

Abstract
Approximately one-third of newly synthesized eukaryotic proteins are targeted to the secretory pathway, which is composed of an organellar network that houses the enzymes and maintains the chemical environment required for the maturation of secreted and membrane proteins. Nevertheless, this diverse group of proteins may fail to achieve their native states and are consequently selected for ER associated degradation (ERAD). Over the past few years, significant effort has been made to dissect the components of the core ERAD machinery that is responsible for the destruction of most ERAD substrates. Interestingly, however, some ERAD substrates associate with dedicated chaperone-like proteins that target them for proteolysis or protect them from destruction. Other substrates fold and function normally but can be selected for ERAD by protein adaptors that identify and transmit regulatory cues.
Introduction
A significant percentage of eukaryotic proteins are destined for the extracellular space or the plasma membrane. Many other proteins ultimately reside within intracellular organelles, such as the endoplasmic reticulum (ER), the Golgi apparatus, and the lysosome/vacuole. Nearly all of the proteins in these compartments are synthesized on ER-bound ribosomes and enter the ER lumen and/or membrane during or soon after their synthesis [1]. Perhaps not surprisingly, enzymes reside within the ER that facilitate protein folding and post-translational modification [2,3]. Among these enzymes are protein disulfide isomerase (PDI), which catalyzes the oxidation of disulfide bonds, the signal peptidase, which cleaves the N-terminal signal peptide, and the oligosaccharyl transferase, which appends a core, N-linked glycan onto secreted proteins that contain an Asn-X-Ser/Thr recognition motif. Other prominent ER residents that facilitate protein maturation are molecular chaperones. There are several classes of molecular chaperones that reside in the ER, but all have the potential to prevent the accumulation of off-pathway folding intermediates and help retain newly synthesized proteins in solution. The activities of some chaperones, such as Hsp70 and Hsp90, are coupled to the binding and hydrolysis of ATP. Protein folding in the ER is also enhanced by the maintenance of a unique chemical environment. For example, the ER is more oxidizing than the cytoplasm and is calcium-rich. These attributes, respectively, favor the formation of disulfide bonds and are necessary for the function of calcium-binding chaperones. The native conformation of a protein is thermodynamically more stable that the unfolded state. Nevertheless, the protein-folding pathway contains energy “hills”, which represent thermodynamic folding barriers, and lower-energy “valleys”, which represent folding intermediates [4]. Thus, polypeptides that enter the ER transiently populate partially folded conformations. Such conformers, if present at high concentrations, may illegitimately associate with other, functional proteins, and exert dominant negative effects on ER homeostasis. In addition, the ER may have to contend with a temporary loss of ER calcium, thermal stress, energy depletion, or the synthesis of mutant proteins. Each of these events may similarly result in the accumulation of toxic folding intermediates. Furthermore, unfolded proteins may arise within the ER due to stochastic errors during transcription and/or translation. To correct folding mistakes and dispose of damaged polypeptides, an ER quality control (ERQC) system has evolved. The synthesis of components of the ERQC machinery, which includes molecular chaperones and enzymes such as PDI, is induced via the Unfolded Protein Response (UPR) [5], and aberrant secreted and membrane proteins can be destroyed via a process termed ER associated degradation (ERAD) [6,7]. Genetic evidence supports the notion that the UPR and ERAD serve as complementary pathways to maintain ERQC.
The degradation of ERAD substrates requires a multi-catalytic, ~2.5Mda, 26S protease, known as the proteasome, which resides in the cytoplasm and nucleus and on the cytoplasmic face of the ER membrane, but not within the ER lumen. The proteasome acts complementarily to the lysosome/vacuole to mediate the destruction of most proteins in eukaryotic cells, and consists of a 19S “cap” (PA700) and a 20S “core” particle [8,9]. The 19S particle contains factors that bind and remove polyubiquitin tags that become attached to proteasome-targeted substrates, a modification that aids in targeting substrates to the proteasome. The 19S particle also contains a ring of AAA-ATPase proteins that helps funnel proteins into the 20S core. The 20S core contains duplicated sets of proteases that possess chymotryptic, tryptic, and peptidyl-glutamyl-like activity.
