

Abstract
Spiraling drug developmental costs and lengthy time-to-market introduction are two critical challenges facing the pharmaceutical industry. The clinical trials success rate for oncology drugs is reported to be 5% as compared to other therapeutic categories (11%) with most failures often encountered late in the clinical development process. PET and SPECT nuclear imaging technologies could play an important role in facilitating the drug development process improving the speed, efficiency and cost of drug development. This review will focus on recent studies of PET and SPECT radioligands in oncology and their application in the investigation of tumor biology. The use of clinically-validated radioligands as imaging-based biomarkers in oncology could significantly impact new cancer therapeutic development.
Keywords: PET, SPECT, radioligands, tumor imaging, tumor biomarkers, drug development
INTRODUCTION
Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT) are nuclear imaging techniques used to map physiological and biological processes in humans and animals following the administration of radiolabeled tracers. A unique advantage of PET and SPECT imaging techniques is their potential for detecting disease-related biochemical and physiologic abnormalities prior to the appearance of anatomical changes which can be visualized by conventional imaging modalities such as CT and MRI. PET uses radioisotopes that decay via emission of positrons, whereas, SPECT radioisotopes decay by electron capture and/or gamma emission.
Table 1 lists some of the most commonly used PET and SPECT radioisotopes and their physical data. The short half lives of the positron-emitters carbon-11, nitrogen-13 and oxygen-15 dictates that radioligand synthesis with these isotopes can only be accomplished in close proximity to a cyclotron. On the other hand, radioisotopes such as fluorine-18, copper-64, indium-111, iodine-123 and iodine-124 are sufficiently long-lived to allow transportation from regional commercial sites. Additionally, the radioisotopes gallium-68, copper-62 and technetium-99m can be conveniently obtained from an in-house generator. At the present time, clinical SPECT imaging is more frequently conducted than PET imaging due to its cost effectiveness and the greater availability of SPECT scanners at most clinical centers.
Table 1.
Commonly used PET and SPECT Radioisotopes
| Isotope | Imaging Mode | Production Method | Half-Life | Decay Mode(s) |
|---|---|---|---|---|
| 11C | PET | Cyclotron | 20.4 min | β+ (99+%) |
| 13N | PET | Cyclotron | 10 min | β+ (100%) |
| 15O | PET | Cyclotron | 2.03 min | β+ (99.9%) |
| 18F | PET | Cyclotron | 110 min | β+ (97%) EC (3%) |
| 124I | PET | Accelerator | 4.2 days | EC (74.4%) β+ (25.6%) |
| 68Ga | PET | Generator | 68.3 min | β+ (90%) EC (10%) |
| 62Cu | PET | Generator | 9.73 min | β+ (98%) EC (2%) |
| 64Cu | PET | Reactor | 12.7 hours | β+ EC |
| 99mTc | SPECT | Generator | 6.02 hours | IT |
| 111In | Gamma Scintigraphy | Accelerator | 2.8 days | EC x-ray |
| 123I | SPECT | Accelerator | 13.3 hours | EC |
β+ = positron emission; EC = electron capture; IT = Isomeric transition.
In PET, positrons (positively charged electrons, β+) ejected from the nucleus during decay travel a few millimeters in tissue, after which, they undergo annihilation by collision with electrons. Each annihilation event releases two γ-ray photons of equal energy (511 keV) in opposite trajectories (180° apart). PET scanners utilize the simultaneous detection of these two photons (coincidence detection) to precisely locate the source of the annihilation event. Subsequently, the event data is processed by computers to reconstruct the spatial distribution of the annihilation events. SPECT scanners on the other hand, use collimators (lead shields containing narrow parallel holes) to acquire only those photons that have a parallel trajectory. Thus, the original path of the detected photon can be linearly extrapolated from knowledge of the collimators orientation. Coincidence detection is significantly more efficient than collimation at recording annihilation events as the latter approach results in discarding a high percentage of useful emitted photons. Thus, PET provides a much better sensitivity (2 – 3 orders of magnitude), quantitation capability and spatial resolution than SPECT.
The process of developing a useful nuclear imaging radiotracer for biological imaging has several requirements that can pose special challenges. Incorporation of the radionuclide (including its chelating functionality in some cases) in a target ligand should have a negligible effect on its binding affinity. Radioligand binding sites (receptor, enzyme, etc.,) usually exist in low concentration (micromolar to nanomolar). Thus, the specific activity of the radioligand should be sufficiently high to represent a high radiative emission from a very small quantity (mass) of radiodiagnostic to avoid producing a pharmacologic effect. Due to the constraints of working with a short radioisotope half-life, the overall synthetic strategy for radioligand preparation should be short, the individual reaction steps rapid and high yielding, and the entire process should be adaptable to microscale manipulation. From an in vivo standpoint, the radioligand should display low or negligible non-specific binding so as to provide a high target-to-background signal and the in vivo kinetics of radioligand target uptake and washout should be compatible with the half-life of the radioisotope. Additionally, the radioligand should not be extensively metabolized and metabolites, if present, should not compete with the radioligand at its intended binding site. Despite these rigorous requirements, many radioligands have been developed which display demonstrated clinical utility for biological imaging (e.g. [18F]fluoro-deoxyglucose ([18F]FDG), [18F]FLT, radiolabeled somatostatin analogs, etc.,).
