

Abstract
Genetic studies in the mouse have demonstrated that conditional cardiac-restricted loss of connexin43 (Cx43), the major ventricular gap junction protein, is highly arrhythmogenic. However, whether more focal gap junction remodeling, as is commonly seen in acquired cardiomyopathies, influences the propensity for arrhythmogenesis is not known. We examined electrophysiological properties and the frequency of spontaneous and inducible arrhythmias in genetically engineered chimeric mice derived from injection of Cx43-deficient embryonic stem cells into wild-type recipient blastocysts. Chimeric mice had numerous well-circumscribed microscopic Cx43-negative foci in their hearts, comprising ~15% of the total surface area as determined by immunohistochemical analysis. Systolic function in the chimeric mice was significantly depressed as measured echocardiographically (19.0% decline in fractional shortening compared with controls, P < 0.05) and by invasive hemodynamics (17.6% reduction in change of pressure over time, P < 0.01). Chimeras had significantly more spontaneous arrhythmic events than controls (P < 0.01), including frequent runs of nonsustained ventricular tachycardia in some of the chimeric mice. However, in contrast to mice with conditional cardiac-resticted loss of Cx43 in the heart, no sustained ventricular tachyarrhythmias were observed. We conclude that focal areas of uncoupling in the myocardium increase the likelihood of arrhythmic triggers, but more widespread uncoupling is required to support sustained arrhythmias.
Keywords: connexin43, ventricular dysfunction, chimera
SUDDEN ARRHYTHMIC DEATH is a common cause of mortality in many forms of heart disease (4, 23, 36, 40). Abnormal inter-cellular coupling in the heart, resulting from defects in expression of connexin43 (Cx43), the major constituent of cardiac gap junctions, plays a key role in the initiation and maintenance of sustained arrhythmias (7, 15, 25, 28, 38). Abnormal Cx43 expression is seen in many common forms of acquired heart disease, although gap junction remodeling in these conditions can be focal in nature and preferentially affect ischemic or injured myocardial segments (9, 19, 22, 35). Nonetheless, these relatively restricted regions of gap junction remodeling may play a key mechanistic role in arrhythmogenesis, as demonstrated in the canine infarct model, where alterations in Cx43 expression in the infarct border zone correlate with the location of reentrant circuits (32).
We have previously established that cardiac-restricted conditional knockout (CKO) of Cx43, in which Cx43 expression is inactivated in the great majority of myocytes, results in a highly arrhythmogenic substrate (7, 13, 15). To investigate the role of focal uncoupling in cardiac function, we have also developed a murine model of heterogeneous gap junction expression by generating chimeric animals from the introduction of Cx43-deficient embryonic stem (ES) cells into wild-type recipient blastocysts (16). Gap junction expression in these hearts is highly abnormal, with well-circumscribed foci of Cx43-deficient myocytes interspersed throughout an otherwise well-coupled myocardial syncitium. Interestingly, these mice had depressed systolic function in the absence of ventricular dilatation or hypertrophy, conceivably a consequence of asynchronous myocardial excitation and contraction (16).
Interestingly, during the course of evaluation of impulse propagation in the chimeras, two of seven isolated-perfused hearts developed spontaneous ventricular tachycardia, as opposed to zero of six control hearts, suggesting increased arrhythmic susceptibility. Accordingly, in the present study, we have performed a detailed analysis of the in vivo electrophys-iological phenotype in Cx43 chimeric mice. We documented a significant increase in spontaneous ventricular ectopy, including some mice with frequent runs of nonsustained ventricular tachycardia, a finding never seen in controls. However, in marked contrast to CKO mice, which have widespread loss of Cx43 in the myocardium (15), these chimeric mice did not develop sustained ventricular tachyarrhythmias, nor were such arrhythmias inducible by programmed electrical stimulation (PES). These data highlight the importance of uncoupled foci as arrhythmic triggers and suggest that additional pathology such as more widespread uncoupling or myocardial injury is required for the maintenance of sustained arrhythmias.
MATERIALS AND METHODS
Generation of chimeric mice and genotyping
Studies involving the Cx43 chimeric mice were approved by the Institutional Animal Care and Use Committees of the New York University School of Medicine, the New York Harbor Veterans Administration Medical Center, and the Mount Sinai School of Medicine (New York, NY) and performed in compliance with National Institutes of Health guidelines. The generation of homozygous Cx43-null ES cells and their injection into wild-type blastocysts was performed as described previously (16).
To compare the incidence of spontaneous ectopy in unanesthetized chimeric and control mice, a pilot series of four chimeric mice (see chimeras 1–4 in Table 3) were generated by injection of 129/Sv ES cells into C57BL/6 host blastocysts, followed by reimplantation into pseudopregnant foster mothers. Controls for this pilot series consisted of 129/Sv × C57BL/6 F1 age- and sex-matched mice (see controls 1 and 2 in Table 3). Data from the pilot series were used only for the comparison of spontaneous ectopic events in telemetry-monitored unanesthetized mice. In addition to the pilot series, additional chimeric mice were generated in which 129/Sv ES cells were injected into 129/Sv host blastocysts. These chimeras were used to compare the incidence of spontaneous ventricular ectopy, as well as for all other comparisons. Purebred age- and sex-matched 129/Sv mice were used as controls for these subsequent series of chimeras.
Table 3.
