

Abstract
Aberrant cytosine methylation in promoter regions leads to gene silencing associated with cancer progression. A number of DNA methyltransferase inhibitors are known to reactivate silenced genes; including 5-azacytidine and 2-(1H)-pyrimidinone riboside (zebularine). Zebularine is a more stable, less cytotoxic inhibitor compared to 5-azacytidine. To determine the mechanistic basis for this difference, we carried out a detailed comparisons of the interaction between purified DNA methyltransferases and oligodeoxyribonucleotides (ODNs) containing either 5-azacytosine or 2-(1H)-pyrimidinone in place of the cytosine targeted for methylation. When incorporated into small ODNs, the rate of C5 DNA methyltransferase inhibition by both nucleosides is essentially identical. However, the stability and reversibility of the enzyme complex in the absence and presence of cofactor differs. 5-Azacytosine ODNs form complexes with C5 DNA methyltransferases that are irreversible when the 5-azacytosine ring is intact. ODNs containing 2-(1H)-pyrimidinone at the enzymatic target site are competitive inhibitors of both prokaryotic and mammalian DNA C5 methyltransferases. We determined that the ternary complexes between the enzymes, 2-(1H)-pyrimidinone inhibitor, and the cofactor S-adenosyl methionine are maintained through the formation of a reversible covalent interaction. The differing stability and reversibility of the covalent bonds may partially account for the observed differences in cytotoxicity between zebularine and 5-azacytidine inhibitors.
Keywords: zebularine, DNA methylation, 2(1H)-Pyrimidinone riboside, DNA methyltransferase, 5-aza-2'-deoxycytidine
1. Introduction
Methylation of cytosine residues in DNA provides epigenetic information that can lead to gene silencing during embryonic development [1-3] and the progression of cancer [4]. Since these processes mediated by DNA methylation can be reversed when DNA replication occurs in the absence of maintenance methylation, inhibitors of DNA methyltransferases are attractive candidate drugs with the potential to reestablish antiproliferative signals and other critical cellular functions that are abnormally silenced in tumor cells by DNA methylation.
Among the first specific inhibitors of DNA (cytosine C5)-methyltransferases (DNA C5-MTases) described were cytidine analogs with nitrogen replacing C5 of the pyrimidine ring (5-azacytidine (ZCyd) and 5-aza-2'-deoxycytidine (ZdCyd) [5, 6]). When incorporated into DNA in place of the cytidine (Cyd) targeted for methylation, these compounds are efficient suicide inhibitors of DNA C5-MTases both in vivo [6-12] and in vitro [10-12]. ZCyd is phosphorylated by uridine-cytidine kinase and can be incorporated into both RNA and DNA whereas ZdCyd, which is phosphorylated by deoxycytidine kinase, is only incorporated into DNA [13, 14]. Once incorporated into DNA, azanucleoside analogs can effectively deplete the cell of active enzyme by forming irreversible covalent adducts with DNA C5-MTases, resulting in global hypomethylation. Despite the initial success of these agents for treating sickle cell anemia, myelodysplastic syndrome, and a number of other cancers [15-20], there are serious treatment-associated side effects. These include myelotoxicity and DNA mutations due to incorporation of the nucleosides into genomic DNA [21], a potential factor in cancer recurrence. Zebularine [2-(1H) pyrimidinone riboside] is a highly stable cytidine analog that is significantly less toxic than ZdCyd (Figure 1). First identified as a bacteriostat [22], zebularine was later found to act as a transition state inhibitor of cytidine deaminase (CDA) [23-27].
Figure 1.
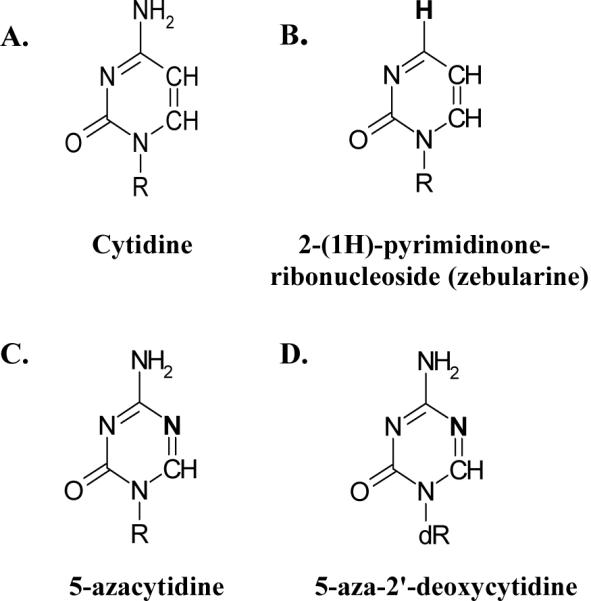
The structure of (A) cytidine (Cyd), and its analogs (B) 2-(1H)-pyrimidinone ribonucleoside (zebularine), (C) 5-azacytidine (ZCyd), and (D) 5-aza-2'-deoxycytidine (ZdCyd). R=ribose and dR=2'-deoxyribose.
At high micromolar concentrations zebularine has been shown to inhibit mammalian DNA C5-MTases in cultured cells and mammalian tumors after prolonged exposure [28-30]. In vitro studies have demonstrated the formation of stable inhibitory complexes between the bacterial DNA C5-MTases M.HgaI and M.MspI and synthetic oligodeoxyribonucleotides (ODNs) with the 2-(1H) pyrimidinone (2P) replacing C in their recognition site [30, 31].
In vivo experiments revealed several logs difference in potency between ZdCyd and zebularine on mammalian DNA methylation [28, 29], but no published studies have directly compared the interactions between Dnmt1 and DNA containing the two analogs. Therefore, we sought to directly compare the potency and inhibitory mechanisms of DNA containing ZdCyt or 2P on Dnmt1. We synthesized small ODN inhibitors containing either ZdCyt or 2P in place of the target C study DNA C5-MTase inactivation. Our analysis included determination of the kinetics of inhibition, thermal stability of complexes, and rate of dissociation in the presence and absence of cofactors. Here, we report that ODNs containing 2P at the enzymatic target site (Figure 2) are competitive inhibitors of both prokaryotic (M.HhaI) and mammalian DNA C5-MTases (Dnmt1). Moreover, we determined that the ternary complexes between M.HhaI:2P-ODN:S-adenosyl methionine (AdoMet) and Dnmt1:DM72P18:AdoMet are maintained through the formation of a reversible covalent interaction.
