

Abstract
Gene therapy offers the potential of mediating disease through modification of specific cellular functions of target cells. However, effective transport of nucleic acids to target cells with minimal side effects remains a challenge despite the use of unique viral and non-viral delivery approaches. Here we present a non-viral nanoparticle gene carrier that demonstrates effective gene delivery and transfection both in vitro and in vivo. The nanoparticle system (NP-CP-PEI) is made of a superparamagnetic iron oxide nanoparticle (NP), which enables magnetic resonance imaging, coated with a novel copolymer (CP-PEI) comprised of short chain polyethylenimine (PEI) and poly(ethylene glycol) (PEG) grafted to the natural polysaccharide, chitosan (CP), which allows efficient loading and protection of the nucleic acids. The function of each component material in this nanoparticle system is illustrated by comparative studies of three nanoparticle systems of different surface chemistries, through material property characterization, DNA loading and transfection analyses, and toxicity assessment. Significantly, NP-CP-PEI demonstrates an innocuous toxic profile and a high level of expression of the delivered plasmid DNA in a C6 xenograft mouse model, making it a potential candidate for safe in vivo delivery of DNA for gene therapy.
Keywords: biomaterials, superparamagnetic nanoparticles, DNA, drug delivery, gene therapy
1. Introduction
The use of viral vectors for gene therapy has been met with some success in recent clinical trials,[1-3] but there is still a major concern about their safety causing researchers to turn to non-viral vector solutions.[4-6] The growing interest in non-viral vector systems for in vivo gene therapy has provided a strong incentive to develop advanced materials for delivery of DNA and siRNA with high efficiency, stability, and minimal toxicity.[7-11] Cationic magnetic nanoparticles are one class of such materials that have been investigated for the transfection of plasmid DNA (pDNA)[12] and oligonucleotides[13] in mammalian cells. These nanoparticles, owing to their superparamagnetism, are desirable because they enable non-invasive monitoring of gene delivery in real-time through magnetic resonance imaging (MRI).[14] Of the materials utilized to engineer such nanovectors, polyethylenimine (PEI), a synthetic polymer, is most widely investigated because of its ability to effectively complex and condense DNA and transfect a broad range of cell lines with high efficiency.[15-17] However, the in vivo applicability of these nanoparticles has been limited due to concerns over toxicity, stability in biological fluids, and sufficient protection of the payload. This has led to a recent interest in research aimed towards the development of alternative magnetic nanoparticles with more favorable properties for in vivo gene delivery.[18, 19]
Materials developed for in vivo gene delivery need to be engineered to overcome several challenges imposed by biological systems. To achieve successful transfection, the nanoparticle system must (1) be sufficiently cationic to complex and condense DNA, (2) remain colloidally stable after DNA loading, (3) protect the DNA from external molecules such as nucleases in complex biological solvents, and (4) be able to escape the harsh environment of the late endosome to preserve and allow expression of the DNA.[20, 21] Furthermore, for magnetic nanoparticle based delivery system, the nanoparticle:DNA complex must maintain the detectability by MRI after DNA loading. Much of these properties are dictated by the design of the polymeric coatings used to functionalize and stabilize the nanoparticles engineered for gene therapy.
Here we describe the development and characterization of a superparamagnetic iron oxide nanoparticle (SPION) system with a novel polymeric coating consisting of chitosan, polyethylene glycol (PEG), and low molecular weight PEI (1.2 kDa). Chitosan, a natural, biocompatible polysaccharide, can serve as an effective coating to stabilize SPIONs, preventing particle agglomeration. While chitosan coated SPIONs have not been used in binding and delivering DNA, free polymeric chitosan has been shown to be able bind and deliver DNA to cells for transfection,[22] and grafting PEI to chitosan polymer could dramatically improve the transfection efficiencies due to the high positive charge of PEI.[23-27] Furthermore, the PEI provides a means of escaping the late endosome through the proton sponge effect wherein the influx of protons and counter-ions into the endosome increases the osmotic pressure leading to swelling and rupture of endosomes and the release of the PEI:DNA complex.[28] However, as noted above, the large positive charge that makes PEI so efficient is also highly toxic to cells through disruption of the cellular membrane.[29, 30] Grafting poly(ethylene glycol) (PEG) to PEI to create a PEI-PEG copolymer has been shown to lessen PEI’s toxicity by providing a physical barrier between the cell and PEI.[31-34] Furthermore, the presence of PEG may increase the nanoparticle’s colloidal stability through steric hindrance and provide non-fouling properties.[35, 36] Here, by grafting PEG and low molecular weight PEI to chitosan, we combine the biocompatibility of chitosan and the steric stabilization of PEG with the large positive charge of PEI to create a nanoparticle system capable of stably binding, protecting, and delivering DNA for gene expression while maintaining superparamagnetic properties and high biocompatibility, as established through the material characterization of nanoparticle:DNA complexes, DNA binding assay, toxicity study, and MR phantom imaging. Lastly, we investigate the potential use of this new nanoparticle system in vivo in a C6 xenograft tumor mouse model.
2. Results and Discussion
2.1. Nanovector Development
To better illustrate the design scheme and evaluate the efficacy of our DNA delivery system, three nanoparticle (NP) systems were prepared and their abilities to bind and protect DNA and enable DNA transfection were compared: NP coated with (1) high molecular weight PEI (25 kDa) (NP-PEI), (2) PEG grafted chitosan (chitosan-g-PEG) (NP-CP), and (3) a combination of CP and low molecular weight PEI (1.2 kDa) (NP-CP-PEI) (see Fig. 1 for conjugation scheme). NPs coated with low molecular weight PEI only were also prepared, but found to be unstable and thus excluded from further study. Low molecular weight PEI was pursued because of its low cytotoxicity, despite its lower transfection efficiencies compared with high molecular weight PEI.[37-39] Fortunately, transfection efficiencies with low molecular weight PEI has been shown to improve upon crosslinking with biodegradable polymers such as chitosan.[40-43]
Figure 1.
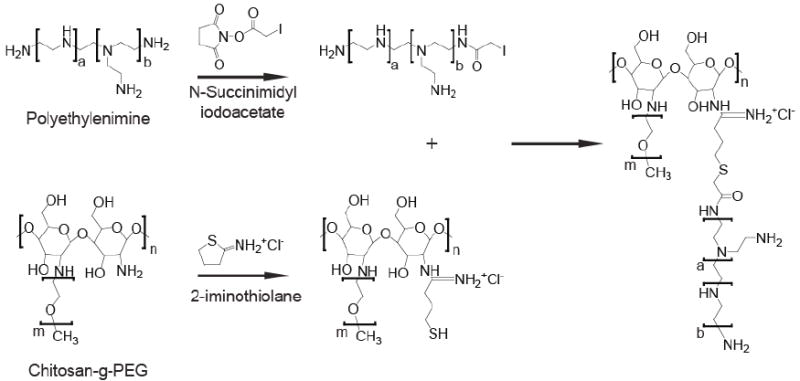
Synthesis of NP-CP-PEI. As synthesized NP-CP was modified with Traut’s Reagent (2-iminothiolane) and then reacted with SIA modified PEI to produce the NP-CP-PEI.
