|
| Thick filament | ||||||
| MYH7 | Cardiac β myosin heavy chain | yes | ↓maximal force generation ↓contractility ↓velocity of actin sliding |
yes | ↑maximal force generation ↑Ca++ sensitivity |
Laing distal myopathy, myosin storage myopathy, scapuloperoneal myopathy, left ventricular non-compaction, endocardial fibroelastosis |
| MYH6 | Cardiac α myosin heavy chain | yes | * | yes | * | Atrial septal defect |
| MYL2 | Regulatory myosin light chain | Not described | yes | ↑Ca++ sensitivity | ||
| MYL3 | Essential myosin light chain | Not described | yes | ↑Ca++ sensitivity | ||
| MYBPC3 | Cardiac myosin-binding protein C | yes | yes | Hypertrophy Diastolic dysfunction | ||
| Thin filament | ||||||
| TNNT2 | Cardiac Troponin T (cTnT) | yes | ↓myofibrillar function ↓Ca++ sensitivity |
yes | ↑myofibrillar function ↑Ca++ sensitivity |
Restrictive cardiomyopathy |
| TNNTI3 | Cardiac troponin I (cTnI) | yes | ↓myofibrillar function ↓binding to cTnT |
yes | ↑myofibrillar function ↑Ca++ sensitivity |
Restrictive cardiomyopathy (↑↑Ca++ sensitivity) |
| TNNC1 | Cardiac troponin C (cTnC) | yes | ↓myofibrillar function ↓Ca++ sensitivity ↓PKC effect |
yes | ↑myofibrillar function | |
| TPM1 | Tropomyosin 1 alpha chain |
yes | ↓Ca++ sensitivity ↓maximum force |
yes | ↑myofibrillar function ↑Ca++ sensitivity |
|
| ACTC1 | α-Cardiac actin | yes | ↓α-cardiac actinin (Z-disk) affinity ↓force transmission |
yes | Impaired actomyosin binding | Restrictive cardiomyopathy |
| Titin filament and Z-disk | ||||||
| TTN | Titin | yes | ↓biding to actinin and Tcap | yes | ↑biding to actinin and Tcap | Tibialis muscular dystrophy or Udd distal myopathy; hereditary myopathy with early respiratory failure (HMERF); recessive limb-girdle muscular dystrophy type 2J (LGMD2J) |
| TCAP | Titin-cap or telethonin | yes | ↓biding to titin, MLP and myozenin-2 | yes | ↑biding to titin, and myozenin-2 | Limb-girdle muscular dystrophy type 2G (LGMD2G) |
| ANKRD1 | Cardiac ankyrin repeat protein (CARP) | yes | ↓biding to talin1 | yes | ↑biding to titin and myopalladin | Total anomalous pulmonary venous return (↑expression) |
| LDB3 | Cypher/ZASP | yes | Cytoskeletal disarray, ↓PKC affinity | Not described | Left ventricular non-compaction, myofibrillar myopathy | |
| CSRP3 | MLP | yes | yes | ↓biding to actinin | ||
| MYOZ2 | Myozenin-2 | Not described | yes | |||
| OBSCN | Obscurin | Not described | yes | ↓biding to titin | ||
Legend: β myosin heavy chain mutations found in HCM and DCM were studied in a myosin heavy chain of knock-in mouse models
However, gain and loss are not the only mechanism involved in the phenotypic heterogeneity of ANKRD1. Indeed, a recently publication by Cinquetti et al. (3) reports the identification of increased CARP expression or protein stability in 3 cases with total anomalous pulmonary venous return (TAPVR), a rare congenital heart defect characterized by failure of the pulmonary veins to connect to the left atrium during development. In this case, CARP overexpression or its increased activity are believed to repress normal cardiac gene expression leading to abnormal heart development.