Spontaneous ectopy in unanesthetized telemetry monitoring of Cx43-/- chimeric and wild-type control mice
| PVCs, 24 h | Couplets, 24 h | Runs of VT, 24 h | |
|---|---|---|---|
| Controls | |||
| 1 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 |
| 3 | 2 | 0 | 0 |
| 4 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 |
| 6 | 8 | 10 | 0 |
| 7 | 51 | 0 | 0 |
| 8 | 21 | 2 | 0 |
| Chimeras | |||
| 1 | 32 | 16 | 0 |
| 2 | 96 | 0 | 16 |
| 3 | 704 | 240 | 912 |
| 4 | 32 | 0 | 0 |
| 5 | 59 | 0 | 0 |
| 6 | 783 | 175 | 49 |
| 7 | 50 | 2 | 0 |
| 8 | 196 | 11 | 821 |
| 9 | 8 | 1 | 0 |
| 10 | 7 | 0 | 0 |
Values are means ± SE. PVC, premature ventricular complex; VT, ventricular tachycardia.
Genomic DNA from chimeric offspring was prepared as described previously (24). Southern blotting was performed by using a 700-bp probe, adjacent to the 3′ end of the Cx43 open reading frame, yielding the expected 6.5-kb wild-type band and the 4.3- and 3.4-kb knockout bands (16).
Histology and immunofluorescence
For the determination of Cx43 expression patterns in chimeric hearts, immunostaining was performed in frozen sections that were blocked and then incubated with a polyclonal anti-Cx43 primary antibody (14). After being washed in PBS, sections were incubated with a FITC-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) and mounted with Vectashield mounting medium (Vector, Berlingame, CA).
Stained sections were visualized on an Axioskop 2 Plus fluorescence microscope, and images were collected by using an Axiocam camera with AxioVision 2.05 software (Carl Zeiss, Munchen-Hall-bergmoos, Germany). IPLab 3.5.5 software (Scanalytic, Fairfax, VA) was used to compare the mean fluorescence of Cx43-positive regions after subtracting the background fluorescent intensity from each sample (14).
To determine the typical sizes of Cx43-deficient regions, 20 shortaxis ventricular sections, separated from each other by 40 μm (for a total survey area of 920 μm in each of 2 Cx43 chimeric hearts), were immunostained for Cx43. The maximum distance across discrete areas devoid of Cx43 staining and the minimal distances between these regions were measured by using AxioVision software.
Immunoblotting and densitometry
Evaluation of Cx43 protein levels was performed in total ventricular lysates from six controls and six chimeras that were prepared by Dounce homogenization in the presence of Complete protease inhibitor cocktail (Roche, Mannheim, Germany). Immunoblotting was performed with equal concentrations of each sample as determined by Bradford assay, and equal loading was confirmed with Coomassie staining. Immunoblots were blocked followed by incubation with the anti-Cx43 polyclonal antibody described above. Horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was then applied, followed by enhanced chemiluminescent processing (Amersham Pharmacia Biotech, Buckinghamshire, UK) and autoradiography. Two separate experiments were quantified by scanning the autoradiograms on a Gel Doc 1000 and calculating band intensity by using Quantity One software (Bio-Rad, Hercules, CA).
Telemetry
Ambulatory electrocardiographic monitoring with implanted telemetry transmitters (Data Sciences International, St. Paul, MN) was performed starting at 8 mo of age. Mice were anesthetized with isoflurane (1.5 vol%), and telemetry transmitters were placed into the peritoneal cavity through a small midline abdominal incision and sutured to the abdominal fascia. The leads were tunneled subcutaneously into each axilla and sutured into place. The animals were allowed at least 48 h to recover from the surgery before telemetry recordings were made. Telemetry signals from PhysioTel Receiver platforms via a Data Exchange Matrix were recorded with Dataquest A.R.T. 2.1 software (Data Sciences International). In the pilot experiment, recordings were made hourly for 5 min at a time for a total of 90 min. Subsequent series of chimeric and control mice were recorded hourly for 5 min at a time 24 h a day over the course of 6 days for approximately 12 h of total telemetry recording time per mouse. After the recording period, telemeters were removed.
Telemetry recordings were analyzed off-line by using a software program custom-made to identify ventricular ectopy. Tracings were reviewed manually to ensure the sensitivity and specificity of the software program for the identification of ventricular ectopy. Ventricular ectopic beats were identified by using well-established criteria (3). Nonsustained ventricular tachycardia was defined a priori as a ventricular arrhythmia consisting of at least three beats and lasting up to 30 s (37).
Electrocardiography and electrophysiological studies
At 10 mo, mice were anesthetized (isoflurane, 1.5 vol%) for a baseline electrocardiogram and electrophysiological studies, as described previously (13). Briefly, a 1-cm incision was made in the epigastric midline through which the diaphragmatic surface of the heart was visualized. A 200-μm platinum monopolar-stimulating electrode (Frederick Haer, Bowdoinham, ME) mounted on a micromanipulator (Fine Science Tools, North Vancouver, BC, Canada) was inserted through the diaphragm directly into the epicardial surface of the heart. PES was performed with a model 2352 Programmable Stimulator (Medtronic, Minneapolis, MN). Signals were amplified by using a Honeywell ECG amplifier (Honeywell, Morristown, NJ), converted from analog to digital with the ACQ-16 Acquisition Interface and recorded with Ponemah Physiology Platform software (Gould, Valley View, OH). Output was set at twice the stimulating threshold. To determine the ventricular effective refractory period (VERP), a train of eight stimuli at cycle lengths of 120, 100, and 80 ms was delivered with single extrastimuli introduced at progressively shorter intervals. The pacing protocol was repeated in each animal at a second pacing site, and the VERP results were averaged. Double extrastimuli at each cycle length and each pacing site were added to assess inducibility of ventricular arrhythmias. After the study, the electrode was removed and the animals were sutured closed and allowed to recover.