The presence of the constituent polymers on the prepared nanoparticles was verified by proton NMR (1H-NMR) (Fig. 2) in D2O. The characteristic 1H-NMR peak of PEG’s ethylene group at δ = 3.65 ppm (peak I) is resolved on the NP-CP spectrum. Peaks associated with chitosan are not visible in the spectra because sample concentrations were low to reduce the amount of peak broadening due to Fe in solution. The characteristic 1H-NMR peaks of the ethylenimine (–NH2–CH2–CH2–) repeat unit of PEI at δ = 3.6–3.0 ppm (peak II) along with the peak of PEG on NP-CP-PEI indicate covalent attachment of PEI to the NP-CP.
Figure 2.
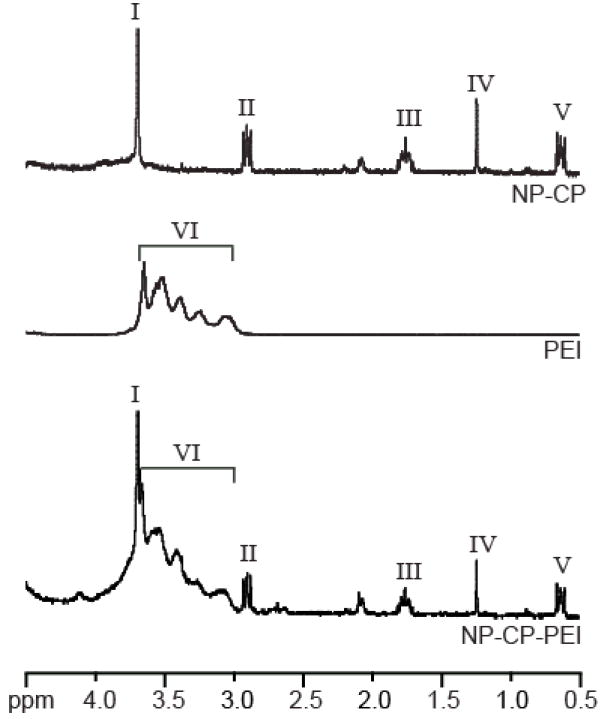
Proton NMR analysis of NP-CP, PEI, and NP-CP-PEI showing the incorporation of PEI onto NP-CP. The characteristic peak of the –O–CH2–CH2– group of PEG (peak I) on NP-CP and –NH2–CH2–CH2– group of PEI (peak II) are all present in the NP-CP-PEI spectrum. All samples were analyzed in D2O.
2.2. NP:DNA Complex Formation
A fundamental requirement for gene delivery is that the vector must be able to efficiently complex with nucleic acids. Here, the abilities of the three NP systems (NP-CP, NP-PEI, and NP-CP-PEI) to condense DNA at varying weight ratios of nanoparticles (iron content) to DNA (pEGFP-CS2) (NP:DNA ratio, wt:wt) were evaluated with a gel retardation assay, as shown in Figure 3a. In this assay, DNA bound to the NPs remained in the loading wells, while unbound DNA migrated down the agarose gel. It was evident that DNA migration was not completely retarded by NP-CP until a NP:DNA ratio of 5:1. Alternatively, NP-PEI and NP-CP-PEI provided complete retardation at a ratio of 0.1:1 and 0.5:1, respectively, illustrating the role of PEI in enhancing DNA binding to NPs.
Figure 3.
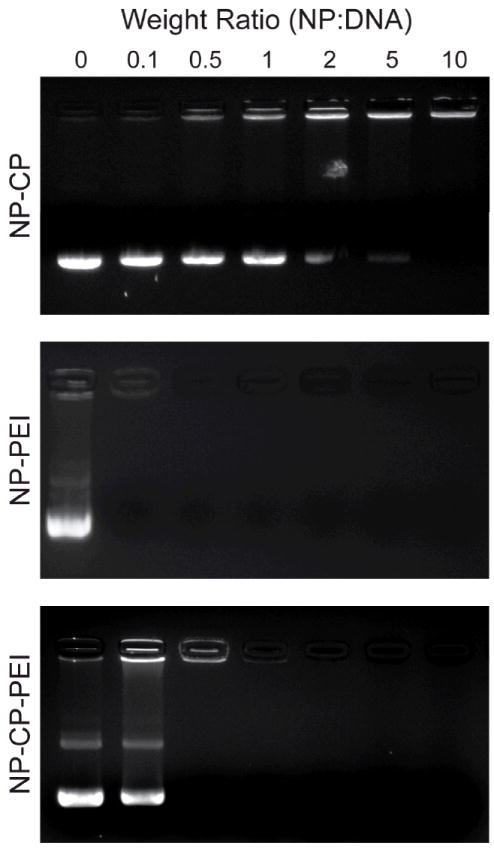
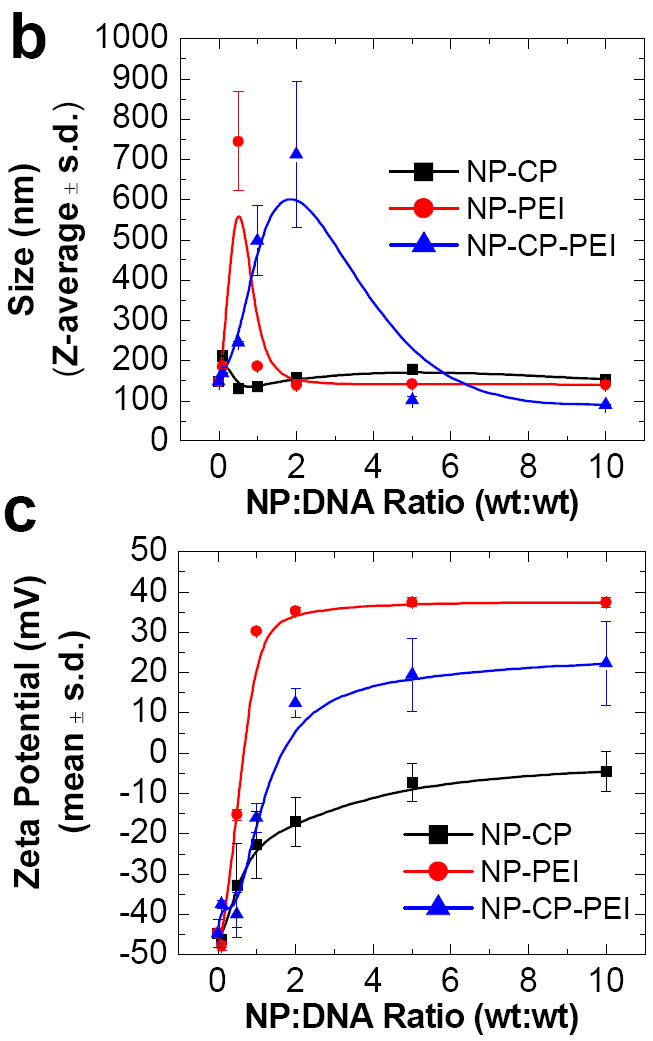
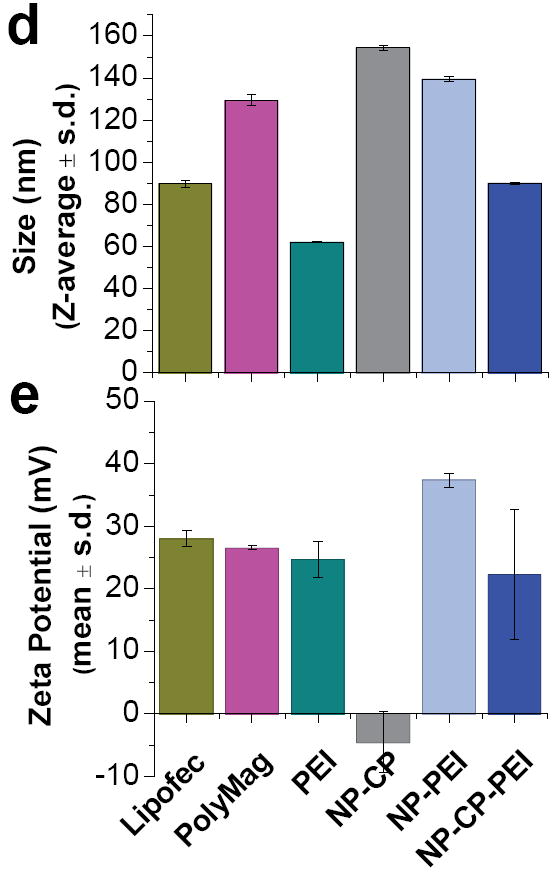
Ability of NP-CP, NP-PEI, and NP-CP-PEI to bind plasmid DNA and their physiochemical properties. a) Gel retardation assay of NPs complexing plasmid DNA at different weight ratios of nanoparticle to DNA. b) and c) Hydrodynamic sizes and zeta potentials, respectively, of NP-CP, NP-PEI, and NP-CP-PEI complexed with plasmid DNA at different weight ratios. d) and e) Comparison of hydrodynamic sizes and zeta potentials, respectively, of NP:DNA prepared in this study and control transfection agents complexed with DNAs.