Currently, [18F]FDG (Fig. (1); a radioligand marker for tumor glucose metabolism), is the workhorse of PET, reportedly used in at least 90% of human PET studies. The majority of these studies are in oncology where [18F]FDG PET is the primary method used for detection and staging of many cancers [1]. However, [18F]FDG is not tumor specific and is known to accumulate in many benign inflammatory processes leading to false-positive interpretation [2]. The past decade has seen the investigation and validation of several alternative radioligands to [18F]FDG that target specific aspects of tumor biology. These targets include molecular biomarkers such as growth factor receptors, protein kinases, specific receptor over-expression or biological events such as angiogenesis, apoptosis, hypoxia and tumor proliferation. This review will focus on selected examples of radioligand validation studies reported in the past three years that target key aspects of tumor biology. The use of clinically-validated radioligands as imaging-based biomarkers in oncology could significantly impact new cancer therapeutic development.
Fig. (1).

Structure of D-glucose and [18F]FDG
RADIOLIGANDS FOR IMAGING ANGIOGENESIS
Angiogenesis, the formation of new blood vessels through capillary sprouting from pre-existing vasculature, plays a key role in the growth and metastatic potential of solid tumors [3, 4]. Tumor growth beyond a 1 – 2 mm3 volume requires an independent vasculature for the cellular supply of oxygen and nutrients and removal of waste products [5]. Consequently, tumors that outgrow their existing blood supply frequently display oxygen deficiency (hypoxia) which can trigger the secretion of various pro-angiogenic growth factors, such as, vascular endothelial growth factors (VEGF’s) for initiating new blood vessel growth [3, 6]. Binding of VEGF’s to the VEGF family of receptors (VEGFR) initiates a signaling cascade that promotes the proliferation, migration and survival of endothelial cells, ultimately leading to angiogenesis [7, 8]. The angiogenic effects of the VEGF family are believed to be primarily mediated through VEGF-A. To date, VEGF-A (also referred to as VEGF) and its receptors are the most characterized signaling pathways in developmental and tumor angiogenesis.
Alternative splicing of RNA has revealed the existence of at least seven different molecular isoforms for VEGF-A, comprising, 121, 145, 148, 165, 189 or 206 amino acids [9]. The angiogenic actions of VEGF-A are mediated primarily via two closely related endothelium-specific receptor tyrosine kinases (VEGFR-1 and VEGFR-2) [10, 11]. All of the VEGF-A isoforms bind to both VEGFR-1 and VEGFR-2, of which, VEGFR-2 is the major mediator of the mitogenic, angiogenic and permeability enhancing effects of VEGF-A [11]. VEGFR’s are over-expressed in a variety of solid tumors with over-expression of VEGFR-2 or VEGF-A in particular, serving as poor prognostic markers [7, 12].
VEGF121 (a molecular isoform of VEGF) radiolabeled with 64Cu has been reported for small animal PET imaging of VEGF receptor expression in vivo [13]. Radiolabeling was achieved via 64Cu chelation to a DOTA-VEGF121 conjugate (DOTA is an abbreviation for 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid). In vivo evaluation of 64Cu-DOTA-VEGF121 using microPET imaging of athymic nude mice bearing U87MG human glioblastoma xenografts showed rapid and high specific accumulation of the radioligand in small U87MG tumors (16% injected dose per gram [ID/g]) at 4 h postinjection. Larger tumors showed significantly lower uptake (1 – 3% ID/g). Differences in tumor localization between large and small tumors showed a good correlation with tumor VEGF receptor expression (VEGR2). In vivo VEGFR2 specificity of the radioligand was also confirmed by pharmacological blocking experiments and ex vivo studies (immunofluorescence staining, western blot analysis). More recently, these authors have also reported on the development of a 64Cu-labeled vasculature-targeting fusion toxin (VEGF121/rGel) composed of a VEGF121 linked recombinant plant toxin gelonin construct (rGel) for multimodality imaging and therapy of glioblastoma [14].
