

Abstract
The pleiotropic cytokine, interleukin-6 (IL-6), has emerged in recent years as a key regulator of the transition from innate to adaptive immunity through its ability to modulate leukocyte recruitment at inflammatory sites. This review highlights a newly identified role for IL-6 transsignaling, initiated by an agonistic complex of IL-6 and a soluble form of IL-6 receptor α, in heightening immune surveillance of peripheral lymphoid organs during febrile inflammatory responses. Inflammatory cues provided by the thermal component of fever trigger IL-6 transsignaling to act at discrete levels in the multistep adhesion cascade that governs the entry of blood-borne lymphocytes across ‘gatekeeper’ high endothelial venules (HEVs) in lymph nodes and Peyer patches. IL-6-trans-signaling–dependent mechanisms have been elucidated during thermal stimulation of primary tethering and rolling of lymphocytes along the lumenal surface of HEVs as well as during secondary firm arrest of lymphocytes in HEVs prior to their migration into the underlying parenchyma. These mechanisms profoundly increase the probability that lymphocytes that continuously patrol the body will engage in productive encounters with target antigens sequestered within lymphoid organs. Findings that the lymphocyte–HEV–IL-6 transsignaling biological axis functions as a thermally-sensitive alert system that promotes immune surveillance provide insight into one of the unresolved mysteries in immunology regarding the benefits of mounting a febrile reaction during inflammation.
Keywords: Fever, lymphocyte trafficking, high endothelial venules, IL-6 trans-signaling, trafficking molecules
1. Overview of immune activity of IL-6
Interleukin-6 (IL-6) plays a complex role in the overall process of inflammation and in the generation of immune responses, exhibiting both pro- and anti-inflammatory activities [1]. Numerous reports have demonstrated that IL-6 is required for optimal antibody production, differentiation of cytokine producing lymphocyte subsets, T cell function, and foremost, in mediating systemic acute-phase reactions [2-5]. Furthermore, in vitro and in vivo studies have shown that IL-6 can contribute to angiogenesis, a critical process in both inflammation and tumor progression, by enhancing the synthesis of vascular endothelial growth factor (VEGF) and fibroblast growth factor [6-10]. IL-6 also indirectly determines anti-inflammatory processes through the activation of acute phase reactants and protects against endotoxin-mediated injury and death by down-regulating the expression of inflammatory mediators such as tumor necrosis factor (TNF), granulocyte macrophage-colony stimulating factor, interferon-γ (IFN-γ) and CXCL1 chemokine [11-15].
The relationship between IL-6 and fever induction during acute inflammation has been well-established. Intravenous or intracerebroventricular injection of IL-6 leads to increased core temperature in rats and rabbits [16, 17]. Alternatively, the ability to induce fever after peripheral challenge with pyrogens is attenuated in experiments using IL-6–neutralizing antibodies or in IL-6–deficient mice [18, 19]. Systemic administration of IL-6 to IL-6–deficient mice can restore the ability to generate fever. However, the role of fever in controlling the expression and activity of IL-6, and consequently its cellular functions, is not fully understood. Recent studies have revealed a novel role of IL-6 in the progression and resolution of inflammation. As discussed in several comprehensive reviews [20-23], IL-6 contributes to the modulation of leukocyte trafficking and apoptosis. This mediates the switch from an innate immune response with predominant neutrophil infiltration, to an adaptive immune response characterized by the infiltration of monocytes and lymphocytes. This review will focus on the IL-6–dependent mechanisms triggered by systemic thermal stress in the context of febrile inflammatory responses that lead to enhanced recruitment of T cells into lymphoid organs and improved immune surveillance.
1.1. IL-6 signaling: classical versus trans-signaling
The complex biological functions of IL-6 are initiated by the engagement of IL-6 with the IL-6 receptor α (IL-6Rα) binding subunit. There are two major naturally occurring forms of IL-6Rα, i.e., a membrane form (mIL-6Rα) and a soluble form (sIL-6Rα) which lacks the trans-membrane domain [24, 25]. Soluble IL-6Rα can be generated by alternative splicing of the IL-6Rα transcript, but it is thought that the major mechanism for generating sIL-6Rα in inflammation is through ectodomain shedding from activated leukocytes by the metalloproteinase ADAM17 (A Disintegrin And Metalloproteinase) and to a lesser extent, by ADAM-10 [26-30]. Glycoprotein 130 (gp130) is the common signal transducing receptor chain for IL-6 family cytokines and is expressed ubiquitously in all cell types [31, 32]. Based on structural similarity and common usage of gp130, several other cytokines, including IL-11, IL-27, oncostatin M (OSM), leukemia inhibitory factor (LIF), ciliary neurotrophic factor, and cardiotrophin-1 are grouped as IL-6 family members [1, 33, 34]. IL-6 signal transduction can occur through a complex consisting of membrane-anchored IL-6Rα and gp130 via a mechanism termed classical signaling (Figure 1) [34, 35]. Alternatively, in trans-signaling, sIL-6Rα pre-associates with IL-6 and then interacts with the ubiquitously expressed gp130 to initiate signaling [22, 36]. IL-6 trans-signaling can expand the range of cells capable of responding to IL-6 from the relatively few types that express the membrane receptor (e.g., hepatocytes, hematopoietic cells and certain types of malignant cells) [26], to virtually all cells in the body [21, 22, 25, 37]. Soluble gp130 (sgp130) is a naturally occurring variant of gp130 that lacks transmembrane and intracellular domains [21, 23, 24, 38]. Sgp130 preferentially interacts with IL-6/sIL-6Rα complexes and thus provides an experimental tool to distinguish trans-signaling from classical signaling events mediated by membrane-anchored IL-6Rα [22, 24, 39]. Several unique roles of IL-6 trans-signaling, including leukocyte recruitment and cancer development associated with inflammation have been dissected utilizing recombinant sgp130 [20, 24, 40-42].
Figure 1. Role of IL-6 in promoting lymphocyte trafficking in acute and chronic inflammation.
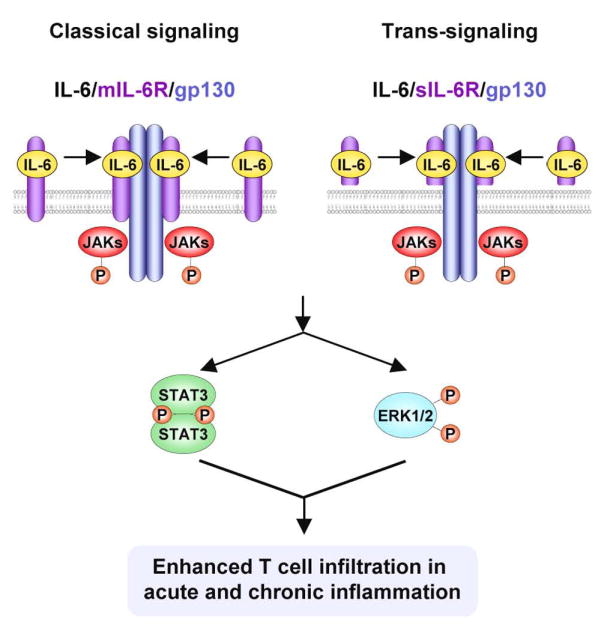
IL-6 transduces signals via membrane-anchored gp130 molecules through a classical pathway initiated by binding to membrane-anchored IL-6Rα (mIL-6Rα) or by a transsignaling mechanism in which IL-6 associates with a soluble form of IL-6Rα (sIL-6Rα). IL-6/IL-6Rα then recruits gp130 into hexameric complexes that initiate activation of the downstream signaling molecules STAT3 and ERK-1/2. IL-6 is causal in increasing the adhesive potential of both lymphocytes and vascular endothelial cells, which leads to enhanced trafficking of lymphocytes to lymphoid organs as well as to sites of acute and chronic inflammation.
The molecular events involved in IL-6 signal transduction include the following steps. The IL-6/mIL-6Rα or IL-6/sIL-6Rα complex recruits and stabilizes gp130 homodimers forming a hexameric signaling complex containing two molecules each of IL-6, IL-6Rα and gp130 (Figure 1) [34, 43]. IL-6/IL-6Rα ligation induces conformational changes in gp130, which result in autophosphorylation of the Janus kinases, JAK1, JAK2 and Tyk2, that are constitutively associated with the membrane-proximal Box1/Box2 region of gp130 [44-46]. Activated JAK kinases trans-phosphorylate themselves and phosphorylate multiple tyrosine residues of the intracellular domain of gp130. These phosphotyrosine residues provide docking sites for downstream signaling molecules including STAT1 and STAT3 and the protein tyrosine phosphatase SHP-2, which communicates signals to the MEK1/ERK-1/2 pathway. More specifically, STAT3 and STAT1 bind to the multiple phospho (p)YXXQ motifs (Y767/814/905/915 in human, Y765/812/904/914 in mouse) of gp130 whereas SHP-2 binds to the more membrane-proximal site (Y759 in human, Y757 in mouse) of gp130 [34, 47]. Phosphorylated STATs hetero-and homo-dimerize and translocate into the nucleus, where they bind to specific DNA sequences (e.g., acute phase response elements, APREs, or IL-6 response elements, IL-6REs) and enhance transcription of target genes [38, 48-50]. STAT3/STAT3 homodimers have a predominant role in gp130 signaling, and mediate a broad range of cellular activities, including mobility, adhesion, proliferation, and survival. Recent studies have shown that STAT3 activation in response to IL-6 is also required for differential leukocyte recruitment during acute inflammatory responses [20, 42]. Phosphorylated SHP2, besides acting as phosphatase at the site of activation, interacts with the Grb2-SOS adaptor complex. The Grb2-SOS complex then activates the MEK1/ERK-1/2 signaling pathway, which leads to phosphorylation of AP1 and the Ets family of transcription factors and downstream activation of immediate growth response genes [51, 52]. Besides transcriptional processes, the MAP kinases ERK-1/2 affect many cellular functions including cell cycle progression as well as cytoskeletal rearrangements and cell motility [1, 24, 37, 53-57].