A significant percentage of cellular proteasomes are found at the cytoplasmic face of the ER [10], which likely facilitates the efficient destruction of ERAD substrates. In principle, ER-bound proteasomes can directly interact with cytoplasmic domains in integral membrane ERAD substrates. It is less clear how integral membrane and ER luminal segments of these proteins are subsequently destroyed, although some integral membrane proteins have been observed to be completely liberated from the ER membrane prior to degradation [11,12]. ER-resident, soluble substrates that are selected for destruction must also be transported to the cytoplasm to engage the proteasome. These “retro-translocation” or “dislocation” events most likely require a proteinaceous channel in the ER, but the identity of this channel remains contentious. What is clear is that the delivery of almost all soluble and integral membrane ERAD substrates to the cytoplasm and their engagement by the proteasome requires another AAA, ATP-hydrolyzing protein complex that resides in the cytoplasm and on the cytoplasmic face of the ER membrane [13]. The energy-requiring component of this complex is Cdc48 (in yeast) or p97 (in mammals). Other ERAD substrates appear to be retro-translocated from the ER directly by the proteasome [14,15].
During retro-translocation most ERAD substrates are ubiquitinylated, which facilitates ER extraction by the Cdc48/p97 complex. Thus, ER-resident E3 ubiquitin ligases, which append ubiquitin onto ERAD substrates, play key roles during degradation. Two conserved ubiquitin ligases, known as Doa10 and Hrd1, have been found to facilitate the degradation of every ubiquitinylated substrate in yeast [16-18]. Because these proteins possess multiple transmembrane-spanning segments, it has also been suggested that they moonlight as the long-sought retro-translocation channel, or comprise a component of the channel [19]. They also may directly contribute to the recognition of some ERAD substrates. A similar, but more complex, situation appears to exist in mammals, where two Hrd1p homologues (Hrd1 and gp78) and a putative Doa10 homologue (TEB4), coexist with several additional ER membrane-located ligases, possibly with more specialized roles (e.g. RMA1/RNF5 and Kf-1) [20,21].
Because approximately one-third of all proteins encounter the secretory pathway, and because each of these proteins can potentially misfold or fail to acquire the proper post-translational modifications, there has been significant effort to understand how ERAD substrates are selected. As noted above, molecular chaperones aid in the folding of nascent polypeptides. Some of these same chaperones may also target misfolded proteins for ERAD. In addition, glycan-binding lectins in the ER act as critical mediators during the selection of some ERAD substrates [22]. Recent evidence indicates that there is a sequential interaction between distinct lectins prior to retro-translocation [23,24].
As the number of ERAD substrates has grown, it has become possible to classify them into distinct groups based on the site of the mis-folding lesion; thus, proteins with lesions in the lumenal space are referred to as “ERAD-L” substrates, proteins with lesions within the ER membrane are referred to as ERAD-M” substrates, and proteins with lesions in the cytoplasm are referred to as “ERAD-C” substrates (Fig. 1). Based on studies in yeast, these classes can also be distinguished by the specific E3 ubiquitin ligase that is required during degradation [25], and to some extent by the requirement for specific chaperones and lectins [16,17,26].
Fig. 1. Degradation of ERAD-L, -M, and —C substrates.
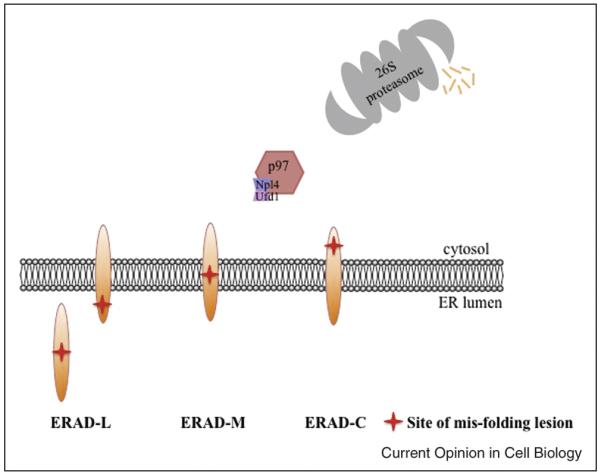
Soluble and integral membrane proteins with mis-folding lesions in the lumen, and integral membrane proteins with mis-folding lesions within the membrane and in the cytoplasmic space are depicted. In nearly all cases, substrates are delivered to the proteasome for degradation in a process that requires the p97 complex (also known as the Cdc48 complex in yeast).