Figure 2.

Sequence of ODNs used in this study. M=5MeC. Inhibitors have modified target sites that contain either 5-azacytosine (Z) or zebularine aglycon (2P) bases in place of the cytosine (C) residue targeted for methylation (underlined).
2. Materials and Methods
2.1 Oligodeoxyribonucleotide synthesis and annealing
Single-stranded (ss) ODNs containing 2P or ZCyt were synthesized as previously described [32-34]. Each ODN was resuspended in Tris-EDTA, pH 8.0 and stored at -20°C (See Figure 2 for sequences). Double stranded (ds) ODN inhibitors of M.HhaI were formed by annealing 1 μg of each 13mer at 65 °C for 10 min and slowly cooling to ~45 °C over a period of 60 min.
2.2 Enzyme purification
M.HhaI was overexpressed in Escherichia coli strain ER1727 containing the pUHE25HhaI plasmid (generously provided by Dr. S. Kumar, New England Biolabs) and purified as described previously [35] using a HiTrap Sepharose SP HP column (cation exchange, Amersham Biosciences, Piscataway, NJ). Purified recombinant Dnmt1 was isolated as described previously [36].
2.3 Assays determining inhibitor potency
The procedure for measuring the rate of inactivation for M.HhaI has been previously described in detail [34]. Briefly, reaction mixtures containing increasing concentrations of substrate (AMp:A') were incubated at 37 °C in the absence or presence of ODN inhibitor (Figure 2). Reactions were terminated after 5 min, a time point within the linear range of the assay. Incorporation of methyl-H3 into DNA was quantified by liquid scintillation counting [37, 38]. Each reaction was performed in duplicate. Error bars indicate standard error from the mean of three independent experiments. The Km and Vmax data were determined using Graph Pad 3.0 software. A Lineweaver-Burk plot of the resulting data was prepared to determine kI values.
Dnmt1 inactivation reactions containing 0.4-0.6 μM enzyme were pre-incubated with 1-80 μM ODN inhibitor (Figure 2) for 0-4 min at 22 °C in imidazole buffer (100 mM imidazole, pH 7.5, 20 mM EDTA) containing 1.3 μM DM7 (substrate), 50 μM AdoMet, and 0.1 mg/ml BSA. Duplicate 1 μl aliquots were removed after specified times of pre-incubation and diluted with 49 μl of assay mixture containing 0.2 mg/ml BSA, 1.2 μM AdoMet, 0.1 μg poly dI-dC:dIdC (~3.45μM dinucleotide), and 0.8 μM 3H-AdoMet (specific activity 3015.5 GBq/mmol) in imidazole buffer. The diluted reactions were incubated for an additional 60 min at 37 °C to determine the amount of Dnmt1 activity remaining. Substrate methylation was quantified as described above.
For each concentration of inhibitor, the amount of remaining enzymatic activity was plotted versus the time of pre-incubation. The t1/2, or time necessary for each concentration of inhibitor to reduce the total activity by one-half was calculated using a fourth order polynomial regression. The t1/2 value was then plotted versus the inverse of the concentration of inhibitor, where kinactivation = ln2/slope. The KI was extrapolated from the resulting graph, with kI = -1/x-intercept.
2.4 Binding Assays
2.4.1 SDS-PAGE analysis of M.HhaI-ODN Complexes
Reaction mixtures containing 32P-end-labeled ODN (50 nM), M.HhaI (2.5 nM), and cofactor (100-200 μM) in Methylation Reaction (MR) buffer (50 mM Tris pH 7.5, 10 mM EDTA, and 5 mM β-mercaptoethanol (BME)) were incubated at 37 °C for 1 h. Reactions were brought to a final concentration of 10% (v/v) glycerol, 1% (v/v) SDS, and 1% (v/v) BME and incubated at the indicated temperatures for 5 min. They were then loaded directly onto a 6% SDS-polyacrylamide gel and electrophoresed at 200 V for 60 min at 4 °C. Dried gels were autoradiographed by exposure to Kodak X-OMAT LS film overnight.
2.4.2 SDS-PAGE analysis of Dnmt1-ODN complexes
Reactions containing 32P-end-labeled ODN (1 μM), Dnmt1 (0.4 μM), and cofactor (100-200 μM) in imidazole buffer were incubated at 37 °C for 1 h. Prior to heating, reactions were brought to concentrations of 1% (v/v) SDS and 1% (v/v) BME in SDS loading buffer and incubated at the indicated temperatures for 5 min. Samples were loaded directly onto a 6%-SDS-polyacrylamide gel, and electrophoresed at 200 V for 3 h (or until the dye front reached the bottom of the gel) at 4 °C. Dried gels were autoradiographed by exposure to Kodak X-OMAT LS film overnight.
2.4.3 Non-denaturing gel shift assay
Binding reaction mixtures containing 45 nM 32P-end labeled ds 2P-ODN (Figure 2), 108 nM M.HhaI, 75 ng poly dAdT:dAdT (6 μM dAdT dinucleotide), and 100 μM cofactor (as indicated in Figure 5) in MR buffer containing 13% (v/v) glycerol were incubated at 22 °C for 30 min as described [38]. After 30 min, one hundred fold excess of unlabeled, ds 2P-ODN (4.5 μM) was added to each binding reaction and incubations were continued at 22 °C for the indicated times. Enzyme-ODN complexes were subjected to electrophoresis at 150 V for 2-3 h on a 10% (w/v) native polyacrylamide gel that had been pre-electrophoresed at 100 V for 1 h in TBE buffer (89 mM Tris borate (pH 8.0), 2 mM EDTA).
Figure 5.

Dissociation of 2P-ODN from M.HhaI. (A) Binding reactions containing 45 nM 32P-end labeled 2P-ODN, 108 nM M.HhaI, 75 ng poly dAdT:dAdT, and 100 μM cofactor (as indicated) were incubated at 22 °C for 30 min. A 100-fold excess unlabeled 2P-ODN was added to all reactions with the exception of those in lanes 1, 8, and 15, and the incubation continued for the indicated times. Zero time points (lanes 2, 9, and 16) represent reactions in which the unlabeled ODN was added immediately before loading onto a non-denaturing polyacrylamide gel. The percent bound ODN (% bound) was determined by imaging of the dried gel on a Typhoon PhosphoImager (Amersham). (% bound = [counts in enzyme-ODN complex/counts in enzyme-ODN complex + counts in free ODN] X 100). (B) The off rate was calculated by plotting the ln (specific binding) versus the time of incubation in the presence of cold competitor. A summary of the data is presented in Table 1.