IL-6 signaling is under tight control, as would be expected in view of its pleiotropic effects. Several negative feedback mechanisms have been shown to down-modulate IL-6 signaling by acting at least at two distinct levels; at the level of receptor complexes and at the level of downstream intracellular signaling molecules. Ligand induced internalization of IL-6 receptor complexes has been identified as one mechanism of down-regulating IL-6 signaling at the receptor level. Gp130 is internalized and degraded upon IL-6 binding [34, 38, 58-60]. Enhanced removal of IL-6Rα and gp130 abrogates signaling capability. Several intracellular molecules have been identified that negatively regulate IL-6 downstream signaling pathways. Protein tyrosine phosphatases such as SHP2 dephosphorylate tyrosine residues on JAKs, gp130, and other recruited proteins, terminating signaling [34, 38]. The protein inhibitor of activated STAT3 (PIAS3) interacts with residues 82-133 of activated STAT3 dimers and blocks STAT3 interactions with target DNA [61, 62]. Suppressor of cytokine signaling (SOCS) proteins are rapidly induced after IL-6 activation via a STAT3-dependent mechanism and interfere with receptor complexes as well as with downstream molecules [63]. Among the SOCS family members, SOCS3 is a particularly effective regulator of IL-6 signaling. SOCS3 binds to the phospho-Y757 docking site on gp130 (in mice) where it acts on both JAKs and gp130 to inhibit phosphorylation of these molecules. [64-67]. Moreover, SOCS proteins can also act as an adaptor for the ubiquitin system by recruiting E3 ubiquitin ligase and may promote ubiqitination and degradation of the IL-6 receptor complex [34]. Taken together, these negative feedback mechanisms ensure that IL-6 signaling is transient in nature. Recovery reactions involve enhanced expression of receptor subunits, reduction of SOCS3 to basal level and dephosphorylation of the signal transducing proteins of the diverse pathways.
2. Molecular determinants of lymphocyte trafficking
The continuous recirculation of lymphocytes from the blood into the secondary lymphoid organs such as lymph nodes (LNs) and Peyer patches (PPs) is necessary for the generation and maintenance of adaptive immune responses. It is estimated that approximately 3×106 blood-borne lymphocytes extravasate into a single LN daily across the specialized post-capillary venules termed high endothelial venules (HEVs) [68]. This highly efficient process maximizes the probability that dendritic cells will present cognate antigen to numerically rare Ag-specific T cells which are present at a frequency of ~1 in 105 - 106 cells. HEVs are present only in the LNs and PPs but not in the spleen or extralymphoid organs [69-71]. HEV-like vessels are also observed in chronically inflamed tissues where they are induced under the influence of local proinflammatory cytokines such as TNF and LT-α [72-76].
HEVs are morphologically and biochemically distinct from the majority of vessels throughout the body that support only limited trafficking under homeostatic conditions [69-71, 77]. In contrast to the squamous endothelial cells that line venules in extralymphoid tissues, HEVs have a near cuboidal morphology, which provide an irregular surface and may contribute to turbulent flow within the vascular microdomains, promoting extravasation of lymphocytes [77]. HEVs present a unique landscape rich in adhesion molecules and chemokines that direct lymphocyte trafficking. HEVs are the only vessels that constitutively possess the full complement of biosynthetic machinery required to elaborate the sulfated carbohydrate structures required for the initial steps of lymphocyte trafficking [73].
The process of lymphocyte trafficking involves a highly integrated sequence of adhesion and signaling events which has been extensively examined in secondary lymphoid organs. The multistep adhesion cascade includes (1) primary tethering and rolling of lymphocytes along the vessel walls, (2) chemokine activation, (3) secondary firm arrest, and (4) transendothelial migration of lymphocytes across vascular endothelial barriers [69-71]. In peripheral LNs (PLNs), primary tethering and rolling is initiated by engagement of C-type lectin L-selectin molecules on lymphocytes by the sulfated glycoprotein complex termed peripheral node addressin (PNAd) which is displayed on the lumenal surface of HEVs [73]. In the case of PPs, this initial step is orchestrated by the interaction of L-selectin and α4β7 integrin on the circulating lymphocyte with mucosal addressin cell adhesion molecule-1 (MAdCAM-1) displayed on the HEVs [69, 70].
Primary adhesive interactions (step 1) result in the reduction of lymphocyte velocity, allowing lymphocytes to sample the vascular microenvironment for the presence of the so-called “homeostatic” chemokine, CCL21 [40, 68]. In human and mouse, CCL21 is constitutively produced by LN stromal cells and is transcytosed to the lumenal surface of the HEVs [78]. Mouse HEVs can also synthesize CCL21 de novo [79, 80]. Engagement of CCL21 by the CCR7 chemokine receptor (step 2) on lymphocytes converts lymphocyte function associated antigen-1 (LFA-1) from an inactive conformer to an intermediate affinity state [81, 82]. The conformational change of LFA-1 from an intermediate affinity to high affinity occurs after engagement by HEV counter-receptors termed intercellular adhesion molecule-1 or -2 (ICAM-1/2) [81, 82]. ICAM-1 and ICAM-2 are members of the immunoglobulin superfamily of genes, and are both constitutively expressed on HEVs [69-71]. High affinity LFA-1/ICAM-1/2 interactions result in firm adhesion of lymphocytes to HEVs (step 3) and subsequent transmigration of lymphocytes into the lymphoid tissue (step 4) [69-71]. Due to their structural similarity, the functions of ICAM-1 and ICAM-2 are largely redundant during homeostatic lymphocyte recirculation through secondary lymphoid organs [69-71, 83].
3. IL-6 trans-signaling mediates thermal control of lymphocyte/endothelial interactions and trafficking
Fever is an ancient, evolutionarily conserved response to infection which has been documented in both endothermic (warm-blooded) and ectothermic (cold-blooded) animals [84-86]. In endotherms, infection results in the production of pyrogenic cytokines, such as TNF, IL-1β and IL-6, which act on the hypothalamus to raise the thermoregulatory setpoint and elevate core temperatures [86]. Disruption of IL-6 function using neutralizing antibodies or IL-6–deficient mice has clearly demonstrated the requirement for IL-6 in mediating the febrile inflammatory response in endothermic animals [19]. Ectotherms, on the other hand cannot regulate their body temperature endogenously, and thus engage in biochemically-regulated heat-seeking behaviors to increase body temperature [84, 85]. Elevated body temperature, whether the result of natural fever or of exogenous heating (defined as hyperthermia), has been demonstrated to contribute to survival during infection and resolution of inflammation in species ranging from endothermic (mammals and birds) and ectothermic vertebrates (fish) to invertebrates (insects) [84-87]. Intriguingly, not only is fever evolutionarily conserved, but the cytokines that mediate fever in mammals are themselves also of ancient origin. IL-6 immuno-reactive proteins and IL-6 activity [88, 89] have been identified in species as diverse as mammals, birds, fishes, mollusks and echinoderms, which arose from a common ancestor ~990 million years ago [90].
Fever-range thermal stress exerts complex effects on cytokine activity and synthesis. [84-86, 91-98]. While heat alone has little effect on the production of pro-inflammatory cytokines, it can act in conjunction with strong inflammatory stimuli like bacterial LPS to increase the synthesis of IL-6, TNF, IFN-α, GM-CSF, G-CSF and CXC chemokines [84, 92, 99-103]. Conversely, thermal stress has also been proposed to play a role in the resolution of inflammatory responses through the downregulation of cytokine production [84, 104-106]. Further investigation has revealed that these apparently paradoxical results reflect complex intracellular signaling networks that integrate cellular stress signals initiated by febrile temperatures and inflammatory signals, such as those initiated by LPS. The crosstalk between these signaling pathways has been studied most extensively in cells of the myeloid lineage [105-109]. Thermal stimulation of macrophages results in the activation of the transcriptional regulatory protein heat shock factor (HSF)-1. HSF-1 activates the transcription of many heat-responsive genes including heat shock proteins by binding to specific recognition sequences in the promoters of target genes [105, 110, 111]. However, in the case of many genes involved in inflammatory responses including TNF and IL-1β, HSF-1 can repress transcription [107, 108, 112, 113]. A functional HSF-1 binding site has been identified in the promoter of the IL-6 gene [114] raising the question as to whether heat stimulates or represses IL-6 responses. HSF-1-mediated transcriptional repression can be lifted, however in the presence of a potent inflammatory stimulus. In this regard, bacterial LPS has been shown to transiently down-modulate HSF-1 activity, allowing transcription of proinflammatory genes [108, 115]. Taken together, these results suggest that the thermal response may have different effects on cytokine regulation early or late during an inflammatory response.
3.1. IL-6 trans-signaling enhances the binding activity of lymphocyte trafficking molecules
One of the remaining mysteries in immunology relates to the potential beneficial mechanism of action of fever. The evolutionary conservation of fever suggests that it is beneficial, since conserved functions point to preservation of organisimal integrity. A series of studies over the past several years has revealed that fever-range temperatures act through a IL-6 trans-signaling mechanism to enhance the adhesive interactions between circulating blood lymphocytes and HEVs, resulting in increased delivery of lymphocytes to immune-relevant sites in the body [40, 41, 116-120]. Remarkably, stimulation with fever-range temperatures was found to exert parallel IL-6 trans-signaling–dependent effects on circulating immune cells as well as on the target vasculature [40, 41, 120]. These studies employed an experimental model originally developed by Repasky and colleagues to raise the core temperature of humans and mice to mimic the febrile component of natural fevers using whole body hyperthermia (WBH) treatment (39.5 ± 0.5°C for 6 hours) [103, 117, 121-123]. Application of systemic fever-range hyperthermia causes a transient decrease in the number of leukocytes circulating in the peripheral blood of mice and of advanced cancer patients [103, 117, 124]. In mice, this decrease has been shown to be associated with an ~2-fold increase in trafficking of lymphocytes into HEV-bearing secondary lymphoid organs (PLNs, MLNs, and PPs) [117, 118, 125]. A physiological consequence of this redirection of circulating lymphocytes into the secondary lymphoid organs is to enhance immune surveillance by increasing the likelihood that rare antigen-specific cells will encounter cognate antigens in organs strategically positioned to be the first line of defense against microbial invaders entering via the skin, lungs, or the gastrointestinal tract.