The gel retardation analysis also provided information about DNA protection by the nanoparticles from the environment. Here, it was observed that DNA bound by NP-CP at a ratio of 10:1 was not protected from staining by ethidium bromide, evidenced by staining in the loading well. On the other hand, NP-PEI and NP-CP-PEI fully protected bound-DNA from staining above ratios of 0.1:1 and 1:1, respectively, obviated by a lack of visualized DNA in the wells. Protection of DNA from agents such as nucleases and destructive enzymes within the endolysosome is critical in preventing degradation of the DNA in transfection experiments. Exposure to these agents may induce nucleic acid breakdown and reduce transfection efficiencies. The addition of PEI to NP-CP was necessary to provide improved binding capacity and protection of DNA. However, complete binding and protection of DNA does not necessarily ensure successful transfection because (1) the size of the nanoparticle:DNA (NP:DNA) complex must be small enough to traverse the body and enter the target cell, and (2) there must be sufficient cationic surface charge to allow cell binding and later induce the proton sponge effect.
The hydrodynamic sizes of NP-CP, NP-PEI, and NP-CP-PEI bound with DNA at different NP:DNA ratios were determined using dynamic light scattering (Fig. 3b). The size of NP-CP:DNA did not change appreciably with increasing NP:DNA ratios, remaining around 150 nm. This can be attributed to the weak interaction between NP-CP and DNA as observed in the gel retardation assay. NP-PEI:DNA formed at a ratio of 0.1:1 had a size of 184 ± 6 nm, but the size increased sharply to 744 ± 122 nm at a ratio of 0.5:1. The size decreased as the NP:DNA ratio further increased, and reached to a small value of 139 ± 2 nm at a ratio of 2:1 and remained stable for higher ratios. A similar trend was observed for NP-CP-PEI:DNA, though occurred at different NP:DNA ratios: the size increased sharply at ratio 0.5:1 (244 ± 2 nm), reached a maximum at 2:1 (712 ± 180 nm), then decreased for further increase in NP:DNA ratio, and stabilized at ratio 5:1 (102 ± 9 nm) and above. This phenomenon can be better understood in terms of electric charges of NP:DNA complexes, as measured by zeta potential. Figure 3c shows the zeta potentials of the NP:DNA complexes prepared in this study as a function of NP:DNA ratio. It is noted that all the sharp changes in size as shown in Figure 3b occurred at NP:DNA ratios when the zeta potential underwent a transition from negative to positive values (Fig. 3c). These represent the unstable states of NP:DNA complexes, at which the complexes are neither completely negatively nor positively charged, leading to the complex agglomeration, and thus the sharp changes in size. We can also see large standard deviations in size at these ratios, further suggesting that the agglomeration occurred because the sizes of agglomerates can vary vastly. The possibility of the agglomeration can be reduced or eliminated when the NP:DNA complexes are fully charged either positively or negatively and thus repeal each other. This is indeed the case as shown in Figures 3b and 3c. Conversely, the zeta potential of NP-CP:DNA remained negative for all the ratios tested, and thus no sharp change in size was observed. It is also noted that the transition of the zeta potential from negative to positive values for NP-PEI:DNA occurred at lower NP:DNA ratio than for NP-CP-PEI:DNA. This is because PEI is highly positive, while introducing CP into the polymer coating partially shields the PEI’s positive charge. We also compared the sizes of nanoparticle systems prepared in this study with three commercially available transfection agents: Lipofectamine 2000, PolyMag, and PEI polymer (Fig. 3d). The size of NP-CP-PEI:DNA is 102 ± 9 nm at a ratio of 5:1 and 90 ± 1 nm at a ratio of 10:1, comparable to the sizes of the Lipofectamine 2000 (90 ± 2 nm), PolyMag (129 ± 3 nm), and PEI (62 ± 0.4 nm) after complexed with DNAs.
The zeta potential of a NP:DNA complex not only determines its colloidal stability, but also influences the effectiveness of its interaction with negatively charged cell membranes and thus the transfection efficiency. The zeta potential of NP-CP:DNA remains negative for all the NP:DNA ratios studied, indicating that NP-CP is probably not a good gene transfection vector. In addition, the lack of protection of DNA by NP-CP, as shown by the gel retardation assay, would increase the chances of DNA degradation. NP-PEI:DNA has positive zeta potentials for all NP:DNA ratios above 0.1:1. The large positive zeta potential and DNA protection provided by NP-PEI suggest that it might provide a high transfection efficiency. NP-CP-PEI:DNA has negative zeta potentials at low NP:DNA ratios, but becomes positively charged at a ratio of 2:1 and above. Like NP-PEI, NP-CP-PEI shows promise as a transfection agent due to its positive zeta potential and protection of DNA. However, as NP-CP-PEI:DNA has a zeta potential lower than NP-PEI:DNA, we expect that it also has a gene transfection efficiency lower than NP-PEI:DNA can provide. The zeta potentials of the NP-CP:DNA, NP-PEI:DNA, and NP-CP-PEI:DNA complexes at DNA binding ratios of 10:1 were also compared to the commercial transfection agents, Lipofectamine 2000 and PolyMag, and 25 kDa PEI, all complexed with DNA (Fig. 3e). Lipofectamine 2000 is a lipid based transfection agent, soluble in the cell membrane. The zeta potential of the Lipofectamine:DNA complex created following the manufacturer’s protocol, is 28 ± 1.3 mV, while PolyMag and 25 kDa PEI loaded with DNA have zeta potentials of 27 ± 0.4 mV and 25 ± 2.9 mV, respectively. Our NP-CP-PEI:DNA has a zeta potential (22.3 ± 10.4) comparable to those of the commercial transfection agents. The transfection efficiency of our NP-CP-PEI:DNA complexes is thus expected to be close to the efficacies of the commercial transfection agents.