Echocardiography
Echocardiography was performed in the same cohort of Cx43 chimeric mice at 10 mo of age, according to a previously described protocol (15, 16). Briefly, mice were anesthetized with tribromoethanol (Avertin, 20 mg/kg ip, 1.2 vol% solution) and imaged by using an Acuson Sequoia echocardiography machine and a 15-MHz linear probe. Measurements were performed online by using customized software generously provided by Acuson.
Hemodynamics
Invasive hemodynamic assessment was performed immediately before death at 11 mo. The mice were anesthetized with Avertin (20 mg/kg ip, 1.2 vol% solution), intubated endotracheally, and ventilated by using a model 687 Mouse Ventilator (Harvard Apparatus, South Natick, MA). A 1.4-Fr Mikro-Tip catheter pressure transducer (Millar, Houston, TX) was placed into the right carotid and advanced retrograde into the left ventricle. Data were recorded by using Ponemah Physiology Platform as described above. After the procedure, mice were given a lethal dose of 500 mg/kg Avertin ip, and their hearts were removed.
Statistics
Data are expressed as means ± SE. Assessment of spontaneous ectopy in controls and chimeras was performed by using the method of Brunner et al. (3), by comparing with a χ2 analysis the number of mice in each group with greater than 5 episodes of spontaneous ventricular ectopy per 24 h of monitoring (Georgetown Linguistics Web Chi Square Calculator). Because of the nonnormal distribution of values for ventricular premature beats and total arrhythmic events, these values were log transformed and then compared with a two-tailed t-test (Microsoft Excel). Percentage of chimerism between epi-, mid- and endocardium was compared with ANOVA by using StatView (SAS Institute, Cary, NC). All other comparisons between groups were performed with a two-tailed t-test (Microsoft Excel). P < 0.05 was considered statistically significant.
RESULTS
Chimerism is detectable on Southern blot analysis
To generate Cx43 chimeric mice, homozygous Cx43-null ES cells derived from 129/Sv mice were injected into wild-type 129/Sv recipient blastocysts and implanted into pseudopregnant females in two separate surgeries. Of the resulting 14 pups, 7 pups had evidence of chimerism determined by Southern blot analysis (Fig. 1A). The chimeric pups were grossly indistin-guishable from the nonchimeric littermates. There were no spontaneous deaths in this cohort of chimeric mice over the 10-mo observation period.
Fig. 1.

Generation and characterization of chimeric connexin43 (Cx43) knockout (KO) mice. Southern blotting (A) of tail DNA samples was performed for the identification of chimeric mice (*). Tail DNA was digested with NcoI, size-fractionated on agarose gels and transferred to a nitrocellulose membrane, which was then probed for the presence of 6.5-kb wild-type allele and 4.3- and 3.4-kb Cx43-null alleles. Immunofluorescent staining for Cx43 in control (B) and chimeric Cx43 KO (C) heart sections shows abundant Cx43 signal at the intercalated discs throughout wild-type hearts. Chimeric hearts, on the other hand, have randomly distributed areas without Cx43 staining juxtaposed to Cx43-expressing regions [demarcation (arrows in C) tends to be rather abrupt in most instances]. Net mean fluorescence (mean fluorescence minus background fluorescence) of Cx43-expressing areas was significantly higher in chimeric hearts than in controls (D). Despite increased expression in Cx43-expressing areas of chimeric hearts, overall Cx43 abundance (representative immunoblots, E) is not significantly elevated in chimeric ventricular lysates compared with controls (F). †P < 0.05.
Cx43 chimerism causes ventricular dysfunction without hypertrophy or dilatation
We utilized echocardiography to non-invasively assess left ventricular function, chamber size, and wall thicknesses. In our previous study of chimeric Cx43 knockout mice (16), we injected ES cells from a C57BL/6 background into 129/Sv host blastocysts. Because the two strains are different sizes in adulthood, we were unable to directly compare chamber sizes. A direct comparison of ventricular size was possible in this series of chimeras because both ES cells and recipient blastocysts were derived from the 129/Sv strain, thus removing strain differences as a complicating factor.
Global ventricular function, as measured by fractional shortening, was significantly reduced in the chimeric mice compared with age- and strain-matched controls (Table 1). Fractional shortening in the chimeras was 33.2 ± 2.08% compared with 41.0 ± 1.27% in the controls (P < 0.05). This difference in fractional shortening represented a 19% decline in global ventricular function in the chimeras compared with the controls. Similarly, fractional shortening was significantly reduced in our previous series of Cx43 chimeras (16). There were no significant differences in end-diastolic left ventricular dimensions or wall thicknesses, indicating that despite a significant decline in global function, there was no echocardiographic evidence of ventricular dilatation or hypertrophy in the chimeras.
Table 1.
Echocardiographic measurements in Cx43-/- chimeric mice
| Controls | Chimeras | |
|---|---|---|
| n | 5 | 5 |
| IDD, mm | 3.51±0.09 | 3.83±0.14 |
| IDS, mm | 2.08±0.08 | 2.55±0.10† |
| FS, % | 41.0±1.27 | 33.2±2.08* |
| AWTs, mm | 0.99±0.02 | 0.85±0.02† |
| AWTd, mm | 0.74±0.04 | 0.74±0.04 |
| PWTs, mm | 1.11±0.03 | 0.97±0.03* |
| PWTd, mm | 0.93±0.06 | 0.80±0.03 |
Values are means ± SE; n, number of mice; IDD, intraventricular dimension in diastole; IDS, intraventricular dimension in systole; FS, fractional shortening; AWTs, anterior wall thickness in systole; AWTd, anterior wall thickness in diastole; PWTs, posterior wall thickness in systole; PWTd, posterior wall thickness in diastole.