Gels were dried and the enzyme-32P-ODN complex quantified by PhosphoImager (Typhoon, Amersham) analysis. The percent ODN in the complex was determined for each sample by dividing the signal in the band containing complex by the sum of the signals from bands containing free ODN and enzyme-ODN complex and multiplying by 100. For dissociation analysis, the natural log of the percent ODN bound in enzyme-ODN complex was plotted against the time of incubation in the presences of unlabeled competitor. The (-) slope of this plot is equal to the koff and t1/2 (=0.693/koff) was calculated.
3. Results
3.1 M.HhaI inhibition by 2P- and ZCyt-ODNs
The ODNs used as substrates or inhibitors in this study (Figure 2) were synthesized by standard, published methods [33-38]. The ability of 2P-ODN to inhibit C5-MTases was compared with that of ZCyt-ODN. Both the bacterial M.HhaI and Dnmt1 were efficiently inhibited by 2P-ODN. The IC50, or concentration at which 50% of the enzymatic activity of was inhibited by 2P-ODN is nearly identical to that of the ZCyt-ODN for both M.HhaI (~20nM or 2.3 nmoles/nmole enzyme) and Dnmt1 (~240nM or 2.5 nmoles/nmol enzyme) (See Figure S1, Supplemental material). This result demonstrates that once 2-(1H)-pyrimidinone (2P) or 5-aza-2'-deoxycytosine (ZCyt) are incorporated into DNA in place of C in CpG sites, they are equally potent inhibitors of both M.HhaI and Dnmt1.
While this type of assay is a convenient method for directly comparing DNA C5-MTase inhibitors, it does not provide information about mechanism. Therefore, rate assays were used to determine Michaelis-Menten kinetic constants for M.HhaI. Increasing concentrations of the substrate AMp:A′ were mixed with the indicated concentrations of inhibitor (0-76 nM). Reactions were initiated by the addition of M.HhaI (8.6 μM), and the rate of incorporation of radiolabeled methyl from [methyl-3H]AdoMet during a 5 min reaction was determined (Materials and Methods). A plot of the rate of incorporation (nM/min) vs. the concentration of AMp:A′ substrate demonstrates that the 2P-ODN is an effective inhibitor of M. HhaI activity in vitro (Figure 3A and Figure S2, Supplemental Material for corresponding Lineweaver-Burk plot). This nonlinear regression model and a Hanes-Woolf plot (Figure 3B) both indicate that the apparent Vmax of the reaction is unchanged while the Km increases with the increase in inhibitor concentration. The apparent equilibrium constant for inhibitor binding (ki) of the 2P-ODN was 9.89 ± 0.56 nM compared to 4.3 ± 0.65 nM for ZCyt ODN. This result is typical of competitive inhibitors that display a high affinity for the catalytic pocket of the targeted enzyme.
Figure 3.

M.HhaI inhibition by 2P-ODN. Reactions containing 8.6 nM M.HhaI, 2.4 μM [methyl-3H]AdoMet, and indicated amounts of 2P-ODN inhibitor were incubated with increasing concentrations of substrate. The rate of incorporation of methyl-3H was determined by liquid scintillation counting. (A) For each concentration the rate was plotted versus the inhibitor concentration. See Figure S2, Supplemental Material for corresponding Lineweaver-Burk plot. (B) A Hanes-Woolf plot of the same rate data. Kinetic determinants extrapolated from each plot are discussed (See Materials and Methods for details). Each reaction was performed in duplicate. Error bars indicate standard error from the mean of three independent experiments.
3.2 Dnmt1 inhibition by 2P- and ZCyt-ODN's
Dnmt1 is a processive enzyme with a catalytic rate 10-100 fold lower than M.HhaI [36, 39]. In addition, interaction of substrate DNA with an allosteric site in the Dnmt1 amino terminal domain can lead to an initial lag in rate of methylation that limits the use of Michaels-Menten kinetic analysis [39]. As an alternative approach, inhibition of Dnmt1 by DM72P18 and DM7ZdCyt18 was characterized by determining kinactivation (maximal rate of enzyme inactivation) and apparent kI (the inhibitor concentration that supports half the maximal rate of enzyme inactivation) [40]. Results for DM72P18 are shown in Figure 4A. Dnmt1 was pre-incubated with increasing amounts of this ODN for the indicated times and the amount of active enzyme remaining was determined by diluting aliquots of the pre-incubation mixture into an assay mixture containing excess DM7 substrate and radiolabeled [methyl-3H]AdoMet. The kI, was determined to be 2.005 ± 0.405 μM (Figure 4B). This is ~ 4.6 fold lower than that observed with DM7ZCyt18 [34] suggesting that Dnmt1 has a somewhat higher affinity for 2P than for ZCyt. The rate of inactivation (kinact) determined by plotting 1/concentration of ODN vs. the time required for half-maximal reduction in activity (t1/2) was 2.65 ± 0.405 min-1 for the DM7 2P18 ODN, in the same range as that reported for DM7ZCyt18 (kinact= 0.96 ± 0.23 min-1) [41]. These results indicate that once 2P is incorporated into a DNA substrate in place of the target C, it displays high affinity for DNA C5-MTases and is comparable to DZCyt in its ability to inhibit DNA methylation[38].
Figure 4.

Dnmt1 inhibition by DM72P18 ODN. Reactions containing 60 nM Dnmt1, 1.3 μM DM7 substrate, and the indicated concentration of DM72P18 inhibitor were incubated at 37 °C. At the specified times, duplicate 1 μl aliquots were removed from the pre-incubation mixture and added to an assay mixture containing 0.71 μM [methyl-3H]AdoMet to detect the remaining enzyme activity as described in Materials and Methods. (A) The percent activity remaining was plotted versus the pre-incubation time for each concentration of inhibitor. (B) The calculated t1/2 was plotted versus 1/concentration of ODN inhibitor. kinactivation=ln2/slope and kI=-1/x-intercept.