Chan and coworkers have reported on the synthesis and evaluation in tumored mice of an 111In-labeled recombinant VEGF isoform VEGF165 (111In-hn-Tf-VEGF) as a tumor angiogenesis marker [15]. VEGF165 was fused through a flexible polypeptide linker to the n-lobe of human transferrin. The latter construct permitted labeling of the radioligand with 111In at a site remote from the VEGF receptor-binding domain. In radioligand stability studies, 111In-hn-Tf-VEGF demonstrated a moderate loss of 111In to transferrin in human plasma in vitro over a 72 h period (21.3% ± 3.4% per day). Radioligand biodistribution studies and whole-body gamma camera imaging were conducted in athymic mice bearing subcutaneous U87MG human glioblastoma xenografts. 111In-hn-Tf-VEGF displayed tumor and blood radioactivity accumulations of 6.7 ± 1.1 %ID/g and 1.6 ± 0.4 %ID/g, respectively, at 72 h post-injection. Co-administration of a 100-fold excess of VEGF led to a 15-fold decrease in tumor uptake of radioactivity. High uptake of radioactivity was also observed in liver (45.5 ± 7.5 %ID/g), kidneys (39.4 ± 7.0 %ID/g) and spleen (35.6 ± 4.4 %ID/g) at this time interval. The authors present evidence to indicate that uptake of radioactivity in these organs is due to 111In-hn-Tf-VEGF and not due to 111In-transferrin via transchelation of 111In from the radioligand to transferrin.
An indirect approach to angiogenesis imaging has focused on radioligands targeting the αvβ3 class of cell adhesion molecule integrins [16]. Integrin αvβ3 receptors are significantly up-regulated in endothelial cells during angiogenesis but not in mature vessels or non-neoplastic epithelium [17, 18, 19]. Integrin αvβ3 is also expressed in a variety of tumor cells, including melanoma, late-stage glioblastoma, ovarian, breast and prostate cancer [20]. The ability to visualize and quantify integrin αvβ3 expression in vivo would allow for appropriate selection of patients for anti-integrin treatment and also monitor treatment efficacy in such patients.
Radioligand development for αvβ3 imaging has focused primarily on small RGD peptide antagonists [21]. The tripeptide sequence motif, arginine-glycine-aspartate (RGD), is found in proteins of the extracellular matrix. Many integrins, including αvβ3 link the intracellular cytoskeleton of cells with the extracellular matrix via recognition and binding to this RGD motif. Wu and coworkers have reported on the enhanced αvβ3 receptor binding characteristics of dimeric and multimeric RGD peptides over monomeric peptides which has been attributed to an increased local concentration of RGD domains at the receptor vicinity (polyvalency effect) [22]. Accordingly, several [18F]- and [64Cu]-labeled, dimeric and tetrameric RGD peptide analogs have been recently synthesized and evaluated by this group for integrin-targeted imaging in lung, brain and breast cancer [22, 23, 24]. As an example, microPET imaging studies with a dimeric RGD peptide coupled to 4-[18F]Fluorobenzoate {[18F]-FB-E[c(RGDyK)]2} showed predominantly renal excretion and twice as much tumor uptake in the same animal model as the monomeric analog [18F]-FB-c(RGDyK) [25]. Binding potentials derived from tracer kinetic modeling studies showed good correlation with tumor integrin expression levels as measured by SDS-PAGE/autoradiography in the six tumor models tested [25].
RADIOLIGANDS FOR IMAGING APOPTOSIS
Apoptosis (programmed cell death) plays a critical role in the homeostasis of multicellular organisms. Initiation of apoptotic cell death leads to activation of a family of cysteine proteases (caspases) which act as central executioners of the apoptotic process [26]. Radiation as well as anticancer drug treatment can induce apoptosis in tumor cells. Consequently, imaging methods that provide information on the rate and extent of apoptosis are of interest in monitoring the efficacy of anticancer treatment. A vast majority of the work on apoptosis-targeted radioligands has focused on Annexin V and its derivatives [27]. Annexin V is a member of the calcium and phospholipid binding superfamily of Annexin proteins that displays selective, nanomolar affinity (Kd ~ 0.5 – 7 nM) toward phosphatidylserine (PS) residues. Induction of apoptosis results in a rapid externalization of PS from the inner leaflet of the plasma membrane to its outer surface [28]. Accordingly, Annexin-mediated imaging of PS has been extensively investigated for identifying cells at the early stages of apoptosis. Annexin V and its derivatives have been radiolabeled with a wide variety of radioisotopes including radioiodine (123I, 124I, 125I), 18F, 99mTc, 111In, 11C, and 64Cu [29]. Radiolabeled caspase substrates, inhibitors and monoclonal antibodies targeted to human Annexin V have also been reported as alternative approaches for apoptosis imaging [30] [31]. An excellent review on these developments has been recently published by Lahorte and coworkers [29].
A recent report by Tait and coworkers have compared the apoptosis-specific liver uptake of several [99mTc]-labeled Annexin V derivatives prepared by amine-directed modification with that labeled site-specifically at the N-terminus [32]. A clear improvement was seen for site-specific labeling as compared to amine-directed modification. Use of [99mTc]-labeled hydrazinenicotinamide-annexin imaging for assessment of response to chemotherapy has also been reported [33]. The reported imaging protocol was able to distinguish responders from non-responders with 94% accuracy (16/17 patients) and a sensitivity and specificity of 86% and 100%, respectively.