One explanation for thermal effects on lymphocyte distribution is that heat acts directly on lymphocytes to affect the binding activity of the primary adhesion molecules L-selectin and α4β7 integrin [41, 116-120]. Short-term (1 hour) homing studies in which murine lymphocytes were incubated at fever temperatures prior to adoptive transfer demonstrated 1.5-2-fold enhanced trafficking to HEV-bearing lymphoid organs [117, 118]. Antibody blockade experiments demonstrated that fever temperatures act on lymphocytes to enhance the binding of L-selectin and α4β7 integrin for their HEV counter-receptors [117, 118]. Considering that the lymphocyte population of a lymph node turns over ~3 times daily under homeostatic (i.e., normothermal) conditions [68], the increase in trafficking observed in response to febrile temperatures represents a profound escalation in the mobilization and delivery of lymphocytes to the anatomical compartment where productive encounters with antigen presenting cells can take place.
A major advance in understanding the underlying mechanisms of thermal regulation on lymphocyte adhesion was the finding that conditioned medium from heat-treated cells can substitute for heat to induce L-selectin and α4β7 integrin binding activity [41, 118, 126, 127]. Lymphocyte adhesion was enhanced by conditioned media from several cell types, including hematopoietic cells (B and T lymphocytes, monocytes) and stromal cells (fibroblasts, endothelial cells), but not parenchymal cells (breast epithelial cells, lung epithelial cells, hepatocytes, melanocytes, neuroblasts) [41, 126]. The identification of pro-adhesive activity in conditioned medium of heat-treated cells suggested that the effects of fever-range thermal stress on lymphocyte primary adhesion are not a direct consequence of heat on membrane fluidity or the conformation of adhesion molecules, but rather is mediated by soluble factors. Several candidate cytokines (IL-6, IFN-α, IFN-γ, TNF, IL-1β) were identified in lymphocyte-conditioned medium that could duplicate the effects of thermal stress on adhesion when added to lymphocytes in vitro. However, no changes were detected in the amounts of any of these factors in response to febrile temperatures, suggesting that thermal stress elevates the bioactivity or bioavailability of selected cytokines [41].
A neutralizing antibody approach identified the specific cytokine present in conditioned medium that is responsible for thermally-regulated changes in L-selectin adhesion [41, 120]. The pro-adhesive effects of fever-range thermal stress or of conditioned medium were inhibited only by functional blockade of IL-6, but not of IFN-α, IFN-γ, TNF, or IL-1β [41]. Multiple leukocyte subsets including B and T lymphocytes, NK cells and monocytes were shown to produce IL-6 during culture in vitro, although no increase in IL-6 production could be detected in response to fever-range thermal stress alone [41]. Additionally, while molecules that mediate cellular responses to IL-6 (i.e., sIL-6Rα and soluble gp130) were identified in lymphocyte conditioned medium, their concentrations were also unaltered by heat. The requirement for IL-6 in mediating thermal stimulation of L-selectin–dependent binding of lymphocytes to HEVs was confirmed by antibody blockade of IL-6Rα or gp130 [41, 120]. Moreover, in vivo experiments in which thermal induction of L-selectin–mediated adhesion of lymphocytes to PLN HEVs was blocked by either IL-6 neutralizing Abs or by sgp130 identifies IL-6 trans-signaling as the crucial event in primary adhesion (Figure 2)[41, 120]. Blockade of IL-6 trans-signaling similarly inhibited thermal stimulation of α4β7 integrin binding activity (SSE, WCW, unpublished observations).
Figure 2. Thermal stimulation of primary adhesion is dependent on IL-6 trans-signaling.
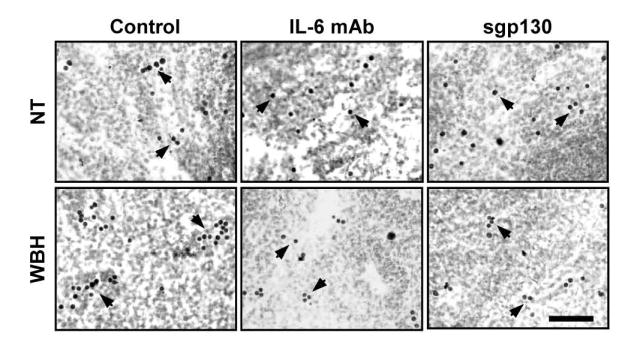
BALB/c mice were maintained at normal body temperature (NT) or treated for 6 hours with whole-body fever-range hyperthermia (WBH, 39.5°C ± 0.5°C). In selected experiments, mice were injected i.v. with function blocking IL-6 monoclonal antibody or with recombinant soluble gp130 (sgp130) prior to thermal treatment. Isolated splenocytes from experimental mice were analyzed for L-selectin–dependent primary adhesion to mouse PLN HEVs in a frozen-section adherence assay as described previously [41, 118, 151]. Photomicrographs show darkly stained exogenous splenocytes bound to HEVs which are below the plane of focus (indicated by arrows). Note that antibody blockade and soluble gp130 markedly reduced the number of WBH-treated lymphocytes adhering to individual HEVs. Bar = 50 μm.
The interpretation that IL-6 was required for thermal stimulation of L-selectin adhesion initially came into question in experiments using IL-6–deficient mice. These results showed paradoxically that lymphocytes from IL-6–deficient mice responded to febrile temperatures by increasing L-selectin binding activity through a gp130–dependent mechanism despite the absence of IL-6 [41]. This conundrum was resolved when the role of IL-6 family members was investigated. In lymphocytes isolated from wild-type C57BL/6 mice, antibody blockade of IL-11, OSM, or LIF had no impact on thermal enhancement of L-selectin adhesion [41]. However, in lymphocytes from IL-6–deficient mice, these cytokines were shown to compensate for the lack of IL-6 and promote lymphocyte adhesion in response to febrile temperatures. Evidence of cytokine redundancy is suggestive of the importance of maintaining gp130 signaling events in order to regulate lymphocyte adhesion and immune surveillance during febrile inflammatory episodes.
Interestingly, the requirement for IL-6 trans-signaling for thermal regulation of L-selectin-like adhesion has recently been documented not only in leukocytes isolated from a broad spectrum of endothermic vertebrates including mammals (human, mouse, rat, rabbit, dog, cattle, elephant, rhinoceros, and tiger) and birds (chicken), but also in ectothermic species including amphibians (Xenopus) and teleosts (rainbow trout) [120]. The evolutionary conservation of this IL-6 transsignaling mechanism of amplifying primary leukocyte adhesion in species that diverged from a common node of ancestry ~450 million years ago [90] strongly supports the hypothesis that this response confers a survival advantage for immune protection in vertebrate animals.
Multiple approaches were used to dissect the molecular mechanisms by which IL-6 transsignaling contributes to enhanced L-selectin function. Direct exposure of lymphocytes to fever-range temperatures does not change L-selectin cell surface density, lectin activity or localization on microvillus processes of lymphocyte surface membranes [41, 116, 118, 119]. Rather, thermal stimulation induces L-selectin to pre-associate with the detergent-insoluble actin-based cytoskeleton [41, 116, 127]. L-selectin–cytoskeletal interactions have similarly been shown to occur after L-selectin crosslinking by natural ligand or antibody reagents and are generally thought to contribute to the tensile strength of adhesive bonds in HEVs under shear [57, 128-130]. Thermal responsiveness was mapped to the C-terminal 11 amino acids of the cytoplasmic domain of L-selectin which contains a binding site for the cytoskeletal linker protein α-actinin [41, 57, 116, 128]. Further investigation demonstrated that the addition of recombinant proinflammatory cytokines (IL-6, IL-1-β, TNF, IFN-α, or IFN-γ) or conditioned medium from heat treated lymphocytes could substitute for thermal stress and drive the association of L-selectin with the cytoskeletal matrix [41]. However, only neutralization of IL-6 blocked induction of L-selectin interactions with the actin-based cytoskeleton and subsequent enhanced binding to HEVs in response to either thermal stimulation or conditioned medium [41].
Combined biochemical and pharmacological strategies placed MEK1/ERK-1/2 in the IL-6 transsignaling pathway that regulates L-selectin association with the cytoskeleton and consequent increased L-selectin binding activity [41]. Thermal stimulation of lymphocytes results in the progressive activation of ERK-1/2. This response occurred over the timeframe during which changes in L-selectin adhesion are observed (i.e., 2-6 hours of heat treatment) and is dependent on IL-6 trans-signaling [41, 118]. Furthermore, pharmacological inhibition of MEK1/ERK-1/2, but not JNK or p38, blocked thermal activation of L-selectin–cytoskeletal interactions and lymphocyte adhesion to HEVs [41]. Thermal stress was also found to activate STAT3, although its role in lymphocyte adherence under febrile conditions remains to be elucidated. Together, these results support a two-stage model for the regulation of L-selectin adhesion in response to the thermal stress associated with infection or inflammation. In the first stage, febrile temperatures cause an increase in the bioactivity or bioavailability of IL-6/sIL-6Rα complexes without detectable changes in the concentrations of IL-6 or sIL-6Rα. While the precise mechanisms are not known, it is likely that heat stabilizes the intermolecular interactions within IL-6/sIL-6Rα complexes and/or changes the threshold sensitivity of cells to IL-6 trans-signaling. In the second stage, these complexes then act on lymphocytes to enhance L-selectin–cytoskeletal interactions and adhesion through a MEK1/ERK-1/2 inside-out signaling pathway [41].