3.3 Enzyme-ODN complex formation
Non-denaturing gel shift assays were used to evaluate the stability of complexes formed between M.HhaI and 32P-radiolabeled-2P-ODN. After a 30 min incubation to allow formation of binary (M.HhaI:ODN) or ternary (M.HhaI:ODN:AdoMet or AdoHcy) complexes, a 100-fold excess of unlabeled 2P-ODN was added to each reaction mixture and incubated for up to 48 h. At the indicated time points (Figure 5A), bound 2P-ODN was separated from unbound ODN by PAGE [34]. In the absence of cofactor, ~80% of the input 2P-ODN was bound in binary complexes within 30 min. Addition of either AdoMet or AdoHcy increased the amount of enzyme-ODN complex to ~90%. However, the stability of these complexes was significantly increased compared to those without cofactor. Addition of 100-fold molar excess of unlabeled 2P-ODN decreased the percent of ODN bound in binary complexes by ~20% within 10 min, and by >80% in 48 h (Figure 5A, lanes 1-7). Both the initial decrease in ODN binding (5-10%) and the rates of dissociation were slower for ternary complexes containing either AdoMet (Figure 5, lanes 8-14) or AdoHcy (Figure 5, lanes 15-21).
The t1/2 dissociation rate calculated from the data plotted in Figure 5B was ~20 h for binary complexes and 70-75 h for ternary complexes (Table 1). No significant difference was detected between the effects of AdoMet and AdoHcy on complex stability. These results contrast with those obtained with ZCyt-ODNs (Table 1). Both binary complexes with ZCyt-ODNs and ternary complexes containing ZCyt-ODNs and AdoMet were ~5 fold more stable than binary and ternary complexes formed with 2P-ODNs. However, ternary complexes containing ZCyt-ODNs and AdoHcy were 15 fold more stable than those formed with AdoHcy and 2P-ODN [12, 34]. The increased stability of M.HhaI:2P-ODN complexes in either the presence of AdoMet or AdoHcy demonstrates that, similarly to what has been reported for both 5,6-dihydro-5-azacytosine (DZCyt) and ZCyt [34, 38], inhibition of M.HhaI by DNA containing 2-(1H)-pyrimidinone in place of the C targeted for methylation does not require transfer of a methyl to the 5-position of the pyrimidine ring.
Table 1.
Summary of dissociation experiments of M.HhaI-ODN complex
| Cofactor | 2P-ODN |
ZCyt-ODN |
|
|---|---|---|---|
| koff(hours-1) | t1/2 (hours) | t1/2 (hours) | |
| None | 0.0349 | 20 | 110 |
| AdoMet | 0.0099 | 70 | 310 |
| AdoHcy | 0.0092 | 75 | 1100 |
For dissociation analysis, the natural log of the percent bound in the M.HhaI:2P-ODN complex was plotted versus the time of competition of pre-formed complexes containing radio labeled 2P-ODN with unlabeled 2P-ODN (Figure 5B).
The koff (= -slope) and the t1/2 (=0.693/koff) were calculated from this plot. The t1/2 values for ZCyt-ODN in the presence and absence of cofactor are presented for comparative purposes [34].
Stability of Dnmt1:DM72P18 complexes could not be accurately evaluated by electrophoresis on non-denaturing gels due to aggregation of enzyme and ODN in the loading wells. However, both M.HhaI and Dnmt1 form tightly closed complexes with ODN inhibitors that are stable to PAGE under denaturing conditions [see references [34] and [42] for detailed description]. This method was used to directly compare the stability 2P-ODN binding to M.HhaI (Figure 6A) and DM72P18 binding to Dnmt1 (Figure 6B). In order to determine complex stability at temperatures greater than 70 °C (a measure of the stability of a covalent bond between DNA methyltransferase and its substrate), each methyltransferase was incubated for 30 min at 37 °C with a 2.5-fold molar excess of radiolabeled 2P-ODN (M.HhaI) or DM72P18 (Dnmt1). Reactions were then heated to the indicated temperatures for 5 min in SDS loading buffer (with a final concentration of 1% (v/v) SDS, 1% (v/v) BME) prior to SDS-PAGE. Both M.HhaI (Figure 6, panel A) and Dnmt1 (Figure 6, panel B) formed detectable complexes. In both cases, a small enhancement of complex formation was observed in the presence of cofactor (Compare lane 1 with lanes 2-3 in both panels). Similarly to complexes formed between M.HhaI and ZCyt-ODNs [34], complexes of M.HhaI with 2P-ODN migrate more rapidly in denaturing gels than free enzyme. Open ODN:enzyme complexes, characterized as having a slower mobility in non-denaturing gels than closed complexes [34, 38], dissociate in the presence of SDS and, unless covalently linked are not detectable in denaturing gels [34, 42]. Heating at ≥50 °C led to almost complete dissociation of binary closed complexes of M.HhaI with 2P-ODN (Figure 6, Panel A, lane 7). At 80 °C, ternary closed complexes containing M.HhaI, 2P-ODN and AdoHcy dissociated, but those containing AdoMet remained intact (Figure 6, Panel A, lanes 14 & 15). This interaction was lost at 95 °C, and substrate binding was no longer detectable. Ternary complex stability after heating at temperatures >50 °C in presence of SDS and reducing agents suggests formation of a stable covalent bond between M.HhaI and 2P-ODN. Ternary complexes between M.HhaI and ZCyt-ODN were similarly stable to heating in the presence of SDS and reducing agents, but dissociated at 75 °C, presumably due to disruption of the integrity of the pyrimidine ring in a manner that prevents rebinding to the enzyme (33, 40).
Figure 6.

Analysis of M.HhaI and Dnmt1 binding to ODNs containing 2P by SDS-PAGE. (A) M.HhaI:2P-ODN complexes were formed in reactions containing 2.5 nM M.HhaI, 50 nM 32P-labeled 2P-ODN, and cofactor (100-200 μM). After a 30 min incubation at 37 °C, reaction mixtures were held at the indicated temperatures in SDS loading buffer (final concentration of 1% SDS, 1% BME) for 5 min prior to electrophoresis. (B) Dnmt1-ODN (DM72P18) complexes were formed in reactions containing 0.4 μM Dnmt1, 1 μM 32P-labeled DM72P18, and cofactor (100-200 μM)) and treated as described above. See text for details. Note that samples in Panel B 1-6 were electrophoresed on a separate gel from samples 7-15. Arrows indicating open (O) and closed (C) complexes in each gel are placed on the closest side of the gel.