RADIOLIGANDS FOR IMAGING HYPOXIA
The growth of solid malignant tumors is frequently accompanied by tissue hypoxia due to outgrowth of its blood supply. Hypoxia in tumor cells leads to resistance to both radiation and anticancer treatment [34]. Noninvasive imaging methods for identification and quantitation of tumor hypoxia status could play a central role in predicting and monitoring treatment response. Initial approaches to hypoxia imaging focused on radiolabeled 2-nitroimidazole derivatives, a class of hypoxia-activated prodrugs [35]. These bioreductive prodrugs undergo reductive metabolism in the hypoxic environment of tumors thereby releasing toxic metabolites that can lead to cell damage [36]. Among the 2-nitroimidazole class of radioligands, [18F]fluoromisonidazole ([18F]FMISO) is currently the most widely used clinical PET hypoxia tracer (Fig. (2)) [37].
Fig. (2).
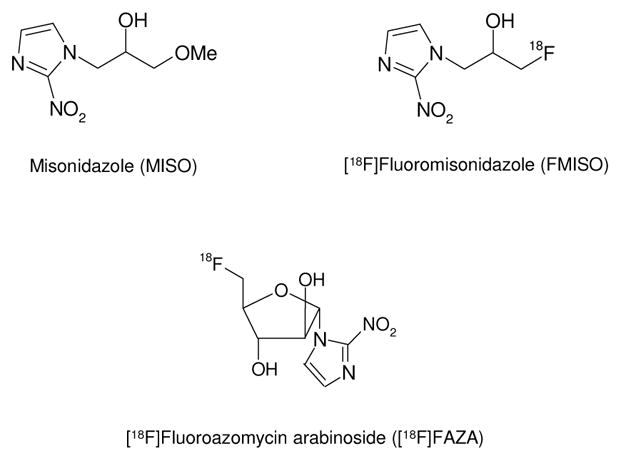
Structure of Misonidazole (MISO), [18F]FMISO and [18F]FAZA
More recently, Rischin et al. have shown that [18F]FMISO PET imaging is a useful method for identifying head and neck cancer patients most likely to benefit from treatment with the hypoxic cell cytotoxin, tirapazamine in a chemoradiotherapy regimen [38]. In an extensive study involving 73 patients with head and neck cancer, Rajendran et al. have shown that [18F]FMISO PET imaging is a predictor of survival prior to radiation therapy [39]. A 18F-labeled 2-nitroimidazole derivative, fluoroazomycin arabinoside ([18F]FAZA; Fig. (2)) that displays enhanced in vivo stability to enzymatic cleavage has been described by Piert and colleagues [40]. Studies indicate that [18F]FAZA may be superior to [18F]FMISO for hypoxia imaging due to its superior biokinetic profile [40]. These authors have also shown that [18F]FAZA imaging has predictive value for the determination of radiotherapy success when used in combination with tirapazamine [41]. Copper-64 labeled Cu-Diacetyl-bis(N4-methylthiosemicarbazone) (64Cu-ATSM) has also been proposed as a PET hypoxia imaging agent. Differences in tumor type selectivity for this radiotracer, however, raises questions regarding its use as a universal PET hypoxia marker [42].
RADIOLIGANDS FOR IMAGING EGF RECEPTORS
The search for radioligands that target the ErbB family of receptor tyrosine kinases is an active area of research. This receptor family includes four members: epidermal growth factor receptor (EGFR/ErbB1/HER1), HER2 (ErbB2/neu), HER3 (ErbB3) and HER4 (ErbB4) [43] [44]. Over-expression of these receptors, particularly HER1 and HER2, has been documented in many epithelial cancers including breast, non-small cell lung (NSCLC), ovarian and bladder cancer [45] [46]. It has also been shown that such over-expression is frequently associated with a poor prognosis [47, 48, 49].