P < 0.05
P < 0.01.
Isolated systolic without diastolic dysfunction in the chimeric mice
Immediately before death, chimeric and control mice were studied with invasive hemodynamic monitoring (Table 2). Consistent with the echocardiographic analysis, overall contractility in the chimeric mice was significantly reduced. Contractility, as measured by maximum change of pressure over time (dP/dtmax), was 7,147 ± 248 mmHg/s in the chimeras compared with 8,673 ± 283 mmHg/s in the controls (P < 0.01), representing a 17.6% decline in the chimeras. Interestingly, indexes of diastolic function, including minimum change of pressure over time (dP/dtmin) and Tau, were not significantly changed in the chimeric mice compared with controls. Although both systemic and left ventricular systolic pressures were decreased in the chimeras, there was no difference in diastolic pressures or heart rate.
Table 2.
Invasive hemodynamic measurements in Cx43-/- chimeric mice
| Controls | Chimeras | |
|---|---|---|
| n | 5 | 5 |
| Aortic pressure, systole, mmHg | 111±6 | 92±4* |
| Aortic pressure, diastole, mmHg | 69±10 | 56±7 |
| LV systolic pressure, mmHg | 125±6 | 97±5† |
| LVEDP, mmHg | 4.3±1.3 | 2.4±1.1 |
| Heart rate, beats/min | 403±3 | 390±7 |
| dP/dtmax, mmHg/s | 8,673±283 | 7,147±248† |
| dP/dtmin, mmHg/s | −6,996±227 | −6,618±1,145 |
| Tau, ms | 7.6±1.4 | 7.2±1.5 |
Values are means ± SE; n, number of mice; LV, left ventricular; LVEDP, LV end-diastolic pressure; dP/dtmax and dP/dtmin, maximum and minimum change in pressure over time, respectively.
P < 0.05
P < 0.01.
Augmented Cx43 expression in nonchimeric areas of chimeric hearts
To assess ventricular expression of Cx43 in the chimeric hearts on an ultrastructural level, immunofluorescent staining was performed (Fig. 1, B and C). As expected, staining for Cx43 in the chimeric hearts showed focal loss of gap junctions throughout the heart, with randomly distributed areas lacking Cx43 staining. Areas devoid of Cx43 (Cx43 negative) were well circumscribed and clearly demarcated from the adjacent Cx43 expressing (Cx43 positive) myocardium (arrows, Fig. 1C). Cx43-negative regions constituted 16.2 ± 5.7% of epicardial sections surveyed in the chimeric hearts, 15.0 ± 3.2% of midmyocardial sections and 12.4 ± 2.2% of endomyocardial sections [6 randomly chosen images from each myocardial level were assayed for each of 6 chimeric hearts; P = not significant (NS) for comparisons between myocardial layers].
Two of the chimeric hearts were surveyed to determine the typical sizes of Cx43-deficient regions. In one of the chimeric hearts, there was an average of 5.5 ± 0.32 Cx43-deficient regions per section (range 4–7) or one Cx43-deficient region per 2.3 mm2 of chimeric myocardium. The maximum dimension of these regions averaged 227.0 ± 11.2 μm (range 73.5–446.4 μm), and they were 330.9 ± 39.7 μm (range 15.6–971.3 μm) from the nearest Cx43-deficient region. In the second chimeric heart surveyed, there was an average of 4.5 ± 0.25 Cx43-deficient regions per section (range 3–6; one Cx43-deficient region per 5.6 mm2 of chimeric myocardium); the maximum dimension averaged 494.8 ± 62.4 μm (range 139.8–1422.2 μm), and they were separated from the nearest Cx43-null region by 161.0 ± 25.2 μm (range 10.0–601.3 μm). Thus Cx43-deficient zones constitute relatively large and well-demarcated areas of the chimeric ventricle, likely accounting for discrete areas of conduction delay and disruption of the activation wave front.
Interestingly, compared with wild-type hearts (n = 6), the abundance of Cx43 in the Cx43-positive regions of the chimeric hearts (n = 6) was significantly greater (126 ± 5.9% of control, P < 0.05; Fig. 1D). Consistent with these immuno-fluorescent findings suggesting a compensatory upregulation in Cx43-positive regions, we found that despite substantial areas devoid of Cx43 in the chimeric hearts, there was no significant difference in overall Cx43 protein content by immunoblotting compared with wild-type hearts (chimeric ventricular Cx43 protein content was 114 ± 11% of control, P = NS; Fig. 1, E and F).
Spontaneous ventricular ectopy in chimeric mice
To analyze the occurrence of spontaneous ventricular arrhythmias in the chimeric mice, we surgically implanted telemeters and recorded electrocardiographic activity. Recordings from unsedated chimeric mice (n = 10) were obtained and compared with age- and sex-matched controls (n = 8). Ventricular ectopy was detected by using custom-made software and was confirmed by manual review. With the use of the criteria of Brunner et al. (3) (>5 ventricular arrhythmic events per 24 h), all 10 of the chimeric mice and only 3 of the 8 controls had evidence of frequent ventricular ectopy (P < 0.01; Table 3). Chimeric mice had significantly more arrhythmic events than controls irrespective of the time of day (P < 0.01; Fig. 2). The incidence of premature ventricular complexes (PVCs) was significantly increased in the chimeras (230.1 ± 95.6 per mouse per 24 h) compared with controls (10.2 ± 6.0; P < 0.05). Interestingly, because PVC morphology was limited to one or two types per control mouse, there were significantly more PVC morphologies in each of the chimeric mice (0.8 ± 0.4 PVC morphologies per control, 6.0 ± 2.0 per chimera, P < 0.05; Fig. 3). This suggests that whereas control mice may have isolated foci generating premature ectopic complexes, chimeric mice have many more such foci. No relationship was detected between the frequency of ventricular ectopy and reduced ventricular function by echocardiography or invasive hemodynamics, suggesting that the cardiomyopathy noted in the chimeras is not tachycardia induced.