In contrast, Dnmt1 forms two distinct complexes with radiolabeled DM72P18 that are stable in the presence of SDS and BME at room temperature, a closed complex with the mobility of an ~145kDa protein and a slower migrating, open complex with the mobility of an ~200kDa protein. Neither complex co-migrates with unbound Dnmt1 (~187kDa). The increased mobility of the closed complex of Dnmt1 bound to DM72P18 indicates a significant conformational change, similar to that seen with M.HhaI. However, formation and stability of the Dnmt1: DM72P18 complex appears to have little dependence on cofactor (Figure 6B). Whether held on ice or at room temperature, the only difference noted was a slightly lower efficiency of ODN binding in the faster migrating closed complexes formed in the absence of cofactor (Figure 6, Panel B, lanes 1-6). Essentially all of the closed complexes were disrupted by heating at 50 °C(Figure 6, Panel B, lanes 7-9) while the low mobility open complexes proved to have a much greater stability. These complexes gradually dissociated as the temperature was raised from 50 to 95 °C (Figure 6, Panel B, lanes 7-15). The persistence of a fraction of open Dnmt1:DM72P18 complex, even at 95 °C, suggests that a small percentage of Dnmt1 is covalently attached to 2P in a manner that disrupts the compact, closed conformation. This pattern of complex distribution after SDS-PAGE is similar to that observed between Dnmt1 and DNA containing 5-fluorocytosine (FCyt) in place of a target C [42] suggesting a disruption of the catalytic pocket by modification of 2P. To further define the characteristics of the open complex, the same binding assay was performed using a catalytic mutant of Dnmt1 (Dnmt1C1228S) in which the active cysteine was converted to a serine residue. The stability of the binary and ternary complexes formed with Dnmt1C1228S was identical to that observed with the wild type Dnmt1 (Figure S3, Supplemental Material). The persistence of the open complex with both Dnmt1 and Dnmt1C1228S suggests that even though the serine is a weaker nucleophile than cysteine, it is capable of forming a stable covalent complex with 2P-ODN.
4. Discussion
The data presented here clearly demonstrate that once incorporated into DNA, zebularine and 5-azacytidine are essentially equivalent as inhibitors of DNA C5-MTases of both bacterial and mammalian origin. Nevertheless, there are some significant mechanistic and structural differences between 2P- and ZCyt-ODNs in their interaction with these enzymes that have implications with regard to their potency in vivo as activators of genes silenced by methylation and their potential toxicity/mutagenicity.
4.1 Formation and stabilization of 2P-ODN complexes
Despite our findings that 2P- and ZCyt-ODNs are equally effective inhibitors of M.HhaI and Dnmt1, ternary complexes with 2P-ODNs have a much shorter half life at 22 °C than ternary complexes with ZCyt-ODNs (t1/2 of 70 and 310 h) [34]. Our results are consistent with the postulates that 1) the covalent bond formed between 2P and Cys81 of M.HhaI is more readily reversible than that with ZCyt and 2) some difference in the interaction of ZCyt and 2P with the amino acids in the catalytic pocket of the enzyme is a critical factor in establishing the stability of the closed ternary complex.
The formation of a covalent bond significantly affects the stability of the enzyme:ODN complex. Several different cytosine analogs have been studied. Ternary closed complexes between M.HhaI:AdoMet and ODNs with DZCyt replacing C (DZCyt-ODNs) are incapable of forming a covalent bond with M.HhaI, have a comparably lower affinity for M.HhaI, and have a t½ of < 8 h under the same conditions. FCyt-ODNs have a high affinity for the enzyme and the complexes they form with DNA methyltransferases are completely stable at 95 °C due to formation of irreversible covalent bonds. A third class of enzyme:DNA complexes studied containing either an abasic furan or a carbocyclic ring replacing C (AP-ODNs) form ternary complexes with high affinity in the presence of AdoMet, that have a t½ of ~180 h [34, 43], despite the fact that a covalent bond cannot be formed. AP-ODN stability is more than twice that of 2P: M.HhaI:AdoMet complexes. This suggests that the difference in stability of ternary complexes containing ZCyt and 2P-ODNs is related to differences in the interactions of these bases with amino acids in the active site pocket in enzyme:DNA:cofactor complex, rather than relying solely on the stability of covalent bonds. Several x-ray crystallographic studies support the proposal that differences in the interaction of bases and sugar moieties with amino acids in the catalytic pocket of DNA methyltransferase can in fact lead to major differences in the stability of the closed enzyme:DNA complex [44-46].
4.2 SDS-PAGE as a tool for examining enzyme/ODN complexes
A primary criterion we have used for identifying stable covalent bond formation with analogs for the target C for a DNA methyltransferase is the ability of the enzyme/DNA complex to withstand heating at ≥75 °C in the presence of SDS and a reducing agent. An ODN with an abasic target site, i.e., one that cannot form a covalent bond, with a DNA methyltransferase is completely dissociated between 50 and 75 °C while an irreversible bond (FCyt) is stable at 95°C. Examination of the temperature stability of M.HhaI/2P-ODN complexes was undertaken to strengthen the evidence for covalent bond formation [47]. However, our results indicate that 2P:M.HhaI complexes are completely dissociated (i.e., substrate is released) upon heating at temperatures ≥50 °C in the absence of cofactors (Figure 6, Panel A). Dissociation is not due to instability of 2P, since 2P:M.HhaI:AdoHcy and 2P:M.HhaI:AdoMet complexes are stable at this temperature. Only the 2P:M.HhaI:AdoMet complex survives at 80 °C, although it is not stable at higher temperatures. Since X-ray crystallographic studies clearly demonstrate that a covalent bond is formed between 2P and the active Cys81 in the catalytic pocket of M. HhaI in vitro, we postulate that the covalent bond is more easily broken than the bond with FCyt, resulting in an enzyme:DNA complex that is unstable under denaturing conditions at temperature above 80 °C.