Preclinical evaluation of a 68Ga-labeled, recombinant human epidermal growth factor DOTA conjugate (68Ga-DOTA-hEGF) has been reported for HER1 imaging [50]. In vitro studies with 68Ga-DOTA-hEGF conducted on EGFR-expressing cell lines, U343 glioma and A431 cervical carcinoma, demonstrated high affinity binding (2 nM), rapid internalization of radioligand and good retention of radioactivity. Radioligand biodistribution in mice bearing A431 tumor xenografts showed a tumor-to-blood ratio of 4.5 at 30 min postinjection (2.7% ID/g in tumor). Interestingly, tumor uptake was dependent on the specific activity of the radioligand: a two-fold increase in tumor uptake was observed with a 10-fold lower specific activity material. Tumors were clearly visualized by microPET imaging in a tumor-bearing mouse although the kinetics of tumor uptake of radioactivity was slow compared to that of liver and kidney. An 111In-labeled human EGF-diethylenetriaminepentaacetic acid (DTPA) conjugate (111In-DTPA-hEGF) is under investigation for future Phase 1 clinical trials as a radiotherapeutic for breast cancer [51]. Preclinical pharmacokinetic, toxicology and dosimetry studies conducted in mice and rabbits with 111In-DTPA-hEGF showed no acute toxicity in female BALB/c mice at 42 times the maximum planned human dose. Highest uptake of radioactivity was seen in liver (41.3% ID/g) and kidney (18.6% ID/g) at 1 h postinjection, although these values had decreased to 4.5 – 4.9% at 72 h. The radiotracer showed fast blood clearance following intravenous injection and no morphologic changes were seen by light microscopy in 19 sampled tissues.
Cetuximab (Erbitux; ImClone Systems, Inc.), a chimeric anti-EGFR monoclonal antibody, is an FDA-approved drug for the treatment of advanced metastatic colorectal cancer. Cai and coworkers describe microPET imaging with a 64Cu-labeled DOTA-cetuximab conjugate (64Cu-DOTA-cetuximab) in seven xenograft tumor mouse models [52]. Uptake of radioactivity for 64Cu-labeled DOTA-cetuximab was similar in major organs and tissues in all seven of the tested tumor models (U87MG human glioblastoma, PC-3 human prostate carcinoma, CT-26 murine colorectal carcinoma, HCT-8, HCT-116, SW620 human colorectal carcinoma and MDA-MB-435 human breast cancer). High uptake of radioactivity was seen for the high EGFR-expression U87MG and PC-3 tumors (13.2%ID/g and 12.8% ID/g, respectively) at 48 h postinjection. Tumor radioactivity uptake determined by microPET imaging showed a good correlation with EGFR expression levels as measured by western blot analysis.
An Affibody dimer, His6-(ZHER2:4)2, has been recently described as a high affinity HER2/neu ligand [53]. Orlova and coworkers have prepared 99mTc- and 125I-labeled His6-(ZHER2:4)2 and compared their biodistribution in tumor-bearing BALB/c nu/nu mice [54]. Significantly, higher levels of radioactivity were observed in tumor as compared to liver for the 125I-labeled ligand. These studies indicate that the radioiodinated ligand may be more suitable than the corresponding [99mTc]-labeled ligand for imaging tumor HER2 expression levels, particularly in liver. A 99mTc-labeled Affibody compound MAG3-(ZHER2:342) labeled via a MAG3 chelator has also been recently reported by this group [55]. Smith-Jones and colleagues have used mouse microPET imaging with a 68Ga-labeled F(ab′)2 fragment of herceptin (68Ga-DOTA-F(ab′)2-herceptin) to image HER2 downregulation after heat shock protein (Hsp90) inhibition [56]. The Ansamycin antibiotic, geldanamycin is known to cause HER2 degradation via inhibition of the Hsp90 chaperone protein. PET imaging was conducted on mice bearing BT474 breast tumor xenografts with 68Ga-DOTA-F(ab′)2-herceptin and [18F]-fluorodeoxyglucose ([18F]FDG) before and after treatment with the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG). A significant decrease of HER2 expression was seen within 24 h of 17-AAG treatment with 68Ga-DOTA-F(ab′)2-herceptin imaging. In contrast, tumor uptake of [18F]FDG (a marker of glycolysis) was unchanged. The authors conclude that PET imaging with the HER2 radioligand, 68Ga-DOTA-F(ab′)2-herceptin is superior to [18F]FDG imaging for evaluating tumor response to 17-AAG therapy.
RADIOLIGANDS FOR IMAGING SOMATOSTATIN RECEPTORS
The utilization of radiolabeled peptide analogs of the hormone somatostatin for the diagnostic imaging and therapy of neuroendocrine tumors (NET) has had notable success [57] [58]. Somatostatin exists in two isoforms: a short peptide having 14 amino acids, and a second peptide with 28 amino acids, both of which bind with high affinity to five receptor subtypes (sst1 – sst5) [59]. A majority of malingnant tumors (e.g., NET, small cell lung cancer, malignant lymphoma and breast tumors) over-express multiple sst receptor subtypes relative to non-tumor tissues, of which the sst2 subtype is frequently more predominantly expressed [60]. Since these G-protein coupled receptors undergo internalization on ligand binding they are uniquely suited for radionuclide imaging. Internalization of the receptor-radioligand complex allows for extended tumor retention times which could enhance diagnostic sensitivity due to improved tumor-to-background ratios. Receptor-radioligand complex internalization could also be an important advantage in targeted radiotherapy applications [61]. A recent elegant study by Cescato and colleagues describes the use of new immunocytochemical methods to quantitatively measure sst2, sst3 and sst5 receptor subtype internalization induced by a variety of somatostatin analogs in human embryonic kidney 293 (HEK293) cells expressing these subtypes [62].