3.2 IL-6 trans-signaling augments endothelial cell adhesion during thermal stress
Fever-range thermal stress has also been shown to have independent effects on the intrinsic binding activity of HEVs. This issue was initially addressed using an in vitro frozen-section adherence assay to measure the adhesive activity of HEVs in lymphoid organs from mice treated with fever-range WBH [40, 117, 131]. In these studies, it was observed that systemic fever-range thermal stress enhances the ability of HEVs in LN and PP cryosections to support lymphocyte adhesion under shear. Intravital microscopic observation of inguinal lymph nodes in live animals further pinpointed the nature of the adhesive interactions regulated by thermal stress [40]. These experiments were designed so that the lymphocyte population undergoing adhesion to HEVs was not exposed to heat treatment. Fever-range hyperthermia increased the capacity of HEVs to support secondary firm adhesion of circulating lymphocytes, without changing the proportion of cells that undergo primary tethering and rolling interactions. Moreover, febrile temperatures caused an ~2-fold increase in the capacity of endothelial cells to support lymphocyte transit across HEVs into the underlying parenchyma in LNs and PPs [40, 117, 125, 131]. As discussed above for the lymphocyte response, augmentation in HEV adhesion is predicted to enhance immune surveillance by favoring chance encounters between rare antigen-specific T cells and dendritic cells presenting cognate antigens within lymphoid organs.
Dissection of the adhesion mechanisms revealed that thermal stress strongly upregulates the HEV surface density of two homing molecules, ICAM-1 and CCL21, which are required for firm adhesion of lymphocytes in HEVs and subsequent transendothelial migration [40]. As predicted by the intravital microscopy studies, heat did not change the intravascular density of adhesion molecules that participate in primary tethering and rolling events in HEVs (i.e., PNAd and MAdCAM-1). Febrile temperatures also did not affect the intravascular display of ICAM-2. ICAM-1 was found to be indispensable for thermally enhanced lymphocyte trafficking across HEVs, while under homeostasis both ICAM-1 and ICAM-2 can support firm arrest and extravasation [40, 83, 132]. These results are interpreted to indicate that display of ICAM-1 at high density enables this ‘gatekeeper’ molecule to become the preferred binding partner of LFA-1 in HEVs. These findings are consistent with earlier observations regarding the obligatory role of ICAM-1, when expressed at high density, in directing lymphocyte trafficking to extralymphoid sites of inflammation [83]. The finding that thermal stress induced the intravascular density of CCL21 [41] was unexpected since CCL21 is generally considered to be a homeostatic chemokine [70]. Thermally enhanced display of CCL21 was deemed to be necessary but not sufficient to support improved trafficking across HEVs without concomitant contributions of ICAM-1 based on data from ICAM-1 neutralizing antibody or ICAM-1–deficient mice [40]. These observations are in line with in vitro studies showing CCL21 and ICAM-1 act cooperatively to promote optimal LFA-1–dependent adhesion in lymphocytes. In this regard, CCL21 activates ‘inside-out’ signaling in a dose-dependent manner and initiates ligand-independent conformational changes in LFA-1 [133, 134]. Further transition of LFA-1 to a high affinity state is triggered by LFA-1 engagement by ICAM-1 through a mechanism that is highly dependent on ICAM-1 density [81, 82].
Proinflammatory cytokines were considered the most likely candidates for regulating the binding function of HEVs in light of the well established role of these cytokines in driving ICAM-1 synthesis in primary endothelial cells in vitro [51, 135]. TNF and IL-1β are generally considered the most potent inducers of ICAM-1 in cultured endothelial cells (e.g., human umbilical vein endothelial cells, HUVEC; human dermal microvascular endothelial cells, HMVEC) whereas IFN-γ and IL-6 have been relegated to a more minor role [51]. Thus, it came as a surprise that a common IL-6 trans-signaling mechanism was identified for thermal stimulation of HEV adhesion, paralleling the mechanism of action reported for lymphocytes discussed above. In this regard, thermal induction of ICAM-1 density as well as ICAM-1–dependent homing across HEVs was abrogated upon treatment of mice with IL-6 neutralizing antibodies [40]. The pro-adhesive effects of heat were not diminished in mice treated with TNF, IL-1β or IFN-γ neutralizing antibodies. The requirement for IL-6 was further validated in IL-6–deficient mice which failed to induce the intravascular display of ICAM-1 and lymphocyte trafficking in response to fever-range thermal stress (Figure 3) [40]. Administration of recombinant soluble gp130 prior to subjecting the mice to fever-range thermal stress prevented ICAM-1 induction, which is indicative of an IL-6 trans-signaling mechanism. In contrast, thermal induction of CCL21 is mediated by an IL-6–independent mechanism revealing a bifurcation in the pathways that integrate thermal stimulation of lymphocyte trafficking across the endothelial barrier [40]. The molecular mechanisms that mediate CCL21 induction remain to be elucidated.
Figure 3. Thermal induction of ICAM-1 on HEVs is dependent on IL-6.

WT or IL-6–deficient (Il6−/−) mice (C57BL/6 background) were exposed to fever-range thermal stress (39.5 ± 0.5°C) for 6 hours and the intravascular density of ICAM-1 on HEVs of peripheral lymph nodes was assessed by immunofluorescence staining as described [40]. The position of near-cuboidal HEVs in tissue sections is demarked by counter-staining for the HEV-restricted molecule, PNAd. Thermal stress strongly upregulates ICAM-1 density on PNAd+ HEVs in WT but not in Il6−/− mice. Bar = 50 μm.
Further insight into the IL-6 trans-signaling mechanism underlying regulation of ICAM-1 is provided by studies utilizing a recombinant fusion protein, termed hyper-IL-6, in which IL-6 is connected to sIL-6Rα by a flexible linker peptide [136]. Systemic administration of hyper-IL-6 increased the intravascular density of ICAM-1, although this response was not restricted to HEVs [40]. Hyper-IL-6 failed to upregulate expression of CCL21 [40], however, supporting the conclusion that thermal induction of CCL21 is independent of IL-6 signaling. Augmentation of ICAM-1 expression on HEVs by thermal stress or hyper-IL-6 in vivo required transcriptional events that could be disrupted by administration of actinomycin D [40].
Evidence that IL-6 family cytokines activate canonical signaling pathways (STAT1, STAT3, or ERK-1/2) in primary human dermal microvascular endothelial cells (HMVEC) in vitro (Figure 4A) suggests that endothelial cells lining HEVs may be a direct target for exogenously administered hyper-IL-6 or endogenous IL-6 trans-signaling during thermal stress in vivo [40]. In macrovascular HUVEC, ICAM-1 gene expression is regulated by STAT3 and NFκB binding elements in the ICAM-1 promoter following activation by IL-6/sIL-6Rα and TNF, respectively [51, 137-140]. Thus, data showing that IL-6 (without sIL-6Rα) and OSM were ineffective at upregulating ICAM-1 protein in HMVEC despite strong activation of PY-STAT3 are suggestive of the complexity of cytokine signaling within endothelial cells (Figure 4B). Hyper-IL-6 generates a pattern of signaling reactions that is distinguishable from that generated by IL-6 alone or other IL-6 type cytokines (Figure 4A). More importantly, the combined action of the hyper-IL-6 recruited signals is effective to induce ICAM-1 in HMVEC (Figure 4B). The result is in line with the notion that endothelial cell expression of membrane-anchored IL-6Rα is below the threshold necessary for efficient signaling [138, 139]. Since similar ICAM-1 induction occurs in HEVs in response to endogenous IL-6 trans-signaling during thermal stress or to systemic administration of hyper-IL-6 or TNF [40], the challenge will be to resolve these in vivo observations with the prominent differences in ICAM-1 detected in endothelial cells in vitro following activation by hyper-IL-6, TNF, or cytokine-rich conditioned medium of LPS-activated macrophages (Figure 4B).
Figure 4. Cytokine-mediated signal transduction and ICAM-1 induction in endothelial cells in vitro.
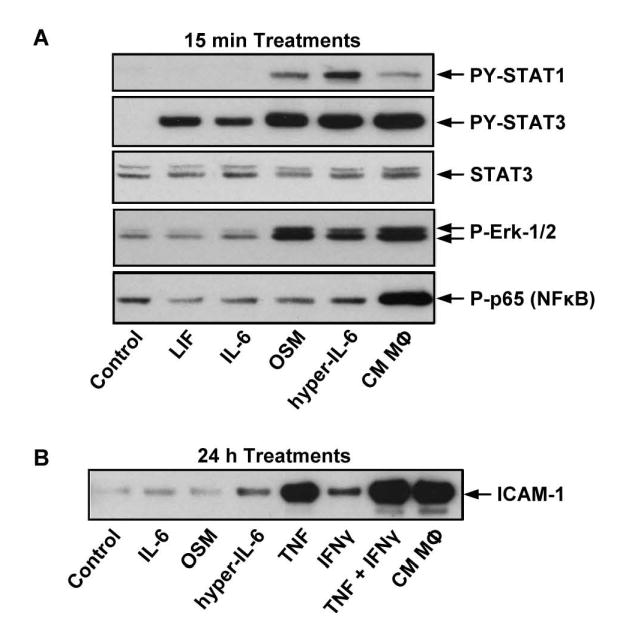
Human derma microvascular endothelial cells (HMVEC) were treated with recombinant cytokines or with conditioned medium from LPS-treated monocytes (CM MΦ) for the indicated time periods. Equal amounts of cell extracts were analyzed by Western blotting for (A) signal transduction molecules, or (B) ICAM-1 expression.