Although the catalytic domains of the mammalian and bacterial methyltransferase are quite homologous, we noted an important difference in their interaction with 2P-ODNs. DM72P18:Dnmt1 complexes were stable at 37 °C in the presence or absence of co-factor (data not shown), yet virtually no closed complexes (Figure 6, Panel B) were detected after heating at 50 °C suggesting that either these closed complexes do not form covalent bonds or that these bonds are readily reversible. Nevertheless, a small population of “closed” complex was still detectable, at 95 °C. We could also detect a small fraction (~1%) of complexes with a similar reduced mobility of open complexes formed by FCyt and M.HhaI or Dnmt1, i.e., complexes that migrate more slowly than the native enzyme alone [34, 42]. This minor fraction of enzyme complexes is stable at 50 °C, and diminishes slowly as the temperature is increased. However, some complexes still persist, even when temperature is raised to 95 °C. The low mobility of these complexes suggests the possibility that the closed complex has been converted into an open, thermally stable complex with the substrate trapped by covalent bond formation with an as yet unidentified electrophile following cleavage of the C-S bond with Cys81 (Figure 7B). Although AdoMet appears to be the most likely electrophile involved and addition of a methyl group to C is sufficient to destabilize the closed complex (33, 42), we and others have not detected any transfer of methyl groups to 2P (data not shown, [47]). Moreover, this conformational change does not require methylation of 2P at the 5-position of the pyrimidine ring, since it occurs in the presence or absence of the AdoHcy and even in the absence of any cofactor. This suggests that at least one of the amino acids other than Cys81 in the catalytic pocket of Dnmt1 plays a role in forming the stable adduct and that this interaction leads to destabilization of the closed complex. Whether the unusual heat sensitivity of the rapidly migrating Dnmt1:DM72P18 complex reflects the fact that 2P and the active site cysteine are in rapid equilibrium between bound and unbound states or that the absence of 4-amino group in the target base leads to the formation of a different or additional covalent bond remains to be determined. Nevertheless, it is clear that some highly stable covalent link(s) links are formed between Dnmt1 and DM72P18 and that they lead to an altered conformation of the enzyme:substrate complex.
Figure 7.

(A) Formation and reversal of 2-(1H)-pyrimidinone adduct with the activated thiol of a DNA C5-MTase. Our data suggests that heating shifts the equilibrium to the left of the reaction. (B) Formation of a stable adduct after reacting with an undetermined electrophile within the catalytic pocket of Dnmt1.
4.3 Zebularine as an epigenetic chemotherapeutic
Zebularine has been under intense study as a potential antitumor agent based on its stability (half life of >500 h at pH 7.4) and low toxicity compared to ZdCyd [48]. The studies reported here demonstrate that zebularine, once incorporated into DNA, is as potent as ZdCyd in activating genes silenced by DNA methylation. However, concentrations of zebularine sufficient to cause significant reduction in DNA methylation in cultured cells are at least 100 fold higher than those needed for inhibition by ZdCyd. Inhibition of DNA methylation and slowing of tumor growth in animals required administration of high doses of zebularine over an extended treatment period[28]. The difference between the two compounds appears to result from the slow conversion of zebularine to 2'-deoxyzebularine-5'-diphosphate by ribonucleotidediphosphate reductase [48]. There are a number of other factors that could contribute to the low toxicity of zebularine in vivo including; zebularine incorporation into RNA is at least 7-fold higher than into DNA [49], rapid inactivation of zebularine by the liver enzyme aldehyde oxidase [50], the slow covalent bond formation resulting in a facile reversal of enzyme:2P-ODN adducts (Section 3.3), and the stability of the pyrimidinone ring relative to ZCyt.
Inactivation of Dnmt1 by ZCyt in DNA occurs through the formation of covalent enzyme:DNA adducts that are potentially mutagenic. ZCyt in this complex is in equilibrium with a ring-open form that can undergo hydrolysis, yielding a structure that can theoretically form a Watson-Crick base pair with cytosine [21] and releasing of a formylated (inactive) enzyme (See Ref. 34 for further detail). 2P is extremely stable, with little or no probability of forming a similar mutagenic lesion. We demonstrate that 2P adducts have a shorter half-life than those formed by ZCyt, a difference that could also decrease toxicity. In contrast to ZdCyt, 2P can only form two hydrogen bonds in a Watson-Crick base pair with G, and forms a “wobble” base when paired with A [51]. Thus, unlike the open ring form of ZCyt, 2P in DNA should not lead to a significant increase in CG → TA transitions during DNA replication relative to that noted in DNA from ZCyd treated cells[52, 53]. Consequently, strategies to increase incorporation of zebularine in a dose dependent manner could theoretically increase the potency of the drug while maintaining a relatively stable enzyme:DNA adduct, thus improving the efficacy of zebularine as an epigenetic drug.
Supplementary Material
Acknowledgements
Partial support for this work was provided by a grant from the NIH/NCI (R21CA91315) to J.K.C. and a fellowship from the Graduate College at UNMC to D.V.B. We are grateful to S. Kumar of New England Biolabs for providing us with Eschericia coli strain ER1727 containing the pUHE25HhaI plasmid. This research was also supported in part with funds from the Intramural Research Program of the NIH, Center for Cancer Research, NCI Frederick. We also thank Dr. Michael Boland for critical reading of the manuscript and careful comments.