2.3. Cytotoxicity and Transfection Efficiency of NP:DNA Complexes
Toxicity effect is a primary concern in development of gene transfection agents for in vivo use. One of designed functions for incorporating CP in the copolymer coating on NP-CP-PEI is to suppress the potential toxicity of PEI. We have shown above that by grafting PEI with CP on nanoparticles, we successfully reduced the zeta potential of NP:DNA complexes (Fig. 3e) while retaining the effectiveness of DNA binding (Fig. 3a). Here we further investigate whether such a measure would reduce or eliminate the toxic effect of PEI on nanoparticles.
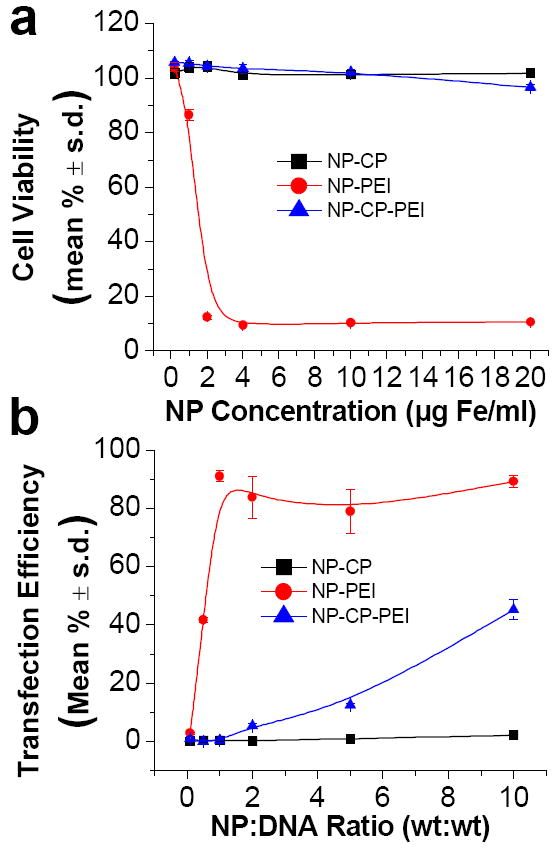
C6 rat glioma cells were incubated with different concentrations of NP:DNA complexes at an NP:DNA ratio of 10:1, and cell viabilities were measured as a function of NP Fe concentration (Fig. 4a). Of the three nanoparticle systems prepared in this study, NP-CP:DNA showed minimal to no toxic effect (e.g., cell viability of 101.8 ± 1.2 % at NP concentration of 20 μg Fe ml−1), as expected, due to the absence of PEI, while NP-PEI:DNA was highly toxic at NP Fe concentration of only 2 μg Fe ml−1 (cell viability of 12.4 ± 0.7 %) and above. NP-CP-PEI:DNA, with the presence of CP coating providing shielding between PEI and cell membranes, effectively inhibited the potential toxicity of PEI, exhibiting a cell viability level comparable to NP:CP:DNA.
Figure 4.
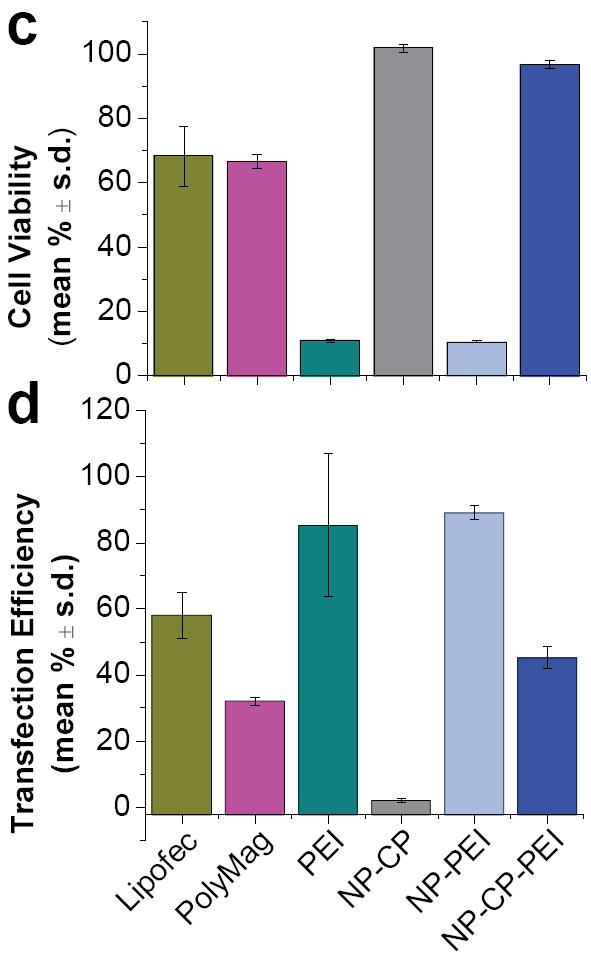
Toxicity and transfection efficiency of NP:DNA complexes. a) Viability of cells treated with different concentrations of NP-CP:DNA, NP-PEI:DNA, and NP-CP-PEI:DNA. b) Transfection efficiencies of cells treated with either NP-CP:DNA, NP-PEI:DNA, or NP-CP-PEI:DNA of different NP:DNA ratios (all with DNA concentration of 2 μg ml−1). c) and d) Viability and transfection efficiency, respectively, of cells treated with NP:DNA complexes prepared in this study in comparison with control transfection agents (NP:DNA ratios of NP-CP:DNA, NP-PEI:DNA, and NP-CP-PEI:DNA are all 10:1).
To evaluate the transfection efficiencies of these NP:DNA complexes, C6 cells were incubated with the complexes at a concentration of 2 μg DNA ml−1 for 48 hrs, and analyzed using flow cytometry. As shown in Figure 4b, the NP-CP:DNA complex did not induce any appreciable transfection, which can be attributed to its negative zeta potential (Fig. 3b). The NP-PEI:DNA complex showed the highest transfection efficiency among the three complexes, as expected, but its highly toxic profile (Fig. 4a) weakens its competency as a gene transfection agent, particularly for in vivo use. The NP-CP-PEI:DNA complex showed low transfection efficiencies at ratios before DNA was completely bound (see Fig. 3a for DNA binding), but the transfection efficiencies increased substantially at higher NP:DNA ratios, and reached 45.2 ± 3.4 % at ratio 10:1.
As references, the toxicities and transfection efficiencies of commercially available transfection agents were compared against those of the nanoparticle systems prepared in this study (Figs. 4c and 4d). NP-CP:DNA and NP-CP-PEI:DNA formed at ratios of 10:1 showed minimal to no toxicities, as compared to the commercial transfection agents and PEI (68.3 ± 9.3% viability for Lipofectamine 2000, 66.6 ± 2.3% viability for PolyMag, and 10.5 ± 0.3% viability for PEI).