Fig. 2.
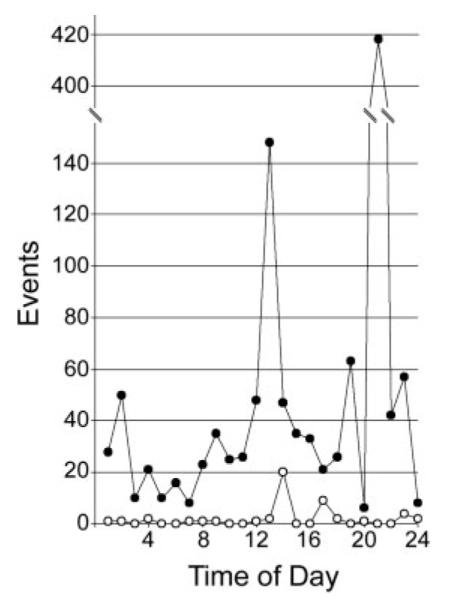
Incidence of ventricular arrhythmic events during 24-h monitoring. Total arrhythmic events in chimeric Cx43 KO (●) and control mice (◯) are arranged by time of day from 1 (1 AM) to 24 (12 AM). Chimeric Cx43 KO mice have significantly more arrhythmic events than controls (P < 0.01).
Fig. 3.

Chimeric Cx43 KO mice display many different premature ventricular complex (PVC) morphologies. In contrast to the controls, each chimeric mouse showed many different PVC morphologies. Representative examples of PVCs from different days in the same control and chimeric mice are shown, demonstrating a consistent PVC morphology in the control over 6 days of monitoring and several different PVC morphologies in the chimera over the same time span.
Complex ventricular ectopy was limited to the chimeric mice. There were frequent runs of nonsustained ventricular tachycardia in 4 of 10 chimeric mice (Fig. 4). Runs of ventricular tachycardia in the chimeras ranged from 3 to 141 beats (maximum of 5.6 s) at cycle lengths of up to 33 ms. In comparison, there was no ventricular tachycardia in any of the controls (P < 0.05 compared with chimeric mice).
Fig. 4.

Telemetry recording from a chimeric Cx43 KO mouse. Examples of frequent nonsustained runs of ventricular tachycardia are seen on a telemetry tracing from an ambulatory, unanesthetized chimeric mouse. Cycle lengths of each of the examples of ventricular tachycardia are ~40 ms.
Electrophysiological indexes are unchanged in chimeric mice
We hypothesized that focal areas of conduction delay within the myocardium caused by heterogeneous loss of gap junctions would provide a substrate for reentrant ventricular arrhythmias. To test this hypothesis, we subjected the chimeric mice and matched controls to electrophysiologic al study with PES. We have previously described an experimental protocol for the determination of electrophysiological indexes in the mouse by using the subdiaphragmatic approach (13). With this approach, we are able to visualize the heart, apply a stimulating electrode directly to the epicardial surface of the heart without intubating the mouse, and allow the mouse to recover after the procedure. Because this procedure yields reproducible results for electrophysiological indexes and reliably induces sustained ventricular arrhythmias in Cx43 CKO mice, we elected to use the same protocol in this study.
No disruption of the diaphragm, respiratory distress, or procedural mortality was noted. Baseline electrocardiographic indexes (Table 4) were not significantly different between chimeric mice and controls. Despite focal areas of conduction delay on optical mapping of isolated chimeric hearts (16), the QRS duration in chimeric mice was not significantly prolonged. There were no significant differences in VERP between the chimeric and control mice at any of the cycle lengths tested (Table 4). Neither nonsustained nor sustained ventricular tachycardia was induced in any of the chimeric mice or age-matched controls.
Table 4.
Electrocardiographic and electrophysiological data from Cx43-/- chimeric mice
| Controls | Chimeras | |
|---|---|---|
| n | 5 | 5 |
| PR interval, ms | 40.8±1.02 | 37.2±1.42 |
| QRS interval, ms | 10.8±0.20 | 11.6±0.40 |
| RR interval, ms | 144.4±5.17 | 147.0±7.30 |
| QRSA, mv | 0.125±0.017 | 0.095±0.009 |
| VERP, 120, ms | 45.8±4.6 | 45.9±2.0 |
| VERP, 100, ms | 45.9±4.9 | 46.0±2.5 |
| VERP, 80, ms | 46.5±5.3 | 46.4±3.7 |
Values are means ± SE; n, number of mice; QRSA, QRS amplitude; VERP, ventricular effective refractory period.