A fundamentally important feature of the IL-6–dependent thermal induction of ICAM-1 is its exquisite site specificity. IL-6 trans-signaling induced by febrile temperatures in the absence of pathogenic stimulation was causal in inducing ICAM-1 expression only in HEVs and not in non-activated squamous endothelial cells [40]. The site-specific nature of IL-6 trans-signaling during fever-range thermal stress is predicted to be essential for optimal immune stimulation since it focuses lymphocyte delivery to immune-relevant sites, i.e., the secondary lymphoid organs. This mechanism prevents the wholesale exodus of lymphocytes from the blood into non-lymphoid organs, thus avoiding dilution of the available lymphocyte repertoire available for antigen-specific recognition in lymphoid organs while at the same time, limiting autoimmune stimulation.
4. Conclusions and perspectives
Studies discussed in this review identify a previously unrecognized role of IL-6 trans-signaling in promoting lymphocyte trafficking across HEVs in the context of the acute febrile inflammatory response. These studies suggest that fever-range thermal stress acts as systemic ‘alarm system’ by triggering IL-6 trans-signaling mechanisms that manipulate discrete events in the multi-step cascade leading to enhanced lymphocyte trafficking across HEVs (Figure 5). IL-6 trans-signaling plays a non-redundant role during thermal stress in promoting primary tethering and rolling interactions (step 1) by increasing the binding activity of L-selectin and α4β7 integrin in lymphocytes [40, 41, 116-118]. While thermal stress increases the availability of CCL21 on HEV surface membranes (step 2) which is required for the transition from primary adhesion to secondary firm arrest, this activity is not mediated by IL-6 [40]. Febrile temperatures further act through an IL-6 trans-signaling mechanism to facilitate the subsequent firm arrest (step 3) and transendothelial migration (step 4) of lymphocytes by increasing the intravascular display of ICAM-1 on HEVs [40]. The newly identified contributions of IL-6 trans-signaling to trafficking in HEVs may be related to its established role in acute and chronic inflammation at extralymphoid sites. Numerous studies in in vitro and in vivo inflammatory models have elegantly demonstrated that IL-6 trans-signaling regulates the expression of trafficking molecules that mediate leukocyte primary adhesion (E-selectin and VCAM-1), chemokine activation (CCL2, CXCL10, CCL4, CCL5, CCL11, and CCL17), as well as secondary firm adhesion and transendothelial migration (ICAM-1 and VCAM-1) [20-22, 42, 138, 139, 141-143]. Interestingly, an anti-inflammatory role of IL-6 trans-signaling has also been demonstrated in vitro that involves inhibition of TNF and IL-1β-induced CXCL1, CXCL8 and CX3CL1 [138, 139, 144].
Figure 5. IL-6 trans-signaling targets each step of adhesion cascade controlling leukocyte extravasation during acute and chronic inflammation.

Thermally responsive trafficking molecules are shown in red. All trafficking molecules shown are regulated through an IL-6 trans-signaling mechanism with the exception of CCL21 (*) which is induced by febrile temperatures through an IL-6–independent mechanism.
An open question is whether the site-specific response to IL-6 trans-signaling during thermal stress reflects intrinsic traits of HEVs or is due to extrinsic factors unique to the local microenvironment in lymphoid organs. Endothelial cells are known to express proteins of the temperature-sensing transient receptor potential (TRP) Ca2+ channel family, some of which can detect changes in temperature within the febrile range [145]. It is tempting to speculate that signaling through these receptor systems lowers the threshold sensitivity to IL-6/sIL-6Rα selectively in the highly differentiated subset of endothelial cells the line HEVs. Alternatively, the heat response of HEVs could be dictated by the bioavailability of IL-6 and/or sIL-6Rα within the local tissue microenvironment. Multiple cell types including vascular endothelial cells, leukocytes and fibroblasts secrete IL-6 constitutively and upon inflammatory stimulation [146-149] whereas the sources for sIL-6Rα are limited to hepatocytes and some hematopoietic cells [20, 26]. Insight into the mechanisms underlying thermal responses is provided by recent evidence from inflammatory systems that neutrophils and fibroblasts regulate endothelial adhesion and leukocyte recruitment by increasing the local availability of IL-6 and/or sIL-6Rα [29, 30, 138, 149]. The cellular source of IL-6 or sIL-6Rα operative in the lymphoid organs during febrile inflammatory responses is yet to be elucidated.
Further investigation is needed to dissect the downstream molecular mechanisms regulating the IL-6/sIL-6Rα–dependent thermal response on both lymphocytes and HEVs. In lymphocytes, our studies have shown that administration of inhibitors specifically targeting the MEK/ERK-1/2 pathway abrogates thermally enhanced L-selectin bioactivity on lymphocytes in vitro [41]. Thermal stimulation of lymphocytes also results in the activation of STAT3, although its role in L-selectin adherence is unknown. It will be of particular interest to determine whether the downstream pathway responsible for thermal induction of ICAM-1 density involves STAT3, as has been reported for IL-6 trans-signaling responses in endothelial cell cultures in vitro [137, 139, 150]. In this event, it is feasible that the transient nature of IL-6 trans-signaling on the adhesive properties of both lymphocytes and endothelial cells observed during thermal stress responses [117, 118] involves canonical negative feedback loops that dampen IL-6 activity (i.e., mediated by receptor internalization, SOCS3 and PIAS). Repression of IL-6 trans-signaling is integral to the resolution phase of inflammation which aims to restore homeostatic trafficking and limit the metabolic demands and cellular damage of the inflammatory response. Overall, these studies reveal a novel role of the lymphocyte–endothelial–IL-6 trans-signaling axis in the setting of acute febrile inflammatory responses. Understanding the molecular mechanisms regulating the effects of febrile temperatures on lymphocyte recruitment has the potential to lead to the development of therapeutic strategies to redirect lymphocyte trafficking during cancer immunotherapy or in the treatment of chronic inflammatory disorders including autoimmune diseases.
Acknowledgments
We thank Drs Elizabeth Repasky, Jennifer Black and Joseph Skitzki for helpful discussions, and Daniel Fisher and Jason Muhitch for critical review of this manuscript. This work was supported by grants from the NIH (CA79764, CA094045, CA16056) The Department of Defense (DAMD17-98-1-8311, W81XWH-01-1-0354) and the Roswell Park Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Trupti D. Vardam, Email: trupti.vardam@roswellpark.org.
Lei Zhou, Email: lei.zhou@roswellpark.org.
Michelle M. Appenheimer, Email: michelle.appenheimer@roswellpark.org.
Qing Chen, Email: qing.chen@roswellpark.org.
Wang-Chao Wang, Email: w.wang@roswellpark.org.
Heinz Baumann, Email: heinz.baumann@roswellpark.org.
Sharon S. Evans, Email: sharon.evans@roswellpark.org.
