

Abstract
While in vitro observations suggest that cross-presentation of antigens is mediated primarily by CD8α+ dendritic cells, in vivo analysis has been hampered by the lack of systems that selectively eliminate this cell lineage. Here we show that deletion of the transcription factor Batf3 ablated development of CD8α+ dendritic cells, allowing us to examine their role in immunity in vivo. Dendritic cells from Batf3-/- mice were defective in cross-presentation and Batf3-/- mice lacked virus-specific CD8+ T cell responses to West Nile virus. Importantly, rejection of highly immunogenic syngeneic tumors was impaired in Batf3-/- mice. These results suggest an important role for CD8α+ dendritic cells and cross-presentation in responses to viruses and in tumor rejection.
During antigen ‘cross-presentation’ (1), antigens generated in one cell are presented by MHC class I molecules of a second cell. It remains unclear whether all antigen presenting cells (APCs) use cross-presentation and whether this pathway plays a role in immune responses in vivo (2). Dendritic cells (DCs) are a heterogeneous group of APCs with two major subsets, plasmacytoid dendritic cells (pDCs) and conventional CD11c+ dendritic cells (cDCs) (3). Subsets of cDCs include CD8α+, CD4+, and CD8α-CD4- populations that may exert distinct functions in immune responses. Evidence has suggested that CD8α+ cDCs are important for cross-presentation during infections, but is based on ex vivo analysis (4-6) or in vitro antigen loading (7). Evidence both for and against a role for cross-presentation in responses against tumors has been reported (8-10).
Attempts have been made to study the in vivo role of dendritic cells by selective depletion. Diphtheria toxin treatment can deplete all CD11chi cells in one transgenic mouse model (11), but affects splenic macrophages and activated CD8+ T cells (12). Gene targeting of transcription factors (e.g., Irf2, Irf4, Irf8, Stat3 and Id2) has caused broad defects in several DC subsets, T cells and macrophages (13). To identify genes regulating DC development, we performed global gene expression analysis across many tissues and immune cells (fig S1A). Batf3 (p21SNFT) (14) was highly expressed in cDCs, with low to absent expression in other immune cells and non-immune tissues. Thus, we generated Batf3-/- mice lacking expression of the Batf3 protein (fig. S1B-D).
In spleens of Batf3-/- mice we found a selective loss of CD8α+ cDCs, without abnormalities in other hematopoietic cell types or architecture (Fig. 1, fig. S2-S11). CD8α+ cDC coexpress DEC205, CD24, and low levels of CD11b (3,15). Batf3-/- mice lacked splenic CD11chi CD8α+ DEC205+ cells (Fig. 1A), showed a loss of CD11chi CD11bdull cells and CD11chi CD8α+ CD24+ cells (Fig. 1B), but had normal populations of CD4+ and CD8α-CD4- cDC subsets (Fig. 1B). Lymph nodes and thymi of Batf3-/- mice lacked CD8α+ DCs but had normal distributions of CD8α- CD11c+ cells (Fig. 1C). DEC205int and DEC205hi DCs were present in lymph nodes draining the skin of Batf3-/- mice (Fig. 1C), and showed normal migration from skin to lymph node after topical application of fluorescein-5-isothiocyanate (fig. S3A). Batf3-/- mice had normal development of pDCs (CD11cint CD11b- B220+) (fig. S3B), interstitial DCs of pancreatic islets (CD11c+ CD8α-) (fig. S3C, D), monocytes, neutrophils (fig. S3E) and SIGN-R1+ and MOMA-1+ marginal zone macrophages (Fig. 2A). CD8α+ cDCs developed normally in heterozygous Batf3+/- mice (fig. S4A), and were absent in Rag2-/- Batf3-/- mice (fig. S4B).
Fig. 1. Batf3-/- mice selectively lack the CD8α+ DC subset.