Impact of ANKRD1 mutations discovery in clinical care
The discovery of ANKRD1 mutations in cardiomyopathies has several implications. Firstly, it contributes to fill the gap of the large number of patients in whom the cause of cardiomyopathy is still unknown, approximately 40% of cases in HCM and probably around 70% in DCM (11). Secondly, it expands our knowledge on the mechanisms leading to hypertrophy and heart failure to include abnormal stretch-based signaling in response to force: this appears to be another “common pathway” for HCM and DCM, which could be targeted by novel therapeutic strategies. Finally, it raises the question of clinical genetic testing of ANKRD1 in HCM and DCM patients. Although the low prevalence of mutations may currently limit the routine screening of ANKRD1 gene, we may expect that the implementation in resequencing technology will allow a systematic screening of rare cardiomyopathy genes in the patient population in the near future.
Acknowledgments
Supported by the NIH (HL69071-01, MO1 #RR00051-1575), AHA 0150453N, MDA PN0007-056
Abbreviations
- CARP
Cardiac Ankyrin Repeat Protein
- ANKRD1
Cardiac Ankyrin Repeat Domain 1 Gene
- DCM
Dilated cardiomyopathy
- HCM
Hypertrophic cardiomyopathy
Footnotes
There are no conflicts of interest or financial disclosures associated with this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moulik M, Vatta M, Witt SH, et al. ANKRD1-The gene encoding cardiac ankyrin repeat protein is a novel dilated cardiomyopathy gene. J Am Coll Cardiol. 2009 doi: 10.1016/j.jacc.2009.02.076. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arimura T, Bos JM, Sato A, et al. Cardiac Ankyrin Repeat Protein Gene (ANKRD1) Mutations in Hypertrophic Cardiomyopathy. J Amer Coll Cardiol. 2009 doi: 10.1016/j.jacc.2008.12.082. in press. [DOI] [PubMed] [Google Scholar]
- 3.Cinquetti R, Badi I, Campione M, et al. Transcriptional deregulation and a missense mutation define ANKRD1 as a candidate gene for total anomalous pulmonary venous return. Hum Mutat. 2008;29:468–74. doi: 10.1002/humu.20711. [DOI] [PubMed] [Google Scholar]
- 4.Mootha VK, Lepage P, Miller K, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zolk O, Frohme M, Maurer A, et al. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochem Biophys Res Commun. 2002;293:1377–82. doi: 10.1016/S0006-291X(02)00387-X. [DOI] [PubMed] [Google Scholar]
- 6.Itoh-Satoh M, Hayashi T, Nishi H, et al. Titin mutations as the molecular basis of dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291:385–91. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 7.Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nature Genetics. 2002;30:201–4. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto Y, Hayashi T, Inagaki N, et al. Functional analysis of titin/connectin N2-B mutations found in cardiomyopathy. J Muscle Res Cell Motil. 2005;26:367–74. doi: 10.1007/s10974-005-9018-5. [DOI] [PubMed] [Google Scholar]
- 9.McKusick VA. Online Mendelian Inheritance in Man, OMIM (TM) McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University; National Center for Biotechnology Information, National Library of Medicine; Baltimore, MD: Bethesda, MD: 2000. [Google Scholar]
- 10.Haines J, Pericak-Vance M. Genetic analysis of complex diseases. 2nd ed. Wiley; Hoboken, NJ: 2006. [PubMed] [Google Scholar]
- 11.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America Practice Guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 12.Mizra M, Marston S, Willot R, et al. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005;280:28498–506. doi: 10.1074/jbc.M412281200. [DOI] [PubMed] [Google Scholar]
- 13.Kaski JP, Syrris P, Burch M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–84. doi: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- 14.Debold EP, Schmitt JP, Patlak JB, et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284–H291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 15.Rajan S, Ahmed RP, Jagatheesan G, et al. Dilated cardiomyopathy mutant tropomyosin mice developcardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ Res. 2007;101:205–214. doi: 10.1161/CIRCRESAHA.107.148379. [DOI] [PubMed] [Google Scholar]
- 16.Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. 2008;77:659–666. doi: 10.1093/cvr/cvm084. [DOI] [PubMed] [Google Scholar]