References
- 1.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–54. [PubMed] [Google Scholar]
- 2.Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265:621–36. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2:619–26. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- 4.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–45. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 5.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 6.Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem. 1996;271:736–41. doi: 10.1074/jbc.271.2.736. [DOI] [PubMed] [Google Scholar]
- 7.Nakahara H, Song J, Sugimoto M, Hagihara K, Kishimoto T, Yoshizaki K, Nishimoto N. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003;48:1521–9. doi: 10.1002/art.11143. [DOI] [PubMed] [Google Scholar]
- 8.Nilsson MB, Langley RR, Fidler IJ. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005;65:10794–800. doi: 10.1158/0008-5472.CAN-05-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang SP, Wu MS, Shun CT, Wang HP, Lin MT, Kuo ML, Lin JT. Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J Biomed Sci. 2004;11:517–27. doi: 10.1007/BF02256101. [DOI] [PubMed] [Google Scholar]
- 10.Jee SH, Chu CY, Chiu HC, Huang YL, Tsai WL, Liao YH, Kuo ML. Interleukin-6 induced basic fibroblast growth factor-dependent angiogenesis in basal cell carcinoma cell line via JAK/STAT3 and PI3-kinase/Akt pathways. J Invest Dermatol. 2004;123:1169–75. doi: 10.1111/j.0022-202X.2004.23497.x. [DOI] [PubMed] [Google Scholar]
- 11.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–20. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J Biol Chem. 1995;270:933–43. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, Simpson RJ, Cheers C. Recombinant interleukin-6 protects mice against experimental bacterial infection. Infect Immun. 1992;60:4402–6. doi: 10.1128/iai.60.10.4402-4406.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mantovani A, Bussolino F, Dejana E. Cytokine regulation of endothelial cell function. Faseb J. 1992;6:2591–9. doi: 10.1096/fasebj.6.8.1592209. [DOI] [PubMed] [Google Scholar]
- 15.Barton BE, Jackson JV. Protective role of interleukin 6 in the lipopolysaccharide-galactosamine septic shock model. Infect Immun. 1993;61:1496–9. doi: 10.1128/iai.61.4.1496-1499.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakata Y, Morimoto A, Long NC, Murakami N. Fever and acute-phase response induced in rabbits by intravenous and intracerebroventricular injection of interleukin-6. Cytokine. 1991;3:199–203. doi: 10.1016/1043-4666(91)90017-8. [DOI] [PubMed] [Google Scholar]
- 17.LeMay LG, Vander AJ, Kluger MJ. Role of interleukin 6 in fever in rats. Am J Physiol. 1990;258:R798–803. doi: 10.1152/ajpregu.1990.258.3.R798. [DOI] [PubMed] [Google Scholar]
- 18.Chai Z, Gatti S, Toniatti C, Poli V, Bartfai T. Interleukin (IL)-6 gene expression in the central nervous system is necessary for fever response to lipopolysaccharide or IL-1 beta: a study on IL-6-deficient mice. J Exp Med. 1996;183:311–6. doi: 10.1084/jem.183.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozak W, Kluger MJ, Soszynski D, Conn CA, Rudolph K, Leon LR, Zheng H. IL-6 and IL-1 beta in fever. Studies using cytokine-deficient (knockout) mice. Ann N Y Acad Sci. 1998;856:33–47. doi: 10.1111/j.1749-6632.1998.tb08310.x. [DOI] [PubMed] [Google Scholar]
- 20.Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175:3463–8. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 21.Jones SA, Richards PJ, Scheller J, Rose-John S. IL-6 transsignaling: the in vivo consequences. J Interferon Cytokine Res. 2005;25:241–53. doi: 10.1089/jir.2005.25.241. [DOI] [PubMed] [Google Scholar]
- 22.Jones SA, Rose-John S. The role of soluble receptors in cytokine biology: the agonistic properties of the sIL-6R/IL-6 complex. Biochim Biophys Acta. 2002;1592:251–63. doi: 10.1016/s0167-4889(02)00319-1. [DOI] [PubMed] [Google Scholar]
- 23.Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11:613–24. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- 24.Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–36. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 25.Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose-John S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem. 2001;268:160–7. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- 26.Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. Faseb J. 2001;15:43–58. doi: 10.1096/fj.99-1003rev. [DOI] [PubMed] [Google Scholar]
- 27.Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4:96–100. doi: 10.1016/1043-4666(92)90043-q. [DOI] [PubMed] [Google Scholar]
- 28.Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300(Pt 2):281–90. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007 doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 30.Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, Rose-John S. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE) J Biol Chem. 2003;278:38829–39. doi: 10.1074/jbc.M210584200. [DOI] [PubMed] [Google Scholar]
- 31.Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–57. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 32.Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, Hirano T, Kishimoto T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573–81. doi: 10.1016/0092-8674(89)90438-8. [DOI] [PubMed] [Google Scholar]
- 33.Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, Haugen HS, Maurer M, Harder B, Johnston J, Bort S, Mudri S, Kuijper JL, Bukowski T, Shea P, Dong DL, Dasovich M, Grant FJ, Lockwood L, Levin SD, LeCiel C, Waggie K, Day H, Topouzis S, Kramer J, Kuestner R, Chen Z, Foster D, Parrish-Novak J, Gross JA. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–60. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 34.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishihara K, Hirano T. Molecular basis of the cell specificity of cytokine action. Biochim Biophys Acta. 2002;1592:281–96. doi: 10.1016/s0167-4889(02)00321-x. [DOI] [PubMed] [Google Scholar]
- 36.Rose-John S. Interleukin-6 biology is coordinated by membrane bound and soluble receptors. Acta Biochim Pol. 2003;50:603–11. [PubMed] [Google Scholar]
- 37.Rose-John S. Coordination of interleukin-6 biology by membrane bound and soluble receptors. Adv Exp Med Biol. 2001;495:145–51. doi: 10.1007/978-1-4615-0685-0_19. [DOI] [PubMed] [Google Scholar]
- 38.Scheller J, Rose-John S. Updating interleukin-6 classic-and trans-signaling. Signal Transduction. 2006;6:240–259. [Google Scholar]
- 39.Diamant M, Rieneck K, Mechti N, Zhang XG, Svenson M, Bendtzen K, Klein B. Cloning and expression of an alternatively spliced mRNA encoding a soluble form of the human interleukin-6 signal transducer gp130. FEBS Lett. 1997;412:379–84. doi: 10.1016/s0014-5793(97)00750-3. [DOI] [PubMed] [Google Scholar]
- 40.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, Rose-John S, von Andrian UH, Baumann H, Evans SS. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol. 2006;7:1299–308. doi: 10.1038/ni1406. [DOI] [PubMed] [Google Scholar]
- 41.Chen Q, Wang WC, Bruce R, Li H, Schleider DM, Mulbury MJ, Bain MD, Wallace PK, Baumann H, Evans SS. Central role of IL-6 receptor signal-transducing chain gp130 in activation of L-selectin adhesion by fever-range thermal stress. Immunity. 2004;20:59–70. doi: 10.1016/s1074-7613(03)00358-3. [DOI] [PubMed] [Google Scholar]
- 42.McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, Ernst M, Topley N, Jones SA. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9589–94. doi: 10.1073/pnas.0501794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paonessa G, Graziani R, De Serio A, Savino R, Ciapponi L, Lahm A, Salvati AL, Toniatti C, Ciliberto G. Two distinct and independent sites on IL-6 trigger gp 130 dimer formation and signalling. Embo J. 1995;14:1942–51. doi: 10.1002/j.1460-2075.1995.tb07186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giese B, Roderburg C, Sommerauer M, Wortmann SB, Metz S, Heinrich PC, Muller-Newen G. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J Cell Sci. 2005;118:5129–40. doi: 10.1242/jcs.02628. [DOI] [PubMed] [Google Scholar]
- 45.Schroers A, Hecht O, Kallen KJ, Pachta M, Rose-John S, Grotzinger J. Dynamics of the gp130 cytokine complex: a model for assembly on the cellular membrane. Protein Sci. 2005;14:783–90. doi: 10.1110/ps.041117105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skiniotis G, Boulanger MJ, Garcia KC, Walz T. Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor. Nat Struct Mol Biol. 2005;12:545–51. doi: 10.1038/nsmb941. [DOI] [PubMed] [Google Scholar]
- 47.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–53. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 48.Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–11. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- 49.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 50.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–8. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J Leukoc Biol. 1999;66:876–88. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- 52.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond B Biol Sci. 1996;351:127–34. doi: 10.1098/rstb.1996.0008. [DOI] [PubMed] [Google Scholar]
- 53.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 54.Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 55.Evans SS, Collea RP, Appenheimer MM, Gollnick SO. Interferon-alpha induces the expression of the L-selectin homing receptor in human B lymphoid cells. J Cell Biol. 1993;123:1889–98. doi: 10.1083/jcb.123.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leid JG, Steeber DA, Tedder TF, Jutila MA. Antibody binding to a conformation-dependent epitope induces L-selectin association with the detergent-resistant cytoskeleton. J Immunol. 2001;166:4899–907. doi: 10.4049/jimmunol.166.8.4899. [DOI] [PubMed] [Google Scholar]
- 57.Pavalko FM, Walker DM, Graham L, Goheen M, Doerschuk CM, Kansas GS. The cytoplasmic domain of L-selectin interacts with cytoskeletal proteins via alpha-actinin: receptor positioning in microvilli does not require interaction with alpha-actinin. J Cell Biol. 1995;129:1155–64. doi: 10.1083/jcb.129.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dittrich E, Rose-John S, Gerhartz C, Mullberg J, Stoyan T, Yasukawa K, Heinrich PC, Graeve L. Identification of a region within the cytoplasmic domain of the interleukin-6 (IL-6) signal transducer gp130 important for ligand-induced endocytosis of the IL-6 receptor. J Biol Chem. 1994;269:19014–20. [PubMed] [Google Scholar]