NP-PEI exhibits the highest transfection efficiency (Fig. 4d). The NP-CP-PEI:DNA has a gene transfection efficiency (45.2 ± 3.4%) higher than PolyMag (32.1 ± 1.2 %), but lower than Lipofectamine 2000 (58.1 ± 7.1 %) and PEI (85.3 ± 21.5 %). However, since no toxicity was observed with NP-CP-PEI:DNA, NP-CP-PEI:DNA shows promise as a novel transfection agent with minimal adverse side effects while retaining substantial transfection efficiency.
To visualize the gene transfection by each gene transfection agent studied here, the EGFP transfected C6 cells were seeded onto glass coverslips for 12 hours for cell attachment, and then incubated with the transfection agents at a concentration of 2 μg DNA ml−1 for 48 hrs for transfection. The fluorescence images in Figure 5 show the EGFP fluorescence in cells treated with different transfection agents. Cells receiving no treatment (first column, Fig. 5) were also imaged for reference. These images again show the ability of NP-CP-PEI to deliver DNA into cells and induce expression levels similar to those of the commercially available agents, while little gene transfection can be identified for NP-CP.
Figure 5.

Confocal fluorescence images of C6 cells treated with different transfection agents complexed with DNA. The DAPI nuclear stain is shown in blue and EGFP fluorescence is shown in green. The scale bar corresponds to 20 μm.
2.4. Magnetic Properties of NP-CP-PEI:DNA
The superparamagnetic iron oxide core of our NP-CP-PEI transfection agent is expected to also act as a contrast agent for MR imaging, which provides a benefit for monitoring gene delivery. To confirm that NP-CP-PEI would retain sufficient magnetism detectable by MRI after DNA complexing, both NP-CP-PEI and NP-CP-PEI:DNA at various Fe concentrations were mixed with agarose and analyzed by MR phantom imaging. Figures 6a and 6b show the visual and quantitative contrast, respectively, provided by the relaxation (R2) changes of agarose phantoms cast with varying concentrations of NP-CP-PEI and NP-CP-PEI:DNA. The results indicate that the magnetism of the complex was readily datable by MRI. The relaxivities (slopes of the R2 vs. NP concentration curves) of NP-CP-PEI and NP-CP-PEI-DNA were seen to be similar (262 mM−1 s−1 for NP-CP-PEI versus 279 mM−1 s−1 for NP-CP-PEI:DNA), confirming no appreciable change in magnetism after DNA complexing. Previous studies have shown that clustering of NPs lowers R2 resulting in reduced contrast,[4, 44, 45] and here we show that the relaxivity of NP-CP-PEI is not affected by the addition of DNA further indicating, along with dynamic light scattering (DLS) data, that no clusters of NP:DNA complexes were formed.
Figure 6.
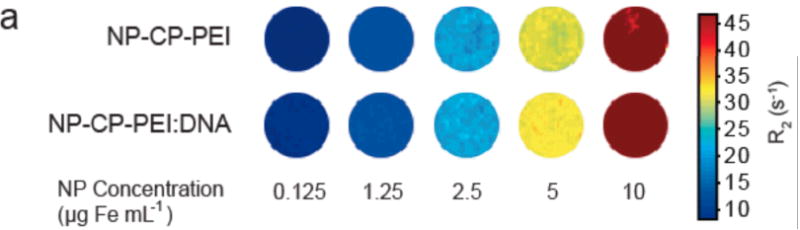
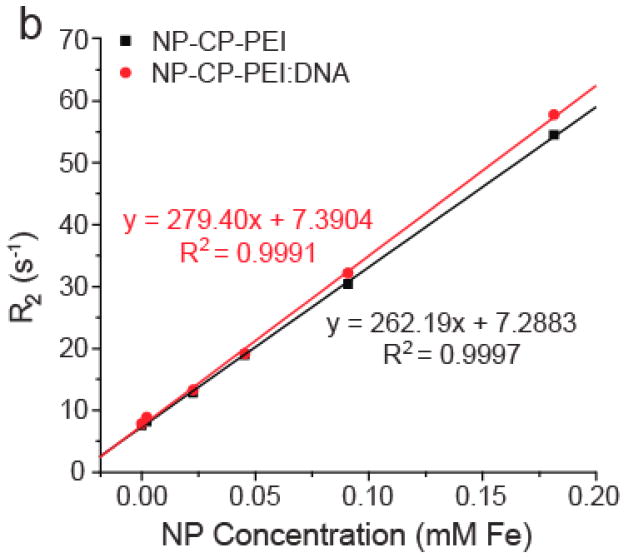
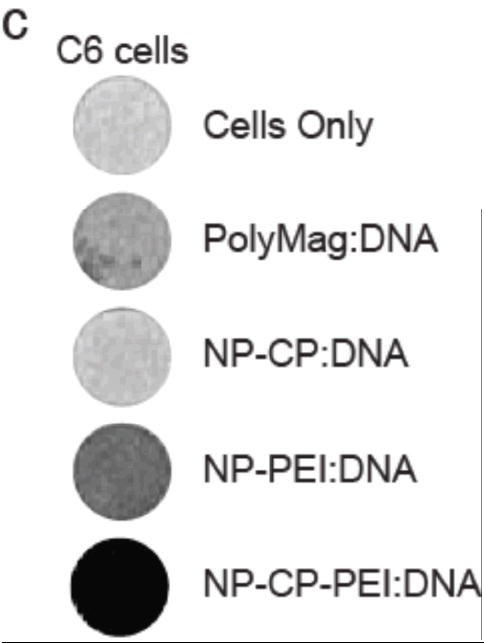
Magnetic properties of NP-CP-PEI and NP-CP-PEI:DNA, and MRI contrast enhancement by cellular uptake of NP:DNA complexes. a) Phantom images of NP-CP-PEI and NP-CP-PEI:DNA samples as a function of nanoparticle concentration. b) Relaxation (R2) plot of NP-CP-PEI and NP-CP-PEI:DNA samples as a function of nanoparticle concentration showing NP-CP-PEI retained the magnetism (magnetic relaxivity, i.e., the slop of the curve) after complexing with DNA (262 mM−1 s−1 for NP-CP-PEI versus 279 mM−1 s−1 for NP-CP-PEI:DNA). c) T2 weighted images (TR = s, TE = 60 ms) of C6 cells incubated with NP:DNA complexes prepared in this study and commercial PolyMag:DNA showing the degree of uptake of these NP:DNA complexes by C6 cells and enhanced contrast provided by cellular uptake.
To investigate the cellular uptake of transfection agents prepared in this study, and the potential MRI contrast enhancement associated with the uptake of these agents, the transfection agents complexed with DNA at NP-CP:DNA and NP-CP-PEI:DNA ratios of 10:1 and NP-PEI ratio of 0.5 were incubated with C6 cells for 24 hrs. For comparison, the commercial PolyMag agent with DNA bound following the manufacturer’s protocol was subjected to the same procedure. After culture with NP:DNA complexes, cells were washed to remove unbound agents and encased in an agarose mold for MRI imaging. T2-weighted MR images of samples containing C6 cells incubated with various complexes are shown in Figure 6c. Cells incubated with NP-CP-PEI: DNA displayed the highest contrast enhancement (darkening) than the cells with NP-PEI:DNA, PolyMag:DNA, and NP-PEI:DNA. This result indicates not only that the addition of PEI onto NP-CP dramatically improves the uptake of the NP:DNA complexes by cells, but also that the amount of Fe used in transfections with NP-CP-PEI:DNA provides much higher contrast that that used with NP-PEI and PolyMag. This shows that NP-CP-PEI would be much more easily detected in MRI when tracking treatment in vivo.