DISCUSSION
Abnormal gap junction expression in the heart has been described in both animal models and clinical pathological studies of heart disease. For example, gap junction remodeling has been observed in both ischemic heart disease and end-stage dilated cardiomyopathy (9, 19, 22, 35). However, in these clinical entities, abnormal expression of Cx43 may be restricted to areas immediately adjacent to the injured myocardium, whereas unaffected areas of the heart maintain relatively normal Cx43 expression (19). Despite the often limited extent of gap junction remodeling, isolated areas of abnormal Cx43 expression have been shown to correlate with the location of reentrant circuits in the canine epicardial infarct border zone (32). In this study, we investigated whether isolated areas devoid of Cx43, as opposed to generalized uncoupling of cardiac myocytes, may increase the propensity for the development of ventricular arrhythmias.
The major finding in this study was our observation that the frequency of ventricular ectopy in the Cx43 chimeric mice was markedly increased compared with the controls. Both the absolute quantity of ectopy as well as the number of unique morphologies was greater, suggesting that the number of ectopic foci was increased in the chimeric hearts. Importantly, despite the increased ectopy, we did not observe a corresponding increase in the frequency of spontaneous or inducible sustained ventricular arrhythmias.
The behavior of the Cx43 chimeras contrasts with that observed in mice with widespread loss of Cx43 in the heart (i.e, CKO mice), which develop sudden arrhythmic death by 2 mo of age and are easily inducible into sustained ventricular arrhythmias (13, 15). Taken together, these studies indicate that the spatial pattern of uncoupling must play a key role in determining the phenotype. In the CKO mice, myocytes devoid of Cx43 expression are dispersed randomly in a microscopic heterogeneous pattern throughout the heart without discrete “knocked-out” areas. As a result, the conduction wave front measured with optical mapping remains smooth, but impulse propagation becomes progressively slower as a function of the extent of the knockout (7, 15). When the abundance of Cx43 falls by at least 60% compared with controls, the likelihood of inducing sustained ventricular arrrhythmias is substantial (7).
In contrast, chimeric Cx43-/- hearts have discrete regions lacking Cx43 expression and corresponding focal areas of conduction delay, rather than global slowing of conduction velocity (16). This focal abnormality is associated with an increased level of ectopy, but the substrate does not appear to support sustained arrhythmias. What accounts for this increase in ectopy? It is well known that diminished coupling isolates pacemaker cells from surrounding tissue to provide the appropriate source-sink relationship for successful impulse propagation from node to working myocardium (2). Primary pacemaker regions of both the sinoatrial and atrioventricular nodes have a reduced or absent expression of Cx43 (6, 10, 18) and correspondingly elevated levels of intercellular resistivity (1). It is conceivable that the discrete areas of uncoupled myocardium, such as those in the chimeric Cx43-/- heart, may mimic this behavior and unmask ectopic foci. This may explain why both the absolute quantity of ectopy and the number of unique morphologies were greater.
Additionally, focal areas of conduction delay caused by chimeric Cx43-deficient clusters of myocytes may disrupt the activation wave front, leading to the formation of wave breaks. These, in turn, may generate reentrant circuits. However, given the lack of sustained arrhythmias, these reentrant circuits must be unstable and short-lived, perhaps in part because global conduction velocity is not significantly slowed (5, 30, 39). Moreover, intervening Cx43-positive regions in the chimeric hearts appear to have supranormal levels of Cx43 expression, which may increase conduction velocity in these areas (8). Theoretically, the increased conduction velocity may lengthen wavelength (the product of conduction velocity and effective refractory period), making sustained reentry less likely.
This study may have important implications for the emerging field of cell transplantation for end-stage heart failure. Transplantation of skeletal myoblasts into the diseased myocardium has been recognized as potentially arrhythmogenic in humans (27), despite positive effects on ventricular function in experimental models of myocardial infarction (11, 17, 27). As grafted myoblasts differentiate into myotubes, they lose expression of both gap junctions and adherens junctions but maintain the ability to contract when electrically stimulated (29, 33). Thus the increased arrhythmogenicity seen in myoblast-derived cardiomyocyte transplantation may result from a myopathic ventricle that is suddenly challenged with an acute increase in the frequency of arrhythmic triggers. Moreover, our observations raise a cautionary note for genetically engineered transplantation strategies in which the proliferative potential of poorly coupled cells is enhanced. It is conceivable that a critical threshold may be reached where foci of such cells reach sufficient size that areas of conduction delay or arrhythmic triggers as well as the substrate for reentrant behavior are more likely.
We previously demonstrated that focal areas of conduction delay are associated with systolic dysfunction, presumably a reflection of diminished synchrony of contraction. This result was confirmed in this study by using strain-matched ES cells and recipient embryos. Interestingly, human clinical trials demonstrate that restoration of synchrony with biventricular pacing can ameliorate contractile deficits (12, 20, 21, 26). These observations raise the possibility that reversal of gap junction remodeling may not only diminish arrhythmic risk but may also lead to improvements in systolic performance. Although the QRS duration was not prolonged in the chimeric mice, recent studies (31, 34) indicate that resynchronization therapy may benefit patients with asynchronous contraction independent of QRS duration.
In summary, these data in concert with our earlier studies suggest that abnormal gap junction expression in the heart serves as a powerful factor for both the initiation and maintenance of arrhythmias (7, 15, 28). Areas of focal uncoupling, as demonstrated in the chimeric mouse, are associated with a significant increase in spontaneous ectopic events, possibly by unmasking intrinsic automaticity or by disrupting the conduction wave front leading to wave breaks. However, our results indicate that more extensive structural and/or functional remodeling may be required in the mouse heart to sustain ventricular arrhythmias.
ACKNOWLEDGMENTS
Blastocyst injection for the production of chimeric Cx43 knockout mice was performed in the Mouse Genetics Shared Resource Facility of the Mount Sinai School of Medicine, New York, NY. F. L. Chen is currently affiliated with the Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA.