- 59.Thiel S, Dahmen H, Martens A, Muller-Newen G, Schaper F, Heinrich PC, Graeve L. Constitutive internalization and association with adaptor protein-2 of the interleukin-6 signal transducer gp130. FEBS Lett. 1998;441:231–4. doi: 10.1016/s0014-5793(98)01559-2. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Fuller GM. Phosphorylation and internalization of gp130 occur after IL-6 activation of Jak2 kinase in hepatocytes. Mol Biol Cell. 1994;5:819–28. doi: 10.1091/mbc.5.7.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 62.Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 63.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–5. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 64.Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, Nicola NA. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. Embo J. 1999;18:375–85. doi: 10.1093/emboj/18.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J Biol Chem. 2000;275:12848–56. doi: 10.1074/jbc.275.17.12848. [DOI] [PubMed] [Google Scholar]
- 66.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–9. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 67.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 68.von Andrian UH. Intravital microscopy of the peripheral lymph node microcirculation in mice. Microcirculation. 1996;3:287–300. doi: 10.3109/10739689609148303. [DOI] [PubMed] [Google Scholar]
- 69.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–6. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 70.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–78. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 71.Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol. 2004;4:360–70. doi: 10.1038/nri1354. [DOI] [PubMed] [Google Scholar]
- 72.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–90. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 73.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–56. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 74.Ruddle NH. Lymphoid neo-organogenesis: lymphotoxin’s role in inflammation and development. Immunol Res. 1999;19:119–25. doi: 10.1007/BF02786481. [DOI] [PubMed] [Google Scholar]
- 75.Pablos JL, Santiago B, Tsay D, Singer MS, Palao G, Galindo M, Rosen SD. A HEV-restricted sulfotransferase is expressed in rheumatoid arthritis synovium and is induced by lymphotoxin-alpha/beta and TNF-alpha in cultured endothelial cells. BMC Immunol. 2005;6:6. doi: 10.1186/1471-2172-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tjew SL, Brown KL, Kannagi R, Johnson P. Expression of N-acetylglucosamine 6-O-sulfotransferases (GlcNAc6STs)-1 and -4 in human monocytes: GlcNAc6ST-1 is implicated in the generation of the 6-sulfo N-acetyllactosamine/Lewis × epitope on CD44 and is induced by TNF-alpha. Glycobiology. 2005;15:7C–13C. doi: 10.1093/glycob/cwi050. [DOI] [PubMed] [Google Scholar]
- 77.Girard JP, Springer TA. High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol Today. 1995;16:449–57. doi: 10.1016/0167-5699(95)80023-9. [DOI] [PubMed] [Google Scholar]
- 78.Carlsen HS, Haraldsen G, Brandtzaeg P, Baekkevold ES. Disparate lymphoid chemokine expression in mice and men: no evidence of CCL21 synthesis by human high endothelial venules. Blood. 2005;106:444–6. doi: 10.1182/blood-2004-11-4353. [DOI] [PubMed] [Google Scholar]
- 79.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:258–63. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baekkevold ES, Yamanaka T, Palframan RT, Carlsen HS, Reinholt FP, von Andrian UH, Brandtzaeg P, Haraldsen G. The CCR7 ligand elc (CCL19) is transcytosed in high endothelial venules and mediates T cell recruitment. J Exp Med. 2001;193:1105–12. doi: 10.1084/jem.193.9.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, Robinson MK, Staunton DE, von Andrian UH, Alon R. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. 2005;6:497–506. doi: 10.1038/ni1194. [DOI] [PubMed] [Google Scholar]
- 83.Lehmann JC, Jablonski-Westrich D, Haubold U, Gutierrez-Ramos JC, Springer T, Hamann A. Overlapping and selective roles of endothelial intercellular adhesion molecule-1 (ICAM-1) and ICAM-2 in lymphocyte trafficking. J Immunol. 2003;171:2588–93. doi: 10.4049/jimmunol.171.5.2588. [DOI] [PubMed] [Google Scholar]
- 84.Hasday JD, Fairchild KD, Shanholtz C. The role of fever in the infected host. Microbes Infect. 2000;2:1891–904. doi: 10.1016/s1286-4579(00)01337-x. [DOI] [PubMed] [Google Scholar]
- 85.Appenheimer MM, Chen Q, Girard R, Wang WC, Evans SS. Impact of fever-range thermal stress on lymphocyte-endothelial adhesion and lymphcoyte trafficking. Immunological Investigations. 2005;34:295–323. doi: 10.1081/imm-200064501. [DOI] [PubMed] [Google Scholar]
- 86.Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev. 1991;71:93–127. doi: 10.1152/physrev.1991.71.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mackowiak PA. Physiological rationale for suppression of fever. Clin Infect Dis. 2000;31(Suppl 5):S185–9. doi: 10.1086/317511. [DOI] [PubMed] [Google Scholar]
- 88.Kaiser P, Rothwell L, Avery S, Balu S. Evolution of the interleukins. Dev Comp Immunol. 2004;28:375–94. doi: 10.1016/j.dci.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 89.Ottaviani E, Franchini A, Franceschi C. Presence of several cytokine-like molecules in molluscan hemocytes. Biochem Biophys Res Commun. 1993;195:984–8. doi: 10.1006/bbrc.1993.2141. [DOI] [PubMed] [Google Scholar]
- 90.Hedges SB. The origin and evolution of model organisms. Nat Rev Genet. 2002;3:838–49. doi: 10.1038/nrg929. [DOI] [PubMed] [Google Scholar]
- 91.Blatteis CM. Fever: pathological or physiological, injurious or beneficial? Journal of Thermal Biology. 2003;28:1. [Google Scholar]
- 92.Hasday JD, Garrison A, Singh IS, Standiford T, Ellis GS, Rao S, He JR, Rice P, Frank M, Goldblum SE, Viscardi RM. Febrile-range hyperthermia augments pulmonary neutrophil recruitment and amplifies pulmonary oxygen toxicity. Am J Pathol. 2003;162:2005–17. doi: 10.1016/S0002-9440(10)64333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roberts NJ, Jr, Sandberg K. Hyperthermia and human leukocyte function. II. Enhanced production of and response to leukocyte migration inhibition factor (LIF) J Immunol. 1979;122:1990–3. [PubMed] [Google Scholar]
- 94.Yoshioka A, Miyachi Y, Imamura S, Hiraoka M, Jo S, Abe M. Suppression of contact sensitivity by local hyperthermia treatment due to reduced Langerhans cell population in mice. Br J Dermatol. 1989;120:493–501. doi: 10.1111/j.1365-2133.1989.tb01322.x. [DOI] [PubMed] [Google Scholar]
- 95.Ostberg JR, Patel R, Repasky EA. Regulation of immune activity by mild (fever-range) whole body hyperthermia: effects on epidermal Langerhans cells. Cell Stress Chaperones. 2000;5:458–61. doi: 10.1379/1466-1268(2000)005<0458:roiabm>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ostberg JR, Kabingu E, Repasky EA. Thermal regulation of dendritic cell activation and migration from skin explants. Int J Hyperthermia. 2003;19:520–33. doi: 10.1080/02656730310001607986. [DOI] [PubMed] [Google Scholar]
- 97.Nahas GG, Tannieres ML, Lennon JF. Direct measurement of leukocyte motility: effects of pH and temperature. Proc Soc Exp Biol Med. 1971;138:350–2. doi: 10.3181/00379727-138-35894. [DOI] [PubMed] [Google Scholar]
- 98.Roberts NJ., Jr Impact of temperature elevation on immunologic defenses. Rev Infect Dis. 1991;13:462–72. doi: 10.1093/clinids/13.3.462. [DOI] [PubMed] [Google Scholar]
- 99.DuBose DA, Balcius J, Morehouse D. Heat stress and/or endotoxin effects on cytokine expression by human whole blood. Shock. 2002;17:217–21. doi: 10.1097/00024382-200203000-00010. [DOI] [PubMed] [Google Scholar]
- 100.Hasday JD, Bannerman D, Sakarya S, Cross AS, Singh IS, Howard D, Drysdale BE, Goldblum SE. Exposure to febrile temperature modifies endothelial cell response to tumor necrosis factor-alpha. J Appl Physiol. 2001;90:90–8. doi: 10.1152/jappl.2001.90.1.90. [DOI] [PubMed] [Google Scholar]
- 101.Jiang Q, Detolla L, Singh IS, Gatdula L, Fitzgerald B, van Rooijen N, Cross AS, Hasday JD. Exposure to febrile temperature upregulates expression of pyrogenic cytokines in endotoxin-challenged mice. Am J Physiol. 1999;276:R1653–60. doi: 10.1152/ajpregu.1999.276.6.R1653. [DOI] [PubMed] [Google Scholar]
- 102.Jiang Q, DeTolla L, van Rooijen N, Singh IS, Fitzgerald B, Lipsky MM, Kane AS, Cross AS, Hasday JD. Febrile-range temperature modifies early systemic tumor necrosis factor alpha expression in mice challenged with bacterial endotoxin. Infect Immun. 1999;67:1539–46. doi: 10.1128/iai.67.4.1539-1546.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ostberg JR, Taylor SL, Baumann H, Repasky EA. Regulatory effects of fever-range whole-body hyperthermia on the LPS-induced acute inflammatory response. J Leukoc Biol. 2000;68:815–20. [PubMed] [Google Scholar]
- 104.Fairchild KD, Viscardi RM, Hester L, Singh IS, Hasday JD. Effects of hypothermia and hyperthermia on cytokine production by cultured human mononuclear phagocytes from adults and newborns. J Interferon Cytokine Res. 2000;20:1049–55. doi: 10.1089/107999000750053708. [DOI] [PubMed] [Google Scholar]
- 105.Singh IS, Viscardi RM, Kalvakolanu I, Calderwood S, Hasday JD. Inhibition of tumor necrosis factor-alpha transcription in macrophages exposed to febrile range temperature. A possible role for heat shock factor-1 as a negative transcriptional regulator. J Biol Chem. 2000;275:9841–8. doi: 10.1074/jbc.275.13.9841. [DOI] [PubMed] [Google Scholar]
- 106.Ensor JE, Wiener SM, McCrea KA, Viscardi RM, Crawford EK, Hasday JD. Differential effects of hyperthermia on macrophage interleukin-6 and tumor necrosis factor-alpha expression. Am J Physiol. 1994;266:C967–74. doi: 10.1152/ajpcell.1994.266.4.C967. [DOI] [PubMed] [Google Scholar]
- 107.Xie Y, Chen C, Stevenson MA, Auron PE, Calderwood SK. Heat shock factor 1 represses transcription of the IL-1beta gene through physical interaction with the nuclear factor of interleukin 6. J Biol Chem. 2002;277:11802–10. doi: 10.1074/jbc.M109296200. [DOI] [PubMed] [Google Scholar]
- 108.Singh IS, He JR, Calderwood S, Hasday JD. A high affinity HSF-1 binding site in the 5’-untranslated region of the murine tumor necrosis factor-alpha gene is a transcriptional repressor. J Biol Chem. 2002;277:4981–8. doi: 10.1074/jbc.M108154200. [DOI] [PubMed] [Google Scholar]