2.5. In Vivo Gene Transfections
To evaluate the ability of NP-CP-PEI to migrate to and transfect cancer cells in vivo, nu/nu mice with C6 xenograft tumors were injected intravenously with NP-CP-PEI:DNA, our candidate transfection agent complexed with DNA, in a pilot study. After 48 hrs, the time allowed for the uptake and expression of the EGFP encoding DNA, the mice receiving NP-CP-PEI:DNA or no injection were sacrificed and the tumors were excised and imaged using a Xenogen IVIS imaging system. Figure 7 shows the images of tumors of different sizes excised from three mice treated with NP-CP-PEI:DNA along with a tumor from the untreated mouse as reference. It is shown that NP-CP-PEI was able to deliver intact DNA to the tumors of various sizes for gene expression as evidenced by the high EGFP signal in the tumors of the NP-CP-PEI:DNA treated mice.
Figure 7.
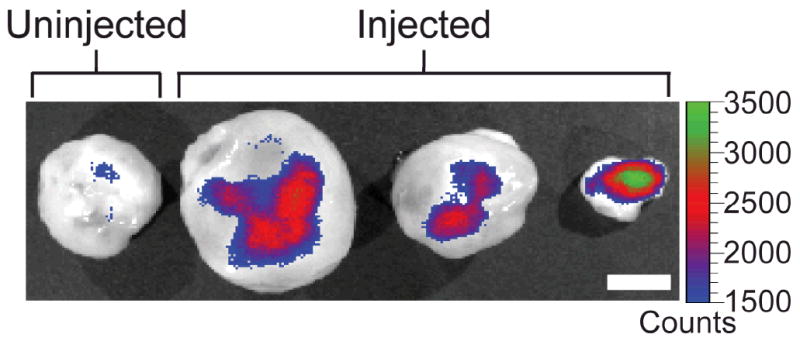
Xenogen IVIS fluorescence images of flank xenograft C6 tumors of different sizes excised from three mice injected with NP-CP-PEI:DNA and a mouse receiving no injection. The scale bar corresponds to 5 mm.
3. Conclusions
We have presented a novel transfection agent, NP-CP-PEI that demonstrates effective gene transfection both in vitro and in vivo. Through comparison studies with NP-PEI and NP-CP prepared in this work, as well as commercially available transfection agents, we illustrated that the presence of PEI on NP-CP-PEI is essential to effective DNA binding and transfection in tumor cells while the incorporation of the copolymer CP into the nanoparticle coating effectively inhibits the toxic effect of PEI. We also showed that the zeta potential of a NP:DNA complex can be a helpful indicator of the transfection efficiency of a gene transfection agent. We further demonstrated that this engineered system is able to function in vivo and deliver DNA for expression in tumor in a xenograft mouse model. As safety is a primary concern in the development of nanomaterials for in vivo applications, the present nanoparticle system is a good candidate for delivering of DNA for gene therapy.
4. Experimental
Materials
Polyethylenimine (PEI; average MW 1.2 kDa and average MW 25 kDa), chitosan, methoxy poly(ethylene glycol) (mPEG; MW 2 kDa) and other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
Plasmid DNA Preparation
The plasmid pEGFP-CS2 containing enhanced green fluorescent protein (EGFP) encoding DNA under control of the cytomegalovirus (CMV) promoter was propagated in DH5-α E. coli and purified using the Plasmid Giga Kit (Qiagen, Valencia, CA). Purified pEGFP-CS2, with a A260/A280 purity between 1.8 and 1.9, was dissolved in TE buffer at 1 mg/ml and stored at −20°C.
Nanoparticle Synthesis
PEG was grafted to depolymerized chitosan (chitosan-g-PEG or CP) by a method described previously[46]. CP coated iron oxide nanoparticles (NP-CP) were prepared in the presence of chitosan-g-PEG by the co-precipitation of ferrous and ferric chlorides with ammonium hydroxide. The nanoparticles were then purified into thiolation buffer (0.1 M sodium bicarbonate, pH 8.0, 5 mM EDTA) through S-200 sephacryl resin (GE Healthcare, Piscataway, NJ). The modification of NP-CP is outlined in Fig. 1. Amine groups on NP-CP were modified with an excess of 2-iminothiolane (Traut’s Reagent, Molecular Biosciences, Boulder, CO) for 1 hr in thiolation buffer before removing unreacted Traut’s Reagent using a PD-10 column (GE Healthcare, Piscataway, NJ) equilibrated with thiolation buffer. Concurrently, 1.2 kDa PEI was modified with succinimidyl iodoacetate (SIA, Molecular Biosciences, Boulder, CO) at a 1:1 molar ratio in 0.1 M sodium bicarbonate buffer (pH 8.5) through N-hydroxy succinimide ester chemistry. The modified PEI was then added in excess to NP-CP-Traut’s for attachment to NPs through the formation a thiol-ether bond. After reaction overnight at 4°C, unreacted PEI was removed through size exclusion chromatography using S-200 sephacryl resin equilibrated with 20 mM HEPES buffer (pH 7.4).
25 kDa PEI coated NPs (NP-PEI) were prepared by co-precipitation of ferrous and ferric chlorides with sodium hydroxide immediately followed by addition of 100 mg of 25 kDa PEI. The produced NP-PEI was then washed 3× with 20 mM HEPES buffer (pH 7.4), using a rare earth magnet.
Nanoparticle Characterization
Nanoparticles for H-NMR analysis were prepared by lyophilizing 50 μg of as synthesized nanoparticles to remove water. 50 μl of DCl and 950 μl of D2O were added to the lyophilized nanoparticles to dissolve the iron core leaving free polymer coating in solution. NMR spectra of polymer coatings were obtained using a Bruker Avance 300 spectrometer operating at 300 MHz (1H) and 325 K (number of scans = 128, acquisition time = 3 s, delay (D1) = 1 s).
Nanoparticle:DNA Complex Formation
Nanoparticles and DNA (pEGFP-CS2) were mixed in 20 mM HEPES buffer (pH 7.4) at concentrations corresponding to the wt:wt ratios tested (0.1, 0.5, 1, 2, 5, and 10:1) and immediately vortexed. The nanoparticle/pEGFP-CS2 solutions were incubated for >10 min with gentle rocking to allow formation of NP:DNA complexes. Size and zeta potential analyses of the complexes in HEPES buffer (pH 7.4) were performed using DTS Zetasizer Nano (Malvern Instruments, Worcestershire, UK).
Gel Retardation Assay
NP:DNA complexes were added to the wells (500 ng DNA per lane) of a 1% agarose gel containing 0.05 μg ml−1 ethidium bromide and run at 100 V for 1 hr. Images were obtained on a Gel Doc XR (Bio-Rad Laboratories, Hercules, CA).
Cell Culture
C6 rat glioma cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic (Invitrogen, Carlsbad, CA). Cultures were maintained at 37°C in a humidified incubator with 5% CO2.