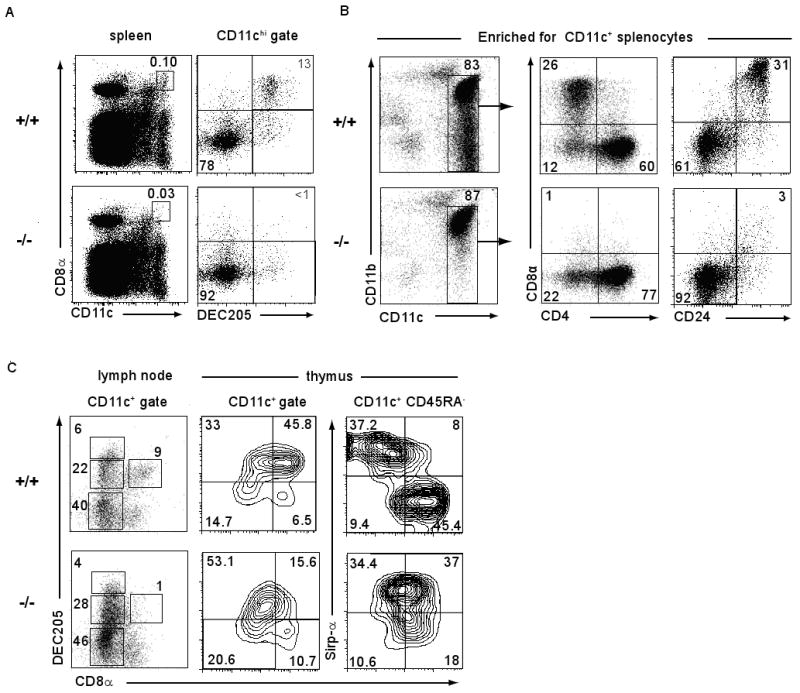
(A) Splenocytes from Batf3+/+ (+/+) or Batf3-/- (-/-) mice were stained for CD11c, CD8α and DEC205. Left panels are gated on live cells. Numbers indicate the percentage of splenocytes within the CD11chiCD8α+ gate. Right panels are gated on CD11chi cells. (B) Splenocytes were depleted of B220+ B cells and Thy1.2+ T cells and positively selected for CD11c expression by antibody coated magnetic beads (MACS). Cells were then stained for CD11c, CD11b, and either CD8α and CD4 or CD8α and CD24, and analyzed by FACS. Numbers represent the percentage of cells within the indicated gates. (C) Lymph node cells pooled from cervical, axillary and inguinal lymph nodes and depleted of Thy1.2+ T cells, or light density cells of the thymus were stained for CD11c, CD45RA CD8α, DEC205 or Sirp-α. Plots are gated on the indicated populations.
Fig. 2. Functional loss of CD8α+ cDCs in Batf3-/- mice is cell-intrinsic to the hematopoietic system.
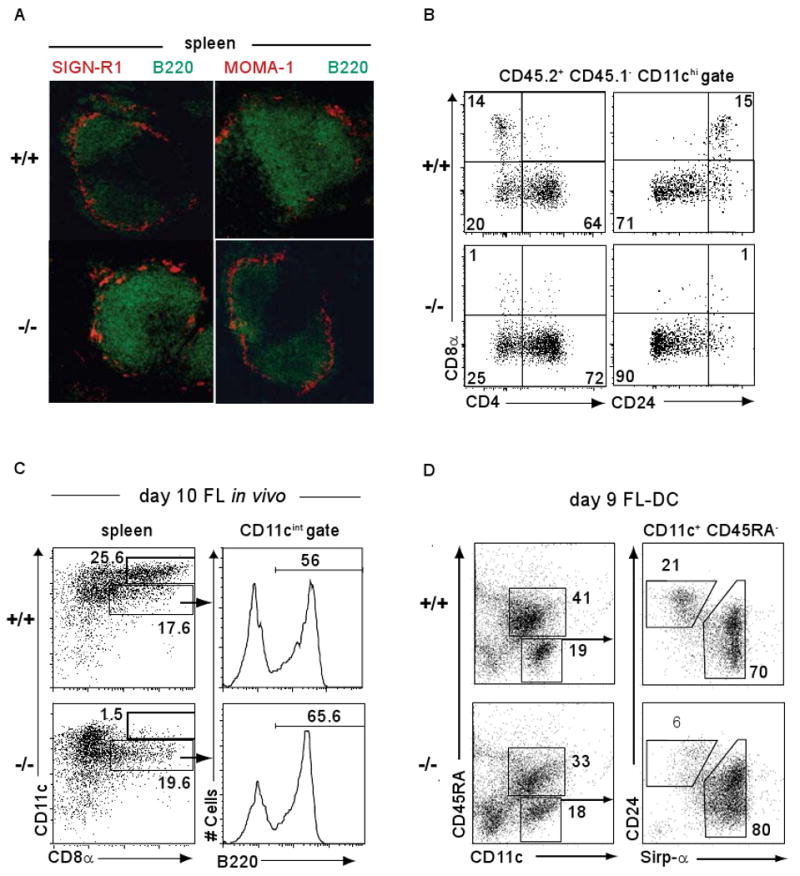
(A) Frozen sections from Batf3+/+ (+/+) or Batf3-/- (-/-) mice were stained for B220 (green) and SIGN-R1 (red) expression (left panels) or for B220 (green) and MOMA-1 (red) (right panels). (B) Irradiated F1(B6.SJL/129SvEv) mice (CD45.1+ CD45.2+) were reconstituted with 2 ×107 bone marrow cells from Batf3+/+ (+/+) or Batf3-/- (-/-) CD45.1-CD45.2+ mice. After 10 weeks, donor cells (CD45.1- CD45.2+) were analyzed for CD11c, CD8α, CD4 and CD24 expression. Shown are plots for CD8α and CD4 (left panels) or CD8α and CD24 (right panels) gated on CD11chi donor-derived cells. Numbers represent the percentage of cells within the indicated gates. (C) Batf3+/+ (+/+) or Batf3-/- (-/-) mice were treated i.p. with 10μg FL-Fc. After 10 days, splenocytes were enriched for CD11c+ by MACS and stained for CD11c, CD8α and B220. Plots are gated on live cells (left) or CD11cintCD8α+ cells (right). Numbers represent the percentage of cells within the indicated gates. (D) Batf3+/+ (+/+) or Batf3-/- (-/-) BM cells were cultured in FL (20 ng/ml) for 9 days, and non-adherent cells analyzed for CD11c, CD45RA, CD24 and Sirp-α expression. Plots are gated on live cells (left) or CD11c+ CD45RA- cells (right).