GRANTS This work was supported by American Heart Association Grant-in-Aid; National Heart, Lung, and Blood Institute Grants HL-04222, HL-081336 (to D. E. Gutstein), HL-30557, and HL-64757; and a Burroughs Wellcome Fund Clinical-Scientist Award in Translational Research (to G. I. Fishman).
REFERENCES
- 1.Anumonwo JM, Wang HZ, Trabka-Janik E, Dunham B, Veenstra RD, Delmar M, Jalife J. Gap junctional channels in adult mammalian sinus nodal cells. Immunolocalization and electrophysiology. Circ Res. 1992;71:229–239. doi: 10.1161/01.res.71.2.229. [DOI] [PubMed] [Google Scholar]
- 2.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. 2000;47:658–687. doi: 10.1016/s0008-6363(00)00135-8. [DOI] [PubMed] [Google Scholar]
- 3.Brunner M, Guo W, Mitchell GF, Buckett PD, Nerbonne JM, Koren G. Characterization of mice with a combined suppression of Ito and IK,slow. Am J Physiol Heart Circ Physiol. 2001;281:H1201–H1209. doi: 10.1152/ajpheart.2001.281.3.H1201. [DOI] [PubMed] [Google Scholar]
- 4.Camm AJ, Katritsis DG. Risk stratification of patients with ventricular arrhythmias. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. 3rd ed Saunders; Philadelphia: 2000. pp. 808–827. [Google Scholar]
- 5.Chow AW, Schilling RJ, Davies DW, Peters NS. Characteristics of wavefront propagation in reentrant circuits causing human ventricular tachycardia. Circulation. 2002;105:2172–2178. doi: 10.1161/01.cir.0000015702.49326.bc. [DOI] [PubMed] [Google Scholar]
- 6.Coppen SR, Kodama I, Boyett MR, Dobrzynski H, Takagishi Y, Honjo H, Yeh HI, Severs NJ. Connexin45, a major connexin of the rabbit sinoatrial node, is co-expressed with connexin43 in a restricted zone at the nodal-crista terminalis border. J Histochem Cytochem. 1999;47:907–918. doi: 10.1177/002215549904700708. [DOI] [PubMed] [Google Scholar]
- 7.Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, Gutstein DE. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darrow BJ, Fast VG, Kleber AG, Beyer EC, Saffitz JE. Functional and structural assessment of intercellular communication. Increased conduction velocity and enhanced connexin expression in dibutyryl cAMP-treated cultured cardiac myocytes. Circ Res. 1996;79:174–183. doi: 10.1161/01.res.79.2.174. [DOI] [PubMed] [Google Scholar]
- 9.Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 10.Efimov IR, Nikolski VP, Rothenberg F, Greener ID, Li J, Dobrzynski H, Boyett M. Structure-function relationship in the AV junction. Anat Rec. 2004;280:952–965. doi: 10.1002/ar.a.20108. [DOI] [PubMed] [Google Scholar]
- 11.Ghostine S, Carrion C, Souza LC, Richard P, Bruneval P, Vilquin JT, Pouzet B, Schwartz K, Menasche P, Hagege AA. Long-term efficacy of myoblast transplantation on regional structure and function after myocardial infarction. Circulation. 2002;106(Suppl 1):I131–I136. [PubMed] [Google Scholar]
- 12.Gras D, Leclercq C, Tang AS, Bucknall C, Luttikhuis HO, Kirstein-Pedersen A. Cardiac resynchronization therapy in advanced heart failure the multicenter InSync clinical study. Eur J Heart Fail. 2002;4:311–320. doi: 10.1016/s1388-9842(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 13.Gutstein DE, Danik SB, Sereysky JB, Morley GE, Fishman GI. Subdiaphragmatic murine electrophysiological studies: sequential determination of ventricular refractoriness and arrhythmia induction. Am J Physiol Heart Circ Physiol. 2003;285:H1091–H1096. doi: 10.1152/ajpheart.00100.2003. [DOI] [PubMed] [Google Scholar]
- 14.Gutstein DE, Liu FY, Meyers MB, Choo A, Fishman GI. The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci. 2003;116:875–885. doi: 10.1242/jcs.00258. [DOI] [PubMed] [Google Scholar]
- 15.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutstein DE, Morley GE, Vaidya D, Liu F, Chen FL, Stuhlmann H, Fishman GI. Heterogeneous expression of Gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation. 2001;104:1194–1199. doi: 10.1161/hc3601.093990. [DOI] [PubMed] [Google Scholar]
- 17.Jain M, DerSimonian H, Brenner DA, Ngoy S, Teller P, Edge AS, Zawadzka A, Wetzel K, Sawyer DB, Colucci WS, Apstein CS, Liao R. Cell therapy attenuates deleterious ventricular remodeling and improves cardiac performance after myocardial infarction. Circulation. 2001;103:1920–1927. doi: 10.1161/01.cir.103.14.1920. [DOI] [PubMed] [Google Scholar]
- 18.Jones SA, Lancaster MK, Boyett MR. Ageing-related changes of connexins and conduction within the sinoatrial node. J Physiol. 2004;560:429–437. doi: 10.1113/jphysiol.2004.072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaprielian RR, Gunning M, Dupont E, Sheppard MN, Rothery SM, Underwood R, Pennell DJ, Fox K, Pepper J, Poole-Wilson PA, Severs NJ. Downregulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation. 1998;97:651–660. doi: 10.1161/01.cir.97.7.651. [DOI] [PubMed] [Google Scholar]