Cell Transfections
C6 rat glioma cells were seeded at 125,000 cells/well in 24-well plates 16 hrs prior to transfection. Nanoparticle:DNA complexes prepared at different wt:wt ratios were added to 1 ml of fully supplemented culture media to give a final DNA concentration of 2 μg DNA ml−1 in each well and gently rocked to mix. The cells were incubated with complexes for 48 hrs and the media were replenished every 12 hrs. Transfections using the commercial agents, Lipofectamine 2000 and PolyMag, were performed following the manufacturer’s protocol. Transfections using 25 kDa PEI were performed at a PEI:DNA weight ratio of 2.5:1. A final concentration of 2 μg DNA ml−1 was used for all the transfection agents for transfection study.
Alamar Blue Viability Assay
The viability of cells treated with the different transfection agents was determined using the Alamar blue viability assay following the manufacture’s protocol (Invitrogen, Carlsbad, CA). Briefly, treated and untreated cells were washed with PBS before adding 1 ml of 10% Alamar blue in phenol-free DMEM to the wells. Cells were incubated for 1 hr, then the Alamar blue solution was transferred to a 96-well plate, and A570 and A600 were read on a SpectraMax M5 microplate reader (Molecular Devices, Union City, CA).
Flow Cytometry
To determine the transfection efficiencies of the nanoparticles, 48 hours after treatment, C6 cells were washed with PBS, trypsinized, and suspended in PBS containing 2% FBS. Analysis of at least 10,000 cells for each sample was performed on a BD FACSCanto flow cytometer (Beckton Dickinson, Franklin Lakes, NJ), and data was analyzed using the FlowJo software package (Tree Star, Ashland, OR).
Confocal Microscopy
For each transfection agent, 125,000 C6 cells were seeded onto 24 mm glass cover slips 12–16 hrs prior to transfection. Cells were transfected as described above, then 48 hrs after transfection, were washed with PBS and fixed with 4% formaldehyde (methanol free, Polysciences Inc., Warrington, PA) in PBS for 30 min. Fixative was then removed and cells were washed with PBS to remove the formaldehyde. The slides were mounted using ProLong Gold antifade solution containing DAPI (Invitrogen, Carlsbad, CA) and imaged using a LSM 510 Meta confocal fluorescence microscope (Carl Zeiss Inc., Peabody, MA) equipped with a 405 nm diode and 488 nm laser for collection of DAPI and FITC emission signals, respectively.
MR Phantom Imaging
NP-CP-PEI and NP-CP-PEI:DNA were diluted into 20 mM HEPES buffer (pH 7.4) to concentrations of 0, 0.25, 2.5, 5, 10, and 20 μg Fe ml−1. 50 μl of the diluted NP-CP-PEI or NP-CP-PEI:DNA were mixed with 50 μl of melted 1% agarose and added to a pre-solidified 1% agarose mold. The mold was then placed at 4°C until the nanoparticle dilutions solidified. For the study of NP uptake by cells, 500,000 cells were treated with each agent to be examined for one day, washed, diluted into 100 μl of melted 1% agarose, and added to the pre-solidified 1% agarose mold. MR images were obtained using a 4.7-T Bruker magnet (Bruker Medical Systems, Karlsruhe, Germany) equipped with a Varian Inova spectrometer (Varian Inc., Palo Alto, CA) and an in-house built 5 cm half volume RF coil in a loop gap resonator type using a conventional multi-spin echo pulse sequence (TR = 000 ms, TE = 13.7, 16, 20, 40, 60, 90, 120, and 170 ms).
In Vivo Studies
All animal experiments were conducted in accordance with IACUC approved protocols. Flank xenograft tumors of C6 cells were prepared by subcutaneous injection of 1 million cells suspended in serum free media and Matrigel (BD Biosciences, MA) into male nu/nu mice (Charles River, MA). Tumors were allowed to grow for 4 weeks before mice were injected intravenously through the tail vein with 200 μl of nanoparticle:DNA complex (0.4 mg Fe ml−1) prepared at a wt:wt ratio of 5:1 for a final dose of 16 μg pEGFP-CS2 per animal. 48 hours after treatment tumors were excised and imaged using a Xenogen IVIS –100 system (Xenogen, CA).
Supplementary Material
Footnotes
This work is supported by NIH grants (R01CA119408, R01EB006043, and R01CA134213). We would like to acknowledge the use of resources at the Department of Immunology’s cell analysis facility and Keck Microscopy Imaging Facility at the University of Washington, Ms. Ni Mu for her laboratory assistance, and Dr. Rajan Paranji for his helpful discussions on proton NMR.
Contributor Information
Mr. Forrest M. Kievit, Department of Materials Science & Engineering, University of Washington, Seattle, WA 98195 (USA)
Mr. Omid Veiseh, Department of Materials Science & Engineering, University of Washington, Seattle, WA 98195 (USA)
Dr. Narayan Bhattarai, Department of Materials Science & Engineering, University of Washington, Seattle, WA 98195 (USA)
Mr. Chen Fang, Department of Materials Science & Engineering, University of Washington, Seattle, WA 98195 (USA)
Dr. Jonathan W. Gunn, Department of Materials Science & Engineering, University of Washington, Seattle, WA 98195 (USA)
Dr. Donghoon Lee, Department of Radiology, University of Washington, Seattle, WA 98195 (USA)
Dr. Richard G. Ellenbogen, Department of Neurological Surgery, University of Washington, Seattle, WA 98195 (USA)
Dr. James M. Olson, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109 (USA)
Prof. Miqin Zhang, Department of Materials Science & Engineering, University of Washington, Seattle, WA 98195 (USA), mzhang@u.washington.edu
References
- 1.Alexander BL, Ali RR, Alton EW, Bainbridge JW, Braun S, Cheng SH, Flotte TR, Gaspar HB, Grez M, Griesenbach U, Kaplitt MG, Ott MG, Seger R, Simons M, Thrasher AJ, Thrasher AZ, Yla-Herttuala S. Gene Ther. 2007;14:1439. doi: 10.1038/sj.gt.3303001. [DOI] [PubMed] [Google Scholar]
- 2.Aiuti A, Bachoud-Levi AC, Blesch A, Brenner MK, Cattaneo F, Chiocca EA, Gao G, High KA, Leen AM, Lemoine NR, McNeish IA, Meneguzzi G, Peschanski M, Roncarolo MG, Strayer DS, Tuszynski MH, Waxman DJ, Wilson JM. Gene Ther. 2007;14:1555. doi: 10.1038/sj.gt.3303033. [DOI] [PubMed] [Google Scholar]
- 3.Edelstein ML, Abedi MR, Wixon J. J Gene Med. 2007;9:833. doi: 10.1002/jgm.1100. [DOI] [PubMed] [Google Scholar]
- 4.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discov. 2005;4:581. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 5.McCormack MP, Rabbitts TH. N Engl J Med. 2004;350:913. doi: 10.1056/NEJMra032207. [DOI] [PubMed] [Google Scholar]