This loss of CD8α+ cDCs could result from a cell-autonomous hematopoietic defect or a cell-extrinsic requirement for Batf3. To distinguish these possibilities, we generated chimeras in which CD45.2+ Batf3+/+ or CD45.2+ Batf3-/- bone marrow (BM) was transplanted into lethally irradiated CD45.1+CD45.2+ recipients (Fig. 2B). Upon reconstitution (fig. S5A), we found CD8α+ cDCs developed only from Batf3+/+ donor BM cell (Fig. 2B), indicating a cell-intrinsic hematopoietic defect in Batf3-/- mice.
Treatment of mice with fms-like tyrosine kinase 3 (flt3) ligand-Fc (FL-Fc) expanded CD8α+ cDCs, CD8α- cDCs and pDCs in Batf3+/+ mice, but failed to expand CD8α+ cDC in Batf3-/- mice (Fig. 2C). In vitro culture of BM with FL generates cell populations corresponding to pDCs (CD11c+CD45RA+) and cDCs (CD11c+CD45RA-) (3,16) (Fig. 2D). These in vitro-derived cDCs do not express CD8α or CD4, but contain a CD24+Sirp-αlo-int population corresponding to CD8α+ cDC (16). Batf3+/+ or Batf3-/- BM cells treated with FL produced similar ratios of pDCs and cDCs (Fig. 2D and fig. S5B). However, Batf3-/- BM generated far fewer CD24+ Sirp-α- cells compared to Batf3+/+ BM (Fig. 2D), corresponding to loss of CD8α+ cDCs. Finally, DCs generated from Batf3-/- BM were selectively deficient in TLR3-induced IL-12 production (fig. S5C), a specific feature of CD8α+ cDCs (16). Similarly, CD11c+ cDCs from the spleens of Batf3-/- mice were selectively deficient in TLR3-induced IL-12 production but had normal responses to TLR4 and TLR9 ligands (fig. S6A).
We next tested whether APCs from Batf3-/- mice could prime CD4+ and CD8+ T cell responses. Similar proliferative responses of OT-II transgenic CD4+ T cells (17) occurred with soluble ovalbumin presented by Batf3+/+ and Batf3-/- cDCs (fig. S6B). However, Batf3-/- cDCs were defective in an assay for cross-presentation of cellular antigen to CD8+ T cells (2,18) (Fig. 3A). OT-I T cells proliferated in response to Batf3+/+ cDCs cocultured with ovalbumin-loaded cells, but failed to proliferate in response to Batf3-/- cDCs in this assay.
Fig. 3. Lack of cross-presentation and antiviral CTL responses in Batf3-/- mice.