Somatostatin has a short plasma half-life (approximately 2 min) making it unsuitable for radioligand development [63]. The discovery of the short octapeptide somatostatin analog, octreotide, which displays a superior pharmacological profile, played a major role in the development of radioligands for somatostatin receptor imaging [64]. Octreotide which displays high affinity for sst2 (IC50 = 2 nM) has a plasma half-life of 1.7 hours and higher metabolic stability than somatostatin [65]. Subsequently, [111In]-labeled DTPA conjugated octreotide (Octreoscan; Mallinkrodt Medical) was developed and introduced for scintigraphic imaging of sst-expressing NET. Octreoscan is currently the gold standard for the localization, staging and management of NET [66]. However, for imaging purposes, use of the 111In radionuclide has certain disadvantages, including high cost, limited availability, less than optimum image quality and elevated patient radiation dose [66]. Consequently, several newer somatostatin analogs labeled with single photon emitters (99mTc, 123I) or positron-emitters (68Ga, 18F, 64Cu) for SPECT or PET application, respectively, have been described [67, 68, 69, 70, 71, 72]. A recent review describes the progress of functional imaging of NET using PET [73].
Current work in sst radioligand development has been directed primarily towards modulation of radioligand subtype specificity or evaluation of different radionuclides for improved tumor-targeted radiotherapy [65]. Ginj et al. have reported on novel 111In-labeled DOTA-conjugated octreotide analogs that display high affinity binding (1.4 – 13 nM) to several sst receptor subtypes (sst2, sst3 and sst5) [61]. Animal biodistribution studies showed high, specific uptake of radioactivity in sst receptor-expressing tumors (AR4-2J) in a rat model. Both radiopeptides were more efficiently internalized than [111In-DOTA,Tyr3]-octreotide. The authors propose that the high-affinity, broad sst specificity of these radioligands will be advantageous for the diagnosis and targeted radiotherapy of a broader range of sst-expressing tumors.
In an extensive study involving 84 patients, Gabriel and coworkers have compared the utility of a new PET somatostatin analog, 68Ga-D-Phe1-Tyr3-octreotide DOTA conjugate (68Ga-DOTA-TOC), for NET imaging with that of SPECT and CT [68]. 68Ga-DOTA-TOC was designed by replacement of the Phe3 residue in the corresponding octapeptide by Tyr which leads to increased hydrophillicity and improved kidney clearance [74, 75]. This modification also provides enhanced affinity for human sst2 [76, 74]. Gabriel et al. compared PET imaging with 68Ga-DOTA-TOC to SPECT imaging with the following radioligands: (111In-DOTA-TOC) and 99mTc-labeled hydrazinonicotinyl-Tyr3-octreotide (99mTc-HYNIC-TOC) [68]. CT imaging was also performed on each patient. Comparison of the three imaging modalities revealed an accuracy for PET imaging of 96% which was significantly higher than that of CT (63%) and SPECT (58%). In addition, 68Ga-DOTA-TOC imaging results were true-positive in 32 patients whose SPECT results were false-negative and it was able to detect more lesions than either SPECT or CT. Moreover, PET detected more metastatic tumors (lymph node, bone and liver) than SPECT thus permitting more accurate disease staging. The authors conclude that PET imaging with 68Ga-DOTA-TOC in conjunction with CT is superior to SPECT in the clinical diagnosis of NET.
Radiolabeled somatostatin analogs that incorporate beta-emitting (90Y, 177Lu) or alpha-emitting (213Bi) radioisotopes for targeted tumor radiotherapy are under active investigation [77, 78]. Preliminary, preclinical data suggest that the radionuclide 90Y (high energy, pure beta-emitter; Emax = 2.25 MeV) may be more effective for treating larger tumors while the use of 177Lu (low energy, beta-emitter; Emax = 0.497 Mev) leads to fewer relapses when treating smaller lesions [79, 80]. These developments have been recently reviewed [65].
RADIOLIGANDS FOR IMAGING TUMOR CELL PROLIFERATION
Tritium-labeled thymidine ([3H]thymidine) has been shown to be rapidly incorporated into newly synthesized DNA. Hence [3H]thymidine has been the gold standard for many years for assessing cell proliferation in cell culture and animal studies [81]. [11C]Thymidine was subsequently developed as a PET radioligand for monitoring cell proliferation in vivo. However, general use of this radioligand is hampered by its rapid catabolism and short half-life [82]. Two PET radiolgands that were subsequently developed to address these limitations include the [18F]-labeled analogs, 3′-deoxy-3′-[18F]fluorothymidine ([18F]FLT) and 1-(2′-deoxy-2′-[18F]fluoro-beta-D-arabinofuranosyl)thymine ([18F]FMAU) (Fig. (3)) [1]. Of these, [18F]FLT has been the most studied to date [1]. [18F]FLT is taken up by cells and phosphorylated by thymidine kinase 1 (TK-1) with subsequent trapping within the cell [81]. Intracellular [18F]FLT retention can thus be used as a measure of cellular TK-1 activity. Since TK-1 enzyme activity closely parallels the DNA synthesis pathway, cellular retention of [18F]FLT is also a measure of cellular proliferation.