- 109.Xie Y, Chen C, Stevenson MA, Hume DA, Auron PE, Calderwood SK. NF-IL6 and HSF1 have mutually antagonistic effects on transcription in monocytic cells. Biochem Biophys Res Commun. 2002;291:1071–80. doi: 10.1006/bbrc.2002.6562. [DOI] [PubMed] [Google Scholar]
- 110.Wu C. Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–69. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- 111.Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. Embo J. 1999;18:5943–52. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cahill CM, Waterman WR, Xie Y, Auron PE, Calderwood SK. Transcriptional repression of the prointerleukin 1beta gene by heat shock factor 1. J Biol Chem. 1996;271:24874–9. [PubMed] [Google Scholar]
- 113.Calderwood SK. Regulatory interfaces between the stress protein response and other gene expression programs in the cell. Methods. 2005;35:139–48. doi: 10.1016/j.ymeth.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 114.Pritts TA, Hungness ES, Hershko DD, Robb BW, Sun X, Luo GJ, Fischer JE, Wong HR, Hasselgren PO. Proteasome inhibitors induce heat shock response and increase IL-6 expression in human intestinal epithelial cells. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1016–26. doi: 10.1152/ajpregu.00492.2001. [DOI] [PubMed] [Google Scholar]
- 115.Singh IS, He JR, Hester L, Fenton MJ, Hasday JD. Bacterial endotoxin modifies heat shock factor-1 activity in RAW 264.7 cells: implications for TNF-alpha regulation during exposure to febrile range temperatures. J Endotoxin Res. 2004;10:175–84. doi: 10.1179/096805104225004851. [DOI] [PubMed] [Google Scholar]
- 116.Evans SS, Schleider DM, Bowman LA, Francis ML, Kansas GS, Black JD. Dynamic association of L-selectin with the lymphocyte cytoskeletal matrix. J Immunol. 1999;162:3615–24. [PubMed] [Google Scholar]
- 117.Evans SS, Wang WC, Bain MD, Burd R, Ostberg JR, Repasky EA. Fever-range hyperthermia dynamically regulates lymphocyte delivery to high endothelial venules. Blood. 2001;97:2727–33. doi: 10.1182/blood.v97.9.2727. [DOI] [PubMed] [Google Scholar]
- 118.Wang WC, Goldman LM, Schleider DM, Appenheimer MM, Subjeck JR, Repasky EA, Evans SS. Fever-range hyperthermia enhances L-selectin-dependent adhesion of lymphocytes to vascular endothelium. J Immunol. 1998;160:961–9. [PubMed] [Google Scholar]
- 119.Evans SS, Bain MD, Wang WC. Fever-range hyperthermia stimulates alpha4beta7 integrin-dependent lymphocyte-endothelial adhesion. Int J Hyperthermia. 2000;16:45–59. doi: 10.1080/026567300285411. [DOI] [PubMed] [Google Scholar]
- 120.Appenheimer MM, Girard RA, Chen Q, Wang WC, Bankert KC, Hardison J, Bain MD, Ridgley F, Sarcione EJ, B S, Kaspers B, Robert J, Rose-John S, Baumann H, Evans SS. Conservation of IL-6 Trans-Signaling Mechanisms Controlling L-Selectin Adhesion by Fever-Range Thermal Stress. Eur J Immunol. doi: 10.1002/eji.200636421. in press. [DOI] [PubMed] [Google Scholar]
- 121.Ostberg JR, Gellin C, Patel R, Repasky EA. Regulatory potential of fever-range whole body hyperthermia on Langerhans cells and lymphocytes in an antigen-dependent cellular immune response. J Immunol. 2001;167:2666–70. doi: 10.4049/jimmunol.167.5.2666. [DOI] [PubMed] [Google Scholar]
- 122.Pritchard MT, Ostberg JR, Evans SS, Burd R, Kraybill W, Bull JM, Repasky EA. Protocols for simulating the thermal component of fever: preclinical and clinical experience. Methods. 2004;32:54–62. doi: 10.1016/s1046-2023(03)00187-7. [DOI] [PubMed] [Google Scholar]
- 123.Burd R, Dziedzic TS, Xu Y, Caligiuri MA, Subjeck JR, Repasky EA. Tumor cell apoptosis, lymphocyte recruitment and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J Cell Physiol. 1998;177:137–47. doi: 10.1002/(SICI)1097-4652(199810)177:1<137::AID-JCP15>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 124.Kraybill WG, Olenki T, Evans SS, Ostberg JR, O’Leary KA, Gibbs JF, Repasky EA. A phase I study of fever-range whole body hyperthermia (FR-WBH) in patients with advanced solid tumours: correlation with mouse models. Int J Hyperthermia. 2002;18:253–66. doi: 10.1080/02656730110116704. [DOI] [PubMed] [Google Scholar]
- 125.Chen Q, Fisher DT, Kucinska SA, Wang WC, Evans SS. Dynamic control of lymphocyte trafficking by fever-range thermal stress. Cancer Immunol Immunother. 2006;55:299–311. doi: 10.1007/s00262-005-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shah A, Unger E, Bain MD, Bruce R, Bodkin J, Ginnetti J, Wang WC, Seon B, Stewart CC, Evans SS. Cytokine and adhesion molecule expression in primary human endothelial cells stimulated with fever-range hyperthermia. Int J Hyperthermia. 2002;18:534–51. doi: 10.1080/02656730210157843. [DOI] [PubMed] [Google Scholar]
- 127.Evans SS, Frey M, Schleider DM, Bruce RA, Wang WC, Repasky EA, Appenheimer MM. Regulation of leukocyte-endothelial cell interactions in tumor immunity. In: Mihich EA, Croce C, editors. The Biology of Tumors. New York: Plenum Press; 1998. pp. 273–286. [Google Scholar]
- 128.Kansas GS, Ley K, Munro JM, Tedder TF. Regulation of leukocyte rolling and adhesion to high endothelial venules through the cytoplasmic domain of L-selectin. J Exp Med. 1993;177:833–8. doi: 10.1084/jem.177.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kahn J, Walcheck B, Migaki GI, Jutila MA, Kishimoto TK. Calmodulin regulates L-selectin adhesion molecule expression and function through a protease-dependent mechanism. Cell. 1998;92:809–18. doi: 10.1016/s0092-8674(00)81408-7. [DOI] [PubMed] [Google Scholar]
- 130.Kishimoto TK, Jutila MA, Butcher EC. Identification of a human peripheral lymph node homing receptor: a rapidly down-regulated adhesion molecule. Proc Natl Acad Sci U S A. 1990;87:2244–8. doi: 10.1073/pnas.87.6.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen Q, Clancy KA, Wang WC, Evans SS. Inflammatory cues controlling lymphocyte-endothelial interactions during fever-range thermal stress. In: Aird W, editor. Endothelial Biomedicine. Cambridge University Press; in press. [Google Scholar]
- 132.Gerwin N, Gonzalo JA, Lloyd C, Coyle AJ, Reiss Y, Banu N, Wang B, Xu H, Avraham H, Engelhardt B, Springer TA, Gutierrez-Ramos JC. Prolonged eosinophil accumulation in allergic lung interstitium of ICAM-2 deficient mice results in extended hyperresponsiveness. Immunity. 1999;10:9–19. doi: 10.1016/s1074-7613(00)80002-3. [DOI] [PubMed] [Google Scholar]
- 133.Carman CV, Springer TA. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol. 2003;15:547–56. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 134.Pachynski RK, Wu SW, Gunn MD, Erle DJ. Secondary lymphoid-tissue chemokine (SLC) stimulates integrin alpha 4 beta 7-mediated adhesion of lymphocytes to mucosal addressin cell adhesion molecule-1 (MAdCAM-1) under flow. J Immunol. 1998;161:952–6. [PubMed] [Google Scholar]
- 135.Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. 2002;4(Suppl 3):S109–16. doi: 10.1186/ar576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A, Grotzinger J, Rose-John S. I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol. 1997;15:142–5. doi: 10.1038/nbt0297-142. [DOI] [PubMed] [Google Scholar]
- 137.Wung BS, Hsu MC, Wu CC, Hsieh CW. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: effects on the inhibition of STAT3 phosphorylation. Life Sci. 2005;78:389–97. doi: 10.1016/j.lfs.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 138.Modur V, Li Y, Zimmerman GA, Prescott SM, McIntyre TM. Retrograde inflammatory signaling from neutrophils to endothelial cells by soluble interleukin-6 receptor alpha. J Clin Invest. 1997;100:2752–6. doi: 10.1172/JCI119821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, Bussolino F, Poli V, Ciliberto G, Mantovani A. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–25. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 140.Chen SC, Chang YL, Wang DL, Cheng JJ. Herbal remedy magnolol suppresses IL-6-induced STAT3 activation and gene expression in endothelial cells. Br J Pharmacol. 2006;148:226–32. doi: 10.1038/sj.bjp.0706647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. 1998;28:2178–87. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 142.Marin V, Montero-Julian FA, Gres S, Boulay V, Bongrand P, Farnarier C, Kaplanski G. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol. 2001;167:3435–42. doi: 10.4049/jimmunol.167.6.3435. [DOI] [PubMed] [Google Scholar]
- 143.Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, Rose-John S, Fuller GM, Topley N, Jones SA. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–14. doi: 10.1016/s1074-7613(01)00151-0. [DOI] [PubMed] [Google Scholar]
- 144.McLoughlin RM, Hurst SM, Nowell MA, Harris DA, Horiuchi S, Morgan LW, Wilkinson TS, Yamamoto N, Topley N, Jones SA. Differential regulation of neutrophil-activating chemokines by IL-6 and its soluble receptor isoforms. J Immunol. 2004;172:5676–83. doi: 10.4049/jimmunol.172.9.5676. [DOI] [PubMed] [Google Scholar]
- 145.Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853–63. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 146.Astaldi GC, Janssen MC, Lansdorp P, Willems C, Zeijlemaker WP, Oosterhof F. Human endothelial culture supernatant (HECS): a growth factor for hybridomas. J Immunol. 1980;125:1411–4. [PubMed] [Google Scholar]
- 147.Helfgott DC, May LT, Sthoeger Z, Tamm I, Sehgal PB. Bacterial lipopolysaccharide (endotoxin) enhances expression and secretion of beta 2 interferon by human fibroblasts. J Exp Med. 1987;166:1300–9. doi: 10.1084/jem.166.5.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Horii Y, Muraguchi A, Suematsu S, Matsuda T, Yoshizaki K, Hirano T, Kishimoto T. Regulation of BSF-2/IL-6 production by human mononuclear cells. Macrophage-dependent synthesis of BSF-2/IL-6 by T cells. J Immunol. 1988;141:1529–35. [PubMed] [Google Scholar]
- 149.Lally F, Smith E, Filer A, Stone MA, Shaw JS, Nash GB, Buckley CD, Rainger GE. A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum. 2005;52:3460–9. doi: 10.1002/art.21394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wung BS, Ni CW, Wang DL. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J Biomed Sci. 2005;12:91–101. doi: 10.1007/s11373-004-8170-z. [DOI] [PubMed] [Google Scholar]
- 151.Stamper HB, Jr, Woodruff JJ. Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med. 1976;144:828–33. doi: 10.1084/jem.144.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]