Abbreviations used
- 2P
2-(1H)-pyrimidinone (zebularine aglycon)
- AdoHcy
S-adenosyl homocysteine
- AdoMet
S-adenosyl methionine
- BME
beta-mercaptoethanol
- bp
base pairs
- Cyd
cytidine
- C
cytosine
- CDA
cytidine deaminase
- DNA C5-MTase
DNA (cytosine C5)-methyltransferase
- Dnmt1
DNA (cytosine C5)-methyltransferase-1
- ds
double stranded
- FCyt
5-fluorocytosine
- M.HhaI
HhaI methyltransferase
- M.HgaI
HgaI methyltransferase
- M.MspI
MspI methyltransferase
- MR buffer
methylation reaction buffer
- ODN
oligodeoxyribonucleotide
- SDS
sodium dodecyl sulfate
- ss
single stranded
- ZCyt
5-azacytosine
- ZCyd
5-azacytidine
- ZdCyd
5-aza-2'-deoxycytidine
- DZCyt
5,6-dihydro-5-azacytosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jaenisch R, Beard C, Lee J, Marahrens Y, Panning B. Mammalian X chromosome inactivation. Novartis Found Symp. 1998;214:200–9. doi: 10.1002/9780470515501.ch12. discussion 9-13, 28-32. [DOI] [PubMed] [Google Scholar]
- [2].Li E, Beard C, Forster AC, Bestor TH, Jaenisch R. DNA methylation, genomic imprinting, and mammalian development. Cold Spring Harb Symp Quant Biol. 1993;58:297–305. doi: 10.1101/sqb.1993.058.01.035. [DOI] [PubMed] [Google Scholar]
- [3].Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- [4].Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10:687–92. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- [5].Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- [6].Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2'-deoxycytidine. J Biol Chem. 1982;257:2041–8. [PubMed] [Google Scholar]
- [7].Taylor SM, Constantinides PA, Jones PA. 5-Azacytidine, DNA methylation, and differentiation. Curr Top Microbiol Immunol. 1984;108:115–27. doi: 10.1007/978-3-642-69370-0_8. [DOI] [PubMed] [Google Scholar]
- [8].Christman JK, Mendelsohn N, Herzog D, Schneiderman N. Effect of 5-azacytidine on differentiation and DNA methylation in human promyelocytic leukemia cells (HL-60) Cancer Res. 1983;43:763–9. [PubMed] [Google Scholar]
- [9].Christman JK. DNA methylation in friend erythroleukemia cells: the effects of chemically induced differentiation and of treatment with inhibitors of DNA methylation. Curr Top Microbiol Immunol. 1984;108:49–78. doi: 10.1007/978-3-642-69370-0_5. [DOI] [PubMed] [Google Scholar]
- [10].Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A. 1984;81:6993–7. doi: 10.1073/pnas.81.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Friedman S. The irreversible binding of azacytosine-containing DNA fragments to bacterial DNA(cytosine-5)methyltransferases. J Biol Chem. 1985;260:5698–705. [PubMed] [Google Scholar]
- [12].Christman JK, Schneiderman N, Acs G. Formation of highly stable complexes between 5-azacytosine-substituted DNA and specific non-histone nuclear proteins. Implications for 5- azacytidine-mediated effects on DNA methylation and gene expression. J Biol Chem. 1985;260:4059–68. [PubMed] [Google Scholar]
- [13].Vesely J. Mode of action and effects of 5-azacytidine and of its derivatives in eukaryotic cells. Pharmacol Ther. 1985;28:227–35. doi: 10.1016/0163-7258(85)90012-9. [DOI] [PubMed] [Google Scholar]
- [14].Li LH, Olin EJ, Buskirk HH, Reineke LM. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 1970;30:2760–9. [PubMed] [Google Scholar]
- [15].Goldberg J, Gryn J, Raza A, Bennett J, Browman G, Bryant J, et al. Mitoxantrone and 5-azacytidine for refractory/relapsed ANLL or CML in blast crisis: a leukemia intergroup study. AmJHematol. 1993;43:286–90. doi: 10.1002/ajh.2830430411. [DOI] [PubMed] [Google Scholar]
- [16].Pinto A, Zagonel V. 5-Aza-2'-deoxycytidine (Decitabine) and 5-azacytidine in the treatment of acute myeloid leukemias and myelodysplastic syndromes: past, present and future trends. Leukemia. 1993;7(Suppl 1):51–60. [PubMed] [Google Scholar]
- [17].Thibault A, Figg WD, Bergan RC, Lush RM, Myers CE, Tompkins A, et al. A phase II study of 5-aza-2'deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori. 1998;84:87–9. doi: 10.1177/030089169808400120. [DOI] [PubMed] [Google Scholar]
- [18].Momparler RL, Ayoub J. Potential of 5-aza-2'-deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer. Lung Cancer. 2001;34(Suppl 4):S111–5. doi: 10.1016/s0169-5002(01)00397-x. [DOI] [PubMed] [Google Scholar]
- [19].Wijermans P, Lubbert M, Verhoef G, Bosly A, Ravoet C, Andre M, et al. Low-dose 5-aza-2'-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–62. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- [20].Schwartsmann G, Schunemann H, Gorini CN, Filho AF, Garbino C, Sabini G, et al. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients with inoperable non-small cell lung cancer. Invest New Drugs. 2000;18:83–91. doi: 10.1023/a:1006388031954. [DOI] [PubMed] [Google Scholar]
- [21].Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. Mutagenicity of 5-aza-2'-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc Natl Acad Sci U S A. 1997;94:4681–5. doi: 10.1073/pnas.94.9.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Votruba I, Holy A, Wightman RH. The mechanism of inhibition of DNA synthesis in Escherichia coli by pyrimidin-2-one beta-D-ribofuranoside. Biochim Biophys Acta. 1973;324:14–23. doi: 10.1016/0005-2787(73)90246-3. [DOI] [PubMed] [Google Scholar]
- [23].Marquez VE, Liu PS, Kelley JA, Driscoll JS, McCormack JJ. Synthesis of 1,3-diazepin-2-one nucleosides as transition-state inhibitors of cytidine deaminase. J Med Chem. 1980;23:713–5. doi: 10.1021/jm00181a001. [DOI] [PubMed] [Google Scholar]
- [24].Kim CH, Marquez VE, Mao DT, Haines DR, McCormack JJ. Synthesis of pyrimidin-2-one nucleosides as acid-stable inhibitors of cytidine deaminase. J Med Chem. 1986;29:1374–80. doi: 10.1021/jm00158a009. [DOI] [PubMed] [Google Scholar]
- [25].Frick L, Yang C, Marquez VE, Wolfenden R. Binding of pyrimidin-2-one ribonucleoside by cytidine deaminase as the transition-state analogue 3,4-dihydrouridine and the contribution of the 4-hydroxyl group to its binding affinity. Biochemistry. 1989;28:9423–30. doi: 10.1021/bi00450a027. [DOI] [PubMed] [Google Scholar]
- [26].Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr Cytidine deaminase. The 2.3 A crystal structure of an enzyme: transition-state analog complex. J Mol Biol. 1994;235:635–56. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- [27].McCormack JJ, Marquez VE, Liu PS, Vistica DT, Driscoll JS. Inhibition of cytidine deaminase by 2-oxypyrimidine riboside and related compounds. Biochemical Pharmacology. 1980;29:830–2. doi: 10.1016/0006-2952(80)90566-3. [DOI] [PubMed] [Google Scholar]