Fig. (3).

Structure of Thymidine, [18F]FLT and [18F]FMAU
Leyton and coworkers have published several reports describing the use of PET imaging of [18F]FLT uptake as a surrogate marker for early in vivo quantitative imaging of drug-induced changes in cell proliferation. In one example, PET imaging with [18F]FLT was used for noninvasive measurement of the biological activity of a novel histone deacetylase inhibitor (LAQ824) [83]. Histone deacetylase inhibitors have been shown to cause growth inhibition of cancer cells in vitro and in animal models [84]. Treatment of mice bearing HCT116 colon- carcinoma xenografts with LAQ824 resulted in a dose-dependent decrease in [18F]FLT tumor uptake. Dose-dependent decreases of tumor TK1 and protein levels were also observed with LAQ824 pretreatment.
In a second study, [18F]FLT PET imaging was found to be superior to [18F]FDG PET imaging for quantitative measurement of tumor cell proliferation following cisplatin treatment [85]. Cisplatin is a widely used chemotherapeutic drug for cervical, lung, bladder and prostate cancer [86]. Decreased [18F]FLT tumor uptake was seen in mice bearing radiation-induced fibrosarcoma 1 (RIF-1) tumors subject to cisplatin treatment, despite a lack of change in tumor size. The decrease in [18F]FLT uptake was associated with a decrease in cell proliferation determined by immunohistochemical analysis. [18F]FLT PET imaging was also successful in quantification of the activity of an administered replicating oncolytic viral vector (dl922-947) in mice bearing IGROVI ovarian carcinoma xenografts [87].
Waldherr and colleagues have used [18F]FLT PET imaging to measure the effect of the ErbB-selective kinase inhibitor PK1-166 on tumor cell proliferation in SCID mice having ErbB1-over-expressing A431 xenograft tumors [88]. Treatment with PK1-166 markedly lowered tumor [18F]FLT uptake within 48 h of drug exposure and led to a 79% decrease of [18F]FLT uptake within a week of treatment. [18F]FLT PET imaging has also been used to monitor decreased tumor proliferation in a murine squamous cell carcinoma following radiation therapy [89].
Taken together, these studies provide good initial evidence that [18F]FLT PET imaging may be useful for monitoring therapeutic response early in the course of treatment. However, present studies suggest that [18F]FDG may be more useful for tumor detection and staging [90].
SUMMARY AND CONCLUSION
PET and SPECT imaging can be of immense utility in many aspects of the drug development process. At the preclinical stage, noninvasive imaging studies with radiolabeled investigational drugs can provide vital proof-of-concept information to aid proper candidate selection. These studies would allow noninvasive determination of a drug’s pharmacokinetic behavior and target versus non-target accumulation. Importantly, such studies could provide information early in the development process if a drug is, in fact, reaching its target and also identify potential toxicity issues. Drug analogs under investigation could also be screened in vivo in animal models against a validated radioligand selective for the same biological target (e.g., receptor, enzyme binding site, etc.) to provide direct measures of receptor occupancy. Such data could be used to design optimal dosing and timing schedules in clinical trials thereby improving their efficiency and cost-effectiveness.
At the clinical level, microdosing studies with suitable radioligands could provide useful metabolic and toxicology data prior to the conducting of classical Phase I trials. The FDA Critical Path Initiative has emphasized the development of validated imaging-based biomarkers as an important tool for streamlining the drug development process. As highlighted in this review, clinically-validated radioligand probes that image downstream biological effects of cancer treatment such as, apoptosis, angiogenesis and decreased tumor cell proliferation could be useful surrogate markers for monitoring the therapeutic response of new oncology drugs in development.
[18F]FDG PET imaging currently plays an increasingly important role in cancer treatment by virtue of its ability to diagnose, stage and assess tumor response to chemotherapy and chemo-radiotherapy. It is highly likely that the future availability of validated molecularly-targeted radioligands will likewise play a major role in cancer therapeutic development. Such achievements will greatly advance the ultimate goal of personalized medical treatment in providing the right drug to the right patient.
Acknowledgments
The authors acknowledge support from the following National Institutes of Health grants (PO1 CA85878, P50 CA93990, and R24 CA83099) during the preparation of this review.