- 6.Check E. Nature. 2005;433:561. doi: 10.1038/433561a. [DOI] [PubMed] [Google Scholar]
- 7.Sun C, Lee JS, Zhang M. Adv Drug Deliv Rev. 2008;60:1252. doi: 10.1016/j.addr.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mykhaylyk O, Antequera YS, Vlaskou D, Plank C. Nat Protoc. 2007;2:2391. doi: 10.1038/nprot.2007.352. [DOI] [PubMed] [Google Scholar]
- 9.Glover DJ, Lipps HJ, Jans DA. Nat Rev Genet. 2005;6:299. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 10.Ogris M, Wagner E. Drug Discov Today. 2002;7:479. doi: 10.1016/s1359-6446(02)02243-2. [DOI] [PubMed] [Google Scholar]
- 11.Wagner E. J Gene Med. 2003;5:S7. [Google Scholar]
- 12.Schillinger U, Brill T, Rudolph C, Huth S, Gersting S, Krotz F, Hirschberger J, Bergemann C, Plank C. Journal Of Magnetism And Magnetic Materials. 2005;293:501. [Google Scholar]
- 13.Krotz F, Wit Cd, Sohn H-Y, Zahler S, Gloe T, Pohl U, Plank C. Molecular Therapy. 2003;7:700. doi: 10.1016/s1525-0016(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 14.Shah K, Jacobs A, Breakefield XO, Weissleder R. Gene Ther. 2004;11:1175. doi: 10.1038/sj.gt.3302278. [DOI] [PubMed] [Google Scholar]
- 15.Lungwitz U, Breunig M, Blunk T, Gopferich A. Eur J Pharm Biopharm. 2005;60:247. doi: 10.1016/j.ejpb.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Scherer F, Anton M, Schillinger U, Henke J, Bergemann C, Kruger A, Gansbacher B, Plank C. Gene Ther. 2002;9:102. doi: 10.1038/sj.gt.3301624. [DOI] [PubMed] [Google Scholar]
- 17.Huth S, Lausier J, Gersting SW, Rudolph C, Plank C, Welsch U, Rosenecker J. J Gene Med. 2004;6:923. doi: 10.1002/jgm.577. [DOI] [PubMed] [Google Scholar]
- 18.Dobson J. Gene Ther. 2006;13:283. doi: 10.1038/sj.gt.3302720. [DOI] [PubMed] [Google Scholar]
- 19.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. Nat Med. 2007;13:372. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]
- 20.Petri-Fink A, Steitz B, Finka A, Salaklang J, Hofmann H. Eur J Pharm Biopharm. 2008;68:129. doi: 10.1016/j.ejpb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Green JJ, Langer R, Anderson DG. Acc Chem Res. 2008 doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao HQ, Roy K, Troung-Le VL, Janes KA, Lin KY, Wang Y, August JT, Leong KW. J Control Release. 2001;70:399. doi: 10.1016/s0168-3659(00)00361-8. [DOI] [PubMed] [Google Scholar]
- 23.Wong K, Sun G, Zhang X, Dai H, Liu Y, He C, Leong KW. Bioconjug Chem. 2006;17:152. doi: 10.1021/bc0501597. [DOI] [PubMed] [Google Scholar]
- 24.Jiang HL, Kim YK, Arote R, Nah JW, Cho MH, Choi YJ, Akaike T, Cho CS. J Control Release. 2007;117:273. doi: 10.1016/j.jconrel.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Zhao QQ, Chen JL, Han M, Liang WQ, Tabata Y, Gao JQ. J Biosci Bioeng. 2008;105:65. doi: 10.1263/jbb.105.65. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Liu C, Zhao X, Xiang J. Journal of Polymer Research. 2008;15:181. [Google Scholar]
- 27.Jiang HL, Kwon JT, Kim EM, Kim YK, Arote R, Jere D, Jeong HJ, Jang MK, Nah JW, Xu CX, Park IK, Cho MH, Cho CS. J Control Release. 2008;131:150. doi: 10.1016/j.jconrel.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 28.Godbey WT, Wu KK, Mikos AG. J Control Release. 1999;60:149. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 29.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. Mol Ther. 2005;11:990. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Hong S, Leroueil PR, Janus EK, Peters JL, Kober MM, Islam MT, Orr BG, Baker JR, Jr, Banaszak Holl MM. Bioconjug Chem. 2006;17:728. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- 31.Glodde M, Sirsi SR, Lutz GJ. Biomacromolecules. 2006;7:347. doi: 10.1021/bm050726t. [DOI] [PubMed] [Google Scholar]
- 32.Nimesh S, Goyal A, Pawar V, Jayaraman S, Kumar P, Chandra R, Singh Y, Gupta KC. J Control Release. 2006;110:457. doi: 10.1016/j.jconrel.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Pan SR, Hu HM, Wu GF, Feng M, Zhang W, Luo X. J Biomed Mater Res A. 2008;84:795. doi: 10.1002/jbm.a.31343. [DOI] [PubMed] [Google Scholar]
- 34.Veiseh O, Kievit FM, Gunn JW, Ratner BD, Zhang M. Biomaterials. 2009;30:649. doi: 10.1016/j.biomaterials.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veiseh O, Sun C, Gunn J, Kohler N, Gabikian P, Lee D, Bhattarai N, Ellenbogen R, Sze R, Hallahan A, Olson J, Zhang M. Nano Lett. 2005;5:1003. doi: 10.1021/nl0502569. [DOI] [PubMed] [Google Scholar]
- 36.Sun C, Veiseh O, Gunn J, Fang C, Hansen S, Lee D, Sze R, Ellenbogen RG, Olson J, Zhang M. Small. 2008;4:372. doi: 10.1002/smll.200700784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer D, Bieber T, Li Y, Elsasser HP, Kissel T. Pharm Res. 1999;16:1273. doi: 10.1023/a:1014861900478. [DOI] [PubMed] [Google Scholar]
- 38.Godbey WT, Wu KK, Mikos AG. J Biomed Mater Res. 1999;45:268. doi: 10.1002/(sici)1097-4636(19990605)45:3<268::aid-jbm15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 39.Thomas M, Klibanov AM. Proc Natl Acad Sci U S A. 2002;99:14640. doi: 10.1073/pnas.192581499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas M, Ge Q, Lu JJ, Chen J, Klibanov AM. Pharm Res. 2005;22:373. doi: 10.1007/s11095-004-1874-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park MR, Han KO, Han IK, Cho MH, Nah JW, Choi YJ, Cho CS. J Control Release. 2005;105:367. doi: 10.1016/j.jconrel.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Tang GP, Guo HY, Alexis F, Wang X, Zeng S, Lim TM, Ding J, Yang YY, Wang S. J Gene Med. 2006;8:736. doi: 10.1002/jgm.874. [DOI] [PubMed] [Google Scholar]
- 43.Arote R, Kim TH, Kim YK, Hwang SK, Jiang HL, Song HH, Nah JW, Cho MH, Cho CS. Biomaterials. 2007;28:735. doi: 10.1016/j.biomaterials.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 44.Weissleder R, Elizondo G, Wittenberg J, Rabito CA, Bengele HH, Josephson L. Radiology. 1990;175:489. doi: 10.1148/radiology.175.2.2326474. [DOI] [PubMed] [Google Scholar]
- 45.Weissleder R, Moore A, Mahmood U, Bhorade R, Benveniste H, Chiocca EA, Basilion JP. Nat Med. 2000;6:351. doi: 10.1038/73219. [DOI] [PubMed] [Google Scholar]
- 46.Bhattarai N, Ramay HR, Gunn J, Matsen FA, Zhang M. J Control Release. 2005;103:609. doi: 10.1016/j.jconrel.2004.12.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.