The increase in the number of known ERAD substrates has also led to the discovery that some substrates engage proteins that seem to be devoted to either enhancing or preventing their degradation. In the following sections, we will briefly describe how these substrate-specific factors were identified and function.
Substrate-Specific Mediators of ERAD
Model substrates in yeast
Yeast contain an essential plasma membrane H+-ATPase, known as Pma1, which spans the membrane 12 times and oligomerizes and folds within the ER. Perhaps because of its essential function and complex folding pathway, the ERAD of some Pma1 mutants utilizes a committed chaperone [27]. This chaperone, known as Eps1, is one of five PDIs in the yeast ER [28], and thus far it appears that the Pmal mutant protein is the only ERAD substrate that requires Eps1. In contrast to Eps1, which is “pro-degradative”, the folding of amino acid permeases in the ER in yeast require a factor known as Shr3 [29]. In the absence of Shr3, the permeases begin to aggregate in the ER and are targeted for ERAD. Interestingly, the amino acid permeases possess 12 transmembrane segments, and Shr3—which is also a membrane protein—retains the first five segments in a folding-competent conformation. Thus, Shr3 exhibits bona fide chaperone activity for a specific class of proteins.
The regulation of lipid metabolism: ApoB and HMG CoA reductase
Apolipoprotein B (ApoB) is a large, ~550 kD protein that binds to cholesterol, cholesterol esters, and phospholipids as it translocates into the ER in the liver and small intestine. This process is required for the biogenesis and secretion of low- and very low-density lipoproteins, and ApoB is the most abundant protein in these cholesterol-carrying particles. If, however, the ER is cholesterol- or lipid-poor then ApoB only partially enters the ER and is retro-translocated and co-translationally targeted for ERAD [30]. Therefore, lipids and cholesterol prevent ApoB ERAD. In turn, the loading of these metabolites onto ApoB requires a committed chaperone, known as the microsomal triglyceride transfer protein (MTP) [31,32]. MTP is a dimer of PDI and a 97 kDa “M” subunit. If MTP is absent or is inactivated ApoB is targeted for ERAD regardless of whether the synthesis and secretion of LDLs and VLDLs are needed. In fact, reduced levels of circulating cholesterol and triglycerides are evident in individuals who lack MTP activity [33]. This condition, known as abetalipoproteinemia, leads to a defect in the utilization of fat-soluble vitamins and profound developmental defects. In contrast, a more modest reduction in MTP activity through the administration of an inhibitor, known as BMS-201038, has been shown to reduce circulating cholesterol levels [34]. Consequently, this substrate-specific ERAD regulator has emerged as a promising therapeutic target.
Another ERAD substrate linked to lipid metabolism and whose stability is modulated by a committed chaperone-like molecule is the multi-spanning transmembrane protein hydroxy-methylglutaryl (HMG)-coenzyme A (CoA) reductase, which has been the topic of investigation for several decades. Because HMG-CoA reductase catalyzes the rate-limiting step in cholesterol synthesis (HMG-CoA conversion to mevalonate), this enzyme is highly regulated. One mode of control is accelerated ERAD that occurs when sterols or mevalonate metabolites (e.g., farnesol) are in excess. This occurs in both mammalian and yeast cells and has been examined in detail in both systems [35,36]. In mammalian cells, sterols cause the binding of INSIG1 or INSIG2 to the reductase, which in turn appears to recruit an ER membrane E3 ubiquitin ligase, gp78, to initiate ERAD [37]. INSIGs are ER membrane proteins that interact with the transmembrane domains of the reductase and other ER membrane proteins in a sterol-sensitive manner and are adaptors that effect regulation of those proteins. Interestingly, the yeast homologs of INSIGs (NSGs) play a different role; these proteins interact with and stabilize yeast HMG-CoA reductase [38]. In yeast, the reductase appears to be marked for ERAD by a farnesol-induced structural transition [39] that allows for recognition by the Hrd1-containing complex that mediates ERAD-M. Recent data indicate that this interaction represents a rate-limiting step for the Cdc48-dependent retro-translocation of the enzyme to the cytosol [40].