(A) Batf3+/+ (+/+) or Batf3-/- (-/-) splenocytes were depleted of B220+ B cells and Thy1.2+ T cells and enriched for CD11c by MACS and cultured with irradiated MHC-class I-/- splenocytes as indicated that were either untreated (-ovalbumin), pulsed with 10 mg/ml soluble ovalbumin (+ovalbumin), or cultured with 1 μM SIINFEKL peptide. CFSE-labeled CD45.1+ OT-I T cells were cultured with these cells and proliferation determined by FACS after 60 hours. Single-color histograms of CD8+CD45.1+ OT-I T cells show the percentage of cells in the indicated gates. (B) Batf3+/+ (+/+) or Batf3-/- (-/-) mice were infected with 100 PFU of WNV. On day 7, isotype-specific anti-WNV E protein titers were measured. (C) Batf3+/+ (+/+) or Batf3-/- (-/-) mice were infected with 100 PFU of WNV, or left uninfected. After 7 days, splenocytes were stimulated in vitro with the WNV-specific NS4B peptide (P33), OVA peptide, or PMA/ionomycin as described. CD8+ T cells were analyzed for expression of intracellular IFN-γ. Data shown are the mean ± SEM (n=9-10). (D) Batf3+/+ (+/+) or Batf3-/- (-/-) CD8+ T cells were transferred i.v. into Rag2-/- recipients. After 24h, mice were infected with 100 PFU of WNV (+ WNV) or left uninfected (-WNV). After 7 days, splenocytes were harvested and analyzed as described in (C). Data shown are the mean ± SEM (n=6). Three independently performed experiments yielded similar results.
We examined responses of Batf3-/- mice to West Nile virus (WNV) (19,20). Batf3-/- mice showed normal WNV-specific antibody responses (Fig. 3B), memory B cells (fig. S6C) and CD4+ T cell responses (fig. S6D) but had a dramatic reduction in WNV-specific CD8+ T cell responses (Fig. 3C) and in vivo CTL killing of WNV peptide-loaded target cells (fig. S7A, B). Batf3-/- mice lacked WNV-specific memory CD8+ T cells and had impaired formation of CD8+ CD44hi CD62Llow cells (fig. S7). Adoptive transfer of Batf3-/- CD8+ T cells into Rag2-/- mice generated normal WNV-specific CD8+ T cell response (Fig. 3D), but adoptive transfer of Batf3+/+ CD8+ T cells into Batf3-/- Rag2-/- mice generated an impaired WNV-specific CD8+ T cell response (fig. S7C). This shows that impaired WNV-specific CTL responses in Batf3-/- mice results from a defect of DCs rather CD8+ T cells.
We challenged Batf3+/+ and Batf3-/- mice with syngeneic fibrosarcomas that normally are rapidly rejected in a CD4+ and CD8+ T cell-dependent manner (21,22) (fig. S8A). Two independent fibrosarcomas were rapidly rejected by Batf3+/+ mice, but grew progressively in Rag2-/- mice and Batf3-/- mice (Fig. 4A, fig. S8B and C). Moreover, Batf3-/- mice failed to develop tumor-specific CTLs (Fig. 4B). Tumor-infiltrating CD8+ T cells, but not CD4+ T cells, were significantly reduced in Batf3-/- mice (Fig. 4C). The failure of Batf3-/- mice to reject these tumors was not due to defective NK cell development or function (fig. S2B and S9A-C). We considered whether Batf3-/- T cells have an intrinsic dysfunction, since overexpression studies had suggested Batf3 might affect IL-2 transcription (14). While Batf3 overexpression reduces IL-2 reporter activity in Jurkat T cells (fig. S10B), Batf3-/- CD4+ T cells showed normal IL-2 production (fig. S10D) and normal TH1, TH2 and TH17 differentiation (fig. S10C-E, S11B). Finally, Batf3-/- CD8+ T cells showed normal allospecific effector responses (fig. S11A) and cytokine production (fig. S11B).
Fig. 4. Lack of tumor rejection in Batf3-/- mice.

(A) 106 H31m1 fibrosarcoma cells were injected subcutaneously into Batf3+/+ (closed circles), Batf3-/- (open circles), or Rag2-/- (closed triangles) mice and tumor diameter (± SD) (n=10) was measured. (B) Mice were treated as in (A). After 9 days, splenocytes were harvested and cocultured with IFN-γ pre-treated, irradiated H31m1 tumor cells. After 5 days, a CTL killing assay using 51Cr- labeled H31m1 or 1773 tumor cells as target cells was performed. Shown is specific killing activity as described in the Methods. (C) Tumors and spleens from mice treated as in (A) were removed on day 11 and cells analyzed by FACS. Plots are gated on live CD45.2+ cells and show CD3, CD8α and CD4 expression. Numbers represent the percentage of cells within the indicated gate. Results are representative of at least three mice per group.
Other DC subsets may cross-present, although less efficiently than CD8α+ DCs (23-26), suggesting there may be residual cross-presentation capacity in Batf3-/- mice. We therefore challenged mice using reduced tumor-cell numbers which might allow effective responses in the setting of reduced cross-presentation (fig. S8). While 104 and 105 tumor cells grew in all Rag2-/- mice, some Batf3-/- mice controlled this lower tumor burden (fig. S8D, 8E) and developed tumor-specific CTL response (fig. S8F). Whereas adoptive transfer of wild type DCs led to partial control of tumor growth in Batf3-/- mice, transfer of Batf3-/- DCs did not (fig. S12).