- 20.Kass DA, Chen CH, Curry C, Talbot M, Berger R, Fetics B, Nevo E. Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay. Circulation. 1999;99:1567–1573. doi: 10.1161/01.cir.99.12.1567. [DOI] [PubMed] [Google Scholar]
- 21.Kerwin WF, Botvinick EH, O’Connell JW, Merrick SH, DeMarco T, Chatterjee K, Scheibly K, Saxon LA. Ventricular contraction abnormalities in dilated cardiomyopathy: effect of biventricular pacing to correct interventricular dyssynchrony. J Am Coll Cardiol. 2000;35:1221–1227. doi: 10.1016/s0735-1097(00)00555-6. [DOI] [PubMed] [Google Scholar]
- 22.Kitamura H, Ohnishi Y, Yoshida A, Okajima K, Azumi H, Ishida A, Galeano EJ, Kubo S, Hayashi Y, Itoh H, Yokoyama M. Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia. J Cardiovasc Electrophysiol. 2002;13:865–870. doi: 10.1046/j.1540-8167.2002.00865.x. [DOI] [PubMed] [Google Scholar]
- 23.Klein H, Auricchio A, Reek S, Geller C. New primary prevention trials of sudden cardiac death in patients with left ventricular dysfunction: SCD-HEFT and MADIT-II. Am J Cardiol. 1999;83:91D–97D. doi: 10.1016/s0002-9149(99)00040-5. [DOI] [PubMed] [Google Scholar]
- 24.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–552. doi: 10.1161/01.cir.101.5.547. [DOI] [PubMed] [Google Scholar]
- 26.Linde C, Leclercq C, Rex S, Garrigue S, Lavergne T, Cazeau S, McKenna W, Fitzgerald M, Deharo JC, Alonso C, Walker S, Braun-schweig F, Bailleul C, Daubert JC. Long-term benefits of biventricular pacing in congestive heart failure: results from the MUltisite STimulation in cardiomyopathy (MUSTIC) study. J Am Coll Cardiol. 2002;40:111–118. doi: 10.1016/s0735-1097(02)01932-0. [DOI] [PubMed] [Google Scholar]
- 27.Menasche P, Hagege AA, Vilquin JT, Desnos M, Abergel E, Pouzet B, Bel A, Sarateanu S, Scorsin M, Schwartz K, Bruneval P, Benbunan M, Marolleau JP, Duboc D. Autologous skeletal myoblast transplantation for severe postinfarction left ventricular dysfunction. J Am Coll Cardiol. 2003;41:1078–1083. doi: 10.1016/s0735-1097(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 28.Morley GE, Danik SB, Bernstein S, Sun Y, Rosner G, Gutstein DE, Fishman GI. Reduced intercellular coupling leads to paradoxical propagation across the Purkinje-ventricular junction and aberrant myocardial activation. Proc Natl Acad Sci USA. 2005;102:4126–4129. doi: 10.1073/pnas.0500881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murry CE, Wiseman RW, Schwartz SM, Hauschka SD. Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest. 1996;98:2512–2523. doi: 10.1172/JCI119070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohara T, Qu Z, Lee MH, Ohara K, Omichi C, Mandel WJ, Chen PS, Karagueuzian HS. Increased vulnerability to inducible atrial fibrillation caused by partial cellular uncoupling with heptanol. Am J Physiol Heart Circ Physiol. 2002;283:H1116–H1122. doi: 10.1152/ajpheart.00927.2001. [DOI] [PubMed] [Google Scholar]
- 31.Penicka M, Bartunek J, De Bruyne B, Vanderheyden M, Goethals M, De Zutter M, Brugada P, Geelen P. Improvement of left ventricular function after cardiac resynchronization therapy is predicted by tissue Doppler imaging echocardiography. Circulation. 2004;109:978–983. doi: 10.1161/01.CIR.0000116765.43251.D7. [DOI] [PubMed] [Google Scholar]
- 32.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 33.Reinecke H, MacDonald GH, Hauschka SD, Murry CE. Electromechanical coupling between skeletal and cardiac muscle. Implications for infarct repair. J Cell Biol. 2000;149:731–740. doi: 10.1083/jcb.149.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuster P, Faerestrand S, Ohm OJ. Color Doppler tissue velocity imaging can disclose systolic left ventricular asynchrony independent of the QRS morphology in patients with severe heart failure. Pacing Clin Electrophysiol. 2004;27:460–467. doi: 10.1111/j.1540-8159.2004.00464.x. [DOI] [PubMed] [Google Scholar]
- 35.Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovasc Res. 2004;62:368–377. doi: 10.1016/j.cardiores.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Sra J, Dhala A, Blanck Z, Deshpande S, Cooley R, Akhtar M. Sudden cardiac death. Curr Probl Cardiol. 1999;24:461–538. doi: 10.1016/s0146-2806(99)90004-x. [DOI] [PubMed] [Google Scholar]
- 37.Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ Res. 1999;85:174–181. doi: 10.1161/01.res.85.2.174. [DOI] [PubMed] [Google Scholar]
- 38.Van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- 39.Viswanathan PC, Rudy Y. Cellular arrhythmogenic effects of congenital and acquired long-QT syndrome in the heterogeneous myocardium. Circulation. 2000;101:1192–1198. doi: 10.1161/01.cir.101.10.1192. [DOI] [PubMed] [Google Scholar]
- 40.Wu AH, Das SK. Sudden death in dilated cardiomyopathy. Clin Cardiol. 1999;22:267–272. doi: 10.1002/clc.4960220404. [DOI] [PMC free article] [PubMed] [Google Scholar]