The regulation of calcium release from the ER
Inositol trisphosphate (IP3) receptors are polytopic ER membrane proteins that tetramerize to form channels that govern the release of Ca2+ stored within the ER lumen of mammalian cells [41]. Channel opening occurs in response to the binding of IP3 and Ca2+, via an as-yet undefined conformational change. Activation also leads to proteasome-mediated degradation of IP3 receptors [42]. This suggests that the conformational change associated with channel opening exposes motifs, perhaps hydrophobic patches, which allow for the recognition of IP3 receptors as ERAD substrates [43]. Interestingly, a large (~1MDa) ring-shaped complex composed of the ER membrane proteins SPFH1 and SPFH2 has recently been shown to associate with IP3 receptors immediately after their activation. These proteins, in turn, mediate the ubiquitination of the IP3 receptors [44,45]. The SPFH1/2 complex also associates with other proteins that undergo ERAD, e.g. the α1D-adrenergic receptor [46], but has a more modest effect on the stability of model ERAD substrates [44]. Thus, while the SPFH1/2 complex appears to be a selective recognition factor for IP3 receptor ERAD (Fig. 2), it may also play a role in the degradation of other ERAD substrates.
Fig. 2. Model of SPFH1/2 complex-mediated ERAD of activated IP3 receptors.

Upon binding of the co-agonists IP3 and Ca2+, IP3 receptor tetramers undergo a conformational change that both opens the Ca2+ channel to allow for the release of ER Ca2+ stores, and triggers association of the SPFH1/2 complex. The SPFH1/2 complex targets activated IP3 receptors for ERAD, perhaps by recruiting the E2 and E3 that catalyze IP3 receptor polyubiquitination. Polyubiquitinated IP3 receptors are then extracted from the ER membrane through the action of the p97-Ufd1-Npl4 complex, and are delivered to the proteasome for degradation.
ERAD mediators produced by pathogens
Some substrate-specific mediators of degradation are produced by pathogens, which augments pathogen replication or immune system evasion in the host. For example, the US2 and US11 human cytomegalovirus gene products interact with class I major histocompatibility complex heavy chains (MHC-I HCs) in infected host cells. The interaction between US2 and US11 with MHC-I HCs facilitates the p97-dependent retrotranslocation and degradation of this component, which is normally required for the presentation of peptides to the immune system [47-50]. Interestingly, US11 also recruits a factor that anchors p97 at the ER membrane and associates with the “derlins”, conserved proteins that have been suggested to act as retro-translocation channels. In addition, US11 functions with TRAM1, which is thought to facilitate the insertion of transmembrane proteins into the lipid bilayer during synthesis [51]. Thus, US11 might re-engineer TRAM1 activity to catalyze MHC-I HC retro-translocation.
And the list continues to grow: Variations on a theme
Some modifiers may act on unique sub-classes of ERAD substrates. For example, a Cdc48/p97- and Hrd1-interacting factor known as Herp was recently found to enhance the ERAD of only non-glycosylated proteins, and may help transfer these substrates to the proteasome [52]. Also, interestingly, one ERAD substrate contains a built-in chaperone-like domain. Pca1, which is a yeast cadmium transporter, is only targeted for ERAD in the absence of cadmium. In the presence of cadmium, a metal-binding domain imparts a conformational change in the protein that prevents substrate ubiquitinylation and degradation [53]. It is likely that other proteins are regulated in a similar manner.
Conclusions
The existence of substrate-specific ERAD mediators that appear to affect the stability of distinct substrates or distinct classes of substrates has only just begun to be appreciated, and this most likely reflects the fact that the number of known ERAD substrates has grown substantially over the past few years. Thus, it is likely that members of this “new” family will continued to grow. It is also vital that additional work is undertaken to define the molecular mechanisms by which these mediators act. In particular, the key challenges now are to define the structural basis for the interactions between ERAD mediators and their specific substrates, and to establish how lumenal ER proteins and complex polytopic proteins are extracted from the ER and delivered to the proteasome.