Subsets of cDCs have recently been described with functional similarities to CD8α+ cDCs. Migratory Langerin+ dermal and lung DC subsets express DEC205+ and CD103+, and like CD8α+ cDCs, are CD11blo/- (27,28). CD8α+ cDC and migratory CD103+ DC populations share the distinctive properties of TLR3 responsiveness (27) and capacity for cross-presentation (26), further supporting the idea that these CD103+ subsets may be related. In spleen, CD103 is co-expressed with CD8α on cDCs (fig. S13A) (29) and is selectively expressed by the ‘CD8α equivalent’ CD24+Sirp-αlo-int cDC subset derived from FL-treated Baft3+/+ BM (fig. S13C), but is not expressed by Batf3-/- splenic cDCs (fig. S13B) or FL-treated Batf3-/- BM cells. This suggests that CD103 expression may require Batf3. In agreement, Batf3-/- mice showed reduced CD103 expression on DEC205+CD8α-CD11blo/- dermal DCs in skin draining lymph nodes (fig. S14).
This study describes a transcription factor that controls development of CD8α+ cDCs. Batf3-/- mice exhibit impaired antigen cross-presentation, impaired CTL responses against viral infection, and impaired responses to tumor challenge. These results suggest an important role for in vivo cross-presentation in CTL responses and provide support for therapeutic approaches that utilize CD8α+ cDCs for the induction of effective immune responses.
Supplementary Material
Acknowledgments
This work was supported by the Howard Hughes Medical Institute (K.M.M.), the Emmy Noether Program of the German Research Foundation (K.H.) and a Burroughs Wellcome Fund Career Award for Medical Scientists (B.T.E.).
References and Notes
- 1.Bevan MJ. J Exp Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Haan JM, Lehar SM, Bevan MJ. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shortman K, Naik SH. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 4.Allan RS, et al. Science. 2003;301:1925–1928. doi: 10.1126/science.1087576. [DOI] [PubMed] [Google Scholar]
- 5.Belz GT, et al. J Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- 6.Belz GT, Shortman K, Bevan MJ, Heath WR. J Immunol. 2005;175:196–200. doi: 10.4049/jimmunol.175.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulz O, et al. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 8.Huang AY, et al. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- 9.Ochsenbein AF, et al. Nature. 2001;411:1058–1064. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 10.Wolkers MC, Stoetter G, Vyth-Dreese FA, Schumacher TN. J Immunol. 2001;167:3577–3584. doi: 10.4049/jimmunol.167.7.3577. [DOI] [PubMed] [Google Scholar]
- 11.Jung S, et al. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Probst HC, et al. Clin Exp Immunol. 2005;141:398–404. doi: 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zenke M, Hieronymus T. Trends Immunol. 2006;27:140–145. doi: 10.1016/j.it.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Iacobelli M, Wachsman W, McGuire KL. J Immunol. 2000;165:860–868. doi: 10.4049/jimmunol.165.2.860. [DOI] [PubMed] [Google Scholar]
- 15.Dudziak D, et al. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 16.Naik SH, et al. J Immunol. 2005;174:6592–6597. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- 17.Barnden MJ, Allison J, Heath WR, Carbone FR. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 18.Wilson NS, et al. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 19.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. J Virol. 2003;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sitati EM, Diamond MS. J Virol. 2006;80:12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shankaran V, et al. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 22.Dunn GP, et al. Nat. 2005;6:722–729. doi: 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 23.Lin ML, Zhan Y, Villadangos JA, Lew AM. Immunol Cell Biol. 2008 doi: 10.1038/icb.2008.3. [DOI] [PubMed] [Google Scholar]
- 24.Belz GT, et al. Proc Natl Acad Sci USA. 2004;101:8670–8675. doi: 10.1073/pnas.0402644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waithman J, et al. J Immunol. 2007;179:4535–4541. doi: 10.4049/jimmunol.179.7.4535. [DOI] [PubMed] [Google Scholar]
- 26.del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Forster R. J Immunol. 2007;178:6861–6866. doi: 10.4049/jimmunol.178.11.6861. [DOI] [PubMed] [Google Scholar]
- 27.Sung SS, et al. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 28.Bursch LS, et al. J Exp Med. 2007;204:3147–3156. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edwards AD, et al. J Immunol. 2003;171:47–60. doi: 10.4049/jimmunol.171.1.47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.