- [28].Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- [29].Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG, et al. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol. 2004;24:1270–8. doi: 10.1128/MCB.24.3.1270-1278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hurd PJ, Whitmarsh AJ, Baldwin GS, Kelly SM, Waltho JP, Price NC, et al. Mechanism-based inhibition of C5-cytosine DNA methyltransferases by 2-H pyrimidinone. J Mol Biol. 1999;286:389–401. doi: 10.1006/jmbi.1998.2491. [DOI] [PubMed] [Google Scholar]
- [31].Ford K, Taylor C, Connolly B, Hornby DP. Effects of co-factor and deoxycytidine substituted oligonucleotides upon sequence-specific interactions between MspI DNA methyltransferase and DNA. J Mol Biol. 1993;230:779–86. doi: 10.1006/jmbi.1993.1200. [DOI] [PubMed] [Google Scholar]
- [32].Guimil Garcia R, Brank AS, Christman JK, Marquez VE, Eritja R. Synthesis of oligonucleotide inhibitors of DNA (Cytosine-C5) methyltransferase containing 5-azacytosine residues at specific sites. Antisense Nucleic Acid Drug Dev. 2001;11:369–78. doi: 10.1089/108729001753411335. [DOI] [PubMed] [Google Scholar]
- [33].Vives M, Eritja R, Tauler R, Marquez VE, Gargallo R. Synthesis, stability, and protonation studies of a self-complementary dodecamer containing the modified nucleoside 2'-deoxyzebularine. Biopolymers. 2004;73:27–43. doi: 10.1002/bip.10515. [DOI] [PubMed] [Google Scholar]
- [34].Brank AS, Eritja R, Garcia RG, Marquez VE, Christman JK. Inhibition of HhaI DNA (Cytosine-C5) methyltransferase by oligodeoxyribonucleotides containing 5-aza-2'-deoxycytidine: examination of the intertwined roles of co-factor, target, transition state structure and enzyme conformation. J Mol Biol. 2002;323:53–67. doi: 10.1016/s0022-2836(02)00918-x. [DOI] [PubMed] [Google Scholar]
- [35].Kumar S, Cheng X, Pflugrath JW, Roberts RJ. Purification, crystallization, and preliminary X-ray diffraction analysis of an M.HhaI-AdoMet complex. Biochemistry. 1992;31:8648–53. doi: 10.1021/bi00151a035. [DOI] [PubMed] [Google Scholar]
- [36].Brank AS, Van Bemmel DM, Christman JK. Optimization of Baculovirus-Mediated Expression and Purification of Hexahistidine-Tagged Murine DNA (Cytosine-C5)-Methyltransferase-1 in Spodoptera frugiperda 9 Cells. Protein Expr Purif. 2002;25:31–40. doi: 10.1006/prep.2001.1606. [DOI] [PubMed] [Google Scholar]
- [37].Christman JK, Sheikhnejad G, Marasco CJ, Sufrin JR. 5-Methyl-2'-deoxycytidine in single-stranded DNA can act in cis to signal de novo DNA methylation. Proc Natl Acad Sci U S A. 1995;92:7347–51. doi: 10.1073/pnas.92.16.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sheikhnejad G, Brank A, Christman JK, Goddard A, Alvarez E, Ford H, Jr., et al. Mechanism of inhibition of DNA (cytosine C5)-methyltransferases by oligodeoxyribonucleotides containing 5,6-dihydro-5-azacytosine. J Mol Biol. 1999;285:2021–34. doi: 10.1006/jmbi.1998.2426. [DOI] [PubMed] [Google Scholar]
- [39].Svedruzic ZM, Reich NO. DNA cytosine C5 methyltransferase Dnmt1: catalysis-dependent release of allosteric inhibition. Biochemistry. 2005;44:9472–85. doi: 10.1021/bi050295z. [DOI] [PubMed] [Google Scholar]
- [40].Silverman RB. Mechanism-based enzyme inactivators. Methids in Enzymology. 1995:240–83. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- [41].Brank AS. Department of Biochemistry and Molecular Biology. University of Nebraska Medical Center; Omaha: 2000. Development and Chracterization of DNA Methyltransferase Inhibitors; p. 237. [Google Scholar]
- [42].Christman JK. 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- [43].Wang P, Nicklaus MC, Marquez VE, Brank AS, Christman JK, Banavali NK, MacKerell AD., Jr. Use of oligodeoxyribonucleotides with conformationally constrained abasic sugar targets to probe the mechanism of base flipping by HhaI DNA (Cytosine C5)-methyltransferase. JAm Chem Soc. 2000;122:12422–34. [Google Scholar]
- [44].O'Gara M, Klimasauskas S, Roberts RJ, Cheng X. Enzymatic C5-cytosine methylation of DNA: mechanistic implications of new crystal structures for HhaL methyltransferase-DNA-AdoHcy complexes. J Mol Biol. 1996;261:634–45. doi: 10.1006/jmbi.1996.0489. [DOI] [PubMed] [Google Scholar]
- [45].O'Gara M, Horton JR, Roberts RJ, Cheng X. Structures of HhaI methyltransferase complexed with substrates containing mismatches at the target base. Nat Struct Biol. 1998;5:872–7. doi: 10.1038/2312. [DOI] [PubMed] [Google Scholar]
- [46].Kumar S, Horton JR, Jones GD, Walker RT, Roberts RJ, Cheng X. DNA containing 4'-thio-2'-deoxycytidine inhibits methylation by HhaI methyltransferase. Nucleic Acids Res. 1997;25:2773–83. doi: 10.1093/nar/25.14.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–9. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Marquez VE, Eritja R, Kelley JA, Vanbemmel D, Christman JK. Potent inhibition of HhaI DNA methylase by the aglycon of 2-(1H)-pyrimidinone riboside (zebularine) at the GCGC recognition domain. Ann N Y Acad Sci. 2003;1002:154–64. doi: 10.1196/annals.1281.014. [DOI] [PubMed] [Google Scholar]
- [49].Ben-Kasus T, Ben-Zvi Z, Marquez VE, Kelley JA, Agbaria R. Metabolic activation of zebularine, a novel DNA methylation inhibitor, in human bladder carcinoma cells. Biochem Pharmacol. 2005;70:121–33. doi: 10.1016/j.bcp.2005.04.010. [DOI] [PubMed] [Google Scholar]
- [50].Klecker RW, Cysyk RL, Collins JM. Zebularine metabolism by aldehyde oxidase in hepatic cytosol from humans, monkeys, dogs, rats, and mice: influence of sex and inhibitors. Bioorganic & medicinal chemistry. 2006;14:62–6. doi: 10.1016/j.bmc.2005.07.053. [DOI] [PubMed] [Google Scholar]
- [51].Gildea B, McLaughlin LW. The synthesis of 2-pyrimidinone nucleosides and their incorporation into oligodeoxynucleotides. Nucleic Acids Res. 1989;17:2261–81. doi: 10.1093/nar/17.6.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lee G, Wolff E, Miller JH. Mutagenicity of the cytidine analog zebularine in Escherichia coli. DNA Repair (Amst) 2004;3:155–61. doi: 10.1016/j.dnarep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- [53].Cupples CG, Miller JH. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A. 1989;86:5345–9. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.