Acknowledgments
Work in the authors’ laboratories on ERAD is supported by grants from the National Institutes of Health (GM75061 to J.L.B. and DK49194 to R.J.H.W.) and the Cystic Fibrosis Foundation (BRODSK08XX0 to J.L.B.).
Contributor Information
Jeffrey L. Brodsky, Department of Biological Sciences 274 Crawford Hall University of Pittsburgh Pittsburgh, PA 15260 Tel.412-624-4831; Fax.412-624-4759; jbrodsky@pitt.edu.
Richard J.H. Wojcikiewicz, Department of Pharmacology 3307 Weiskotten Hall SUNY Upstate Medical University Syracuse, NY 13210 Tel.315-464-7956; Fax.315-464-8014; wojcikir@upstate.edu
References
- 1.Wickner W, Schekman R. Protein translocation across biological membranes. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- 2.Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- 3.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 4.Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272:5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x. [DOI] [PubMed] [Google Scholar]
- ••5.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. Yeast were transcriptionally profiled to uncover the full spectrum of factors induced by the unfolded protein response (UPR), and the UPR and ERAD were shown to function as complementary legs during ER protein quality control.
- 6.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kostova Z, Wolf DH. NEW EMBO MEMBER’S REVIEW: For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. Embo J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 9.Hanna J, Finley D. A proteasome for all occasions. FEBS Lett. 2007;581:2854–2861. doi: 10.1016/j.febslet.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enenkel C, Lehmann A, Kloetzel PM. Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelop-ER network in yeast. Embo J. 1998;17:6144–6154. doi: 10.1093/emboj/17.21.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •11.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. The first evidence for the extraction of a membrane protein into the cytoplasm was presented.
- •12.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. The pathway for the extraction of a membrane protein into the cytoplasm was reconstituted through the use of an in vitro ERAD assay.
- 13.Jentsch S, Rumpf S. Cdc48 (p97): a ‘molecular gearbox’ in the ubiquitin pathway? Trends Biochem Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, Romisch K, DeMartino GN, Thomas PJ, Brodsky JL. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. Embo J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •15.Mayer TU, Braun T, Jentsch S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. Embo J. 1998;17:3251–3257. doi: 10.1093/emboj/17.12.3251. Evidence was provided that the proteasome was sufficient for both the extraction and degradation of an ERAD substrate.
- 16.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 17.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 18.Gauss R, Sommer T, Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281:4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- 20.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 21.Maruyama Y, Yamada M, Takahashi K, Yamada M. Ubiquitin ligase Kf-1 is involved in the endoplasmic reticulum-associated degradation pathway. Biochem Biophys Res Commun. 2008;374:737–741. doi: 10.1016/j.bbrc.2008.07.126. [DOI] [PubMed] [Google Scholar]
- 22.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 23.Clerc S, Hirsch C, Oggier DM, Deprez P, Jakob C, Sommer T, Aebi M. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol. 2009;184:159–172. doi: 10.1083/jcb.200809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell. 2008;32:870–877. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •25.Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. Data demonstrating the existence of an ERAD-L, ERAD-C, and ERAD-M pathway were presented.
- 26.Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct Machinery Is Required in Saccharomyces cerevisiae for the Endoplasmic Reticulum-associated Degradation of a Multispanning Membrane Protein and a Soluble Luminal Protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- ••27.Wang Q, Chang A. Eps1, a novel PDI-related protein involved in ER quality control in yeast. Embo J. 1999;18:5972–5982. doi: 10.1093/emboj/18.21.5972. Through the use of a yeast genetic screen, Eps1 was identified as the first substrate specific mediator of ERAD.
- 28.Norgaard P, Westphal V, Tachibana C, Alsoe L, Holst B, Winther JR. Functional differences in yeast protein disulfide isomerases. J Cell Biol. 2001;152:553–562. doi: 10.1083/jcb.152.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kota J, Gilstring CF, Ljungdahl PO. Membrane chaperone Shr3 assists in folding amino acid permeases preventing precocious ERAD. J Cell Biol. 2007;176:617–628. doi: 10.1083/jcb.200612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brodsky JL, Fisher EA. The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol Metab. 2008;19:254–259. doi: 10.1016/j.tem.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X, Zhou M, Huang LS, Wetterau J, Ginsberg HN. Demonstration of a physical interaction between microsomal triglyceride transfer protein and apolipoprotein B during the assembly of ApoB-containing particles. J Biol Chem. 271:10277–10281. doi: 10.1074/jbc.271.17.10277. [DOI] [PubMed] [Google Scholar]
- 32.Bakillah A, Hussain MM. Binding of microsomal triglyceride transfer protein to lipids results in increased affinity for apolipoprotein B. J Biol Chem. 2001;276:31466–31473. doi: 10.1074/jbc.M100390200. [DOI] [PubMed] [Google Scholar]
- 33.Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, Schmitz J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- 34.Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–156. doi: 10.1056/NEJMoa061189. [DOI] [PubMed] [Google Scholar]
- 35.DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18:609–621. doi: 10.1038/cr.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hampton RY, Garza RM. Protein Quality Control as a Strategy for Cellular Regulation: Lessons from Ubiquitin-Mediated Regulation of the Sterol Pathway. Chem Rev. 2009 doi: 10.1021/cr800544v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •37.Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19:829–840. doi: 10.1016/j.molcel.2005.08.009. The direct coupling between a substrate-specific ERAD mediator and the uniquitin machinery was obtained in mammalian cells.
- 38.Flury I, Garza R, Shearer A, Rosen J, Cronin S, Hampton RY. INSIG: a broadly conserved transmembrane chaperone for sterol-sensing domain proteins. EMBO J. 2005;24:3917–3926. doi: 10.1038/sj.emboj.7600855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••39.Shearer AG, Hampton RY. Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. Embo J. 2005;24:149–159. doi: 10.1038/sj.emboj.7600498. A limited protease assay was used to detect a lipid-dependent conformational change in HMG-CoA reductase, suggesting that a lipid could turn a functional form of the enzyme into a conformationally altered protein that was subsequently recognized by the ERAD machinery.
- 40.Garza RM, Sato BK, Hampton RH. In vitro analysis of Hrd1-mediated retrotranslocation of its multi-spanning membrane substrate HMG-CoA reductase. J Biol Chem. doi: 10.1074/jbc.M809607200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wojcikiewicz RJ. Regulated ubiquitination of proteins in GPCR-initiated signaling pathways. Trends Pharmacol Sci. 2004;25:35–41. doi: 10.1016/j.tips.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 43.Alzayady KJ, Panning MM, Kelley GG, Wojcikiewicz RJ. Involvement of the p97-Ufd1-Npl4 complex in the regulated endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors. J Biol Chem. 2005;280:34530–34537. doi: 10.1074/jbc.M508890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••44.Pearce MM, Wang Y, Kelley GG, Wojcikiewicz RJ. SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J Biol Chem. 2007;282:20104–20115. doi: 10.1074/jbc.M701862200. An ER-resident substrate recognition factor was identified that acted at an early recognition step in targeting IP3 receptors for ERAD.
- 45.Pearce MM, Wormer DB, Wilkens S, Wojcikiewicz RJ. An ER membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of IP3 receptors. J Biol Chem. doi: 10.1074/jbc.M809801200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyssand JS, DeFino MC, Tang XB, Hertz AL, Feller DB, Wacker JL, Adams ME, Hague C. Blood pressure is regulated by an alpha1D-adrenergic receptor/dystrophin signalosome. J Biol Chem. 2008;283:18792–18800. doi: 10.1074/jbc.M801860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••47.Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. A viral protein product was shown to select a specific membrane protein in host cells and rapidly target this component of the immune system for ERAD.
- 48.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 49.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 50.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 51.Oresic K, Ng CL, Tortorella D. TRAM1 participates in human cytomegalovirus US2- and US11-mediated dislocation of an endoplasmic reticulum membrane glycoprotein. J Biol Chem. 2009;284:5905–5914. doi: 10.1074/jbc.M807568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adle DJ, Lee J. Expressional control of a cadmium-transporting P1B-type ATPase by a metal sensing degradation signal. J Biol Chem. 2008;283:31460–31468. doi: 10.1074/jbc.M806054200. [DOI] [PMC free article] [PubMed] [Google Scholar]