References
- 1.Shields AF. Mol Imaging Biol. 2006;8:141–150. doi: 10.1007/s11307-006-0039-2. [DOI] [PubMed] [Google Scholar]
- 2.Metser U, Even-Sapir E. Semin Nucl Med. 2007;37:206–222. doi: 10.1053/j.semnuclmed.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 4.Sturk C, Dumont D. In: The Basic Science of Oncology. Tannock IF, Hill RP, Bristow RG, Harrington L, editors. McGraw-Hill, Medical Pub. Division; New York: 2005. pp. 231–248. [Google Scholar]
- 5.Bergers G, Benjamin LE. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 6.Safran M, Kaelin WG., Jr J Clin Invest. 2003;111:779–783. doi: 10.1172/JCI18181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrara N. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 8.Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 9.Renner W, Pilger E. J Vasc Res. 1999;36:133–138. doi: 10.1159/000025636. [DOI] [PubMed] [Google Scholar]
- 10.Sato Y, Kanno S, Oda N, Abe M, Ito M, Shitara K, Shibuya M. Ann N Y Acad Sci. 2000;902:201–205. doi: 10.1111/j.1749-6632.2000.tb06314.x. discussion 205–207. [DOI] [PubMed] [Google Scholar]
- 11.Ferrara N. Exs. 2005:209–231. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 12.Rudlowski C, Pickart AK, Fuhljahn C, Friepoertner T, Schlehe B, Biesterfeld S, Schroeder W. Int J Gynecol Cancer. 2006;16 Suppl 1:183–189. doi: 10.1111/j.1525-1438.2006.00307.x. [DOI] [PubMed] [Google Scholar]
- 13.Cai W, Chen K, Mohamedali KA, Cao Q, Gambhir SS, Rosenblum MG, Chen X. J Nucl Med. 2006;47:2048–2056. [PubMed] [Google Scholar]
- 14.Hsu AR, Cai W, Veeravagu A, Mohamedali KA, Chen K, Kim S, Vogel H, Hou LC, Tse V, Rosenblum MG, Chen X. J Nucl Med. 2007;48:445–454. [PubMed] [Google Scholar]
- 15.Chan C, Sandhu J, Guha A, Scollard DA, Wang J, Chen P, Bai K, Lee L, Reilly RM. J Nucl Med. 2005;46:1745–1752. [PubMed] [Google Scholar]
- 16.Danen EH. Curr Pharm Des. 2005;11:881–891. doi: 10.2174/1381612053381756. [DOI] [PubMed] [Google Scholar]
- 17.Hood JD, Cheresh DA. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 18.Kumar CC. Curr Drug Targets. 2003;4:123–131. doi: 10.2174/1389450033346830. [DOI] [PubMed] [Google Scholar]
- 19.Ruoslahti E. Nat Rev Cancer. 2002;2:83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 20.Cai W, Wu Y, Chen K, Cao Q, Tice DA, Chen X. Cancer Res. 2006;66:9673–9681. doi: 10.1158/0008-5472.CAN-06-1480. [DOI] [PubMed] [Google Scholar]
- 21.Chen X. Mini Rev Med Chem. 2006;6:227–234. doi: 10.2174/138955706775475975. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, Zhang X, Xiong Z, Cheng Z, Fisher DR, Liu S, Gambhir SS, Chen X. J Nucl Med. 2005;46:1707–1718. [PubMed] [Google Scholar]
- 23.Chen X, Sievers E, Hou Y, Park R, Tohme M, Bart R, Bremner R, Bading JR, Conti PS. Neoplasia. 2005;7:271–279. doi: 10.1593/neo.04538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai W, Zhang X, Wu Y, Chen X. J Nucl Med. 2006;47:1172–1180. [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, Chen X. J Nucl Med. 2006;47:113–121. [PMC free article] [PubMed] [Google Scholar]
- 26.Hengartner MO. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 27.Blankenberg FG. Curr Pharm Des. 2004;10:1457–1467. doi: 10.2174/1381612043384790. [DOI] [PubMed] [Google Scholar]
- 28.Corsten MF, Hofstra L, Narula J, Reutelingsperger CP. Cancer Res. 2006;66:1255–1260. doi: 10.1158/0008-5472.CAN-05-3000. [DOI] [PubMed] [Google Scholar]
- 29.Lahorte CM, Vanderheyden JL, Steinmetz N, Van de Wiele C, Dierckx RA, Slegers G. Eur J Nucl Med Mol Imaging. 2004;31:887–919. doi: 10.1007/s00259-004-1555-4. [DOI] [PubMed] [Google Scholar]
- 30.Haberkorn U, Kinscherf R, Krammer PH, Mier W, Eisenhut M. Nucl Med Biol. 2001;28:793–798. doi: 10.1016/s0969-8051(01)00247-5. [DOI] [PubMed] [Google Scholar]
- 31.Bauer C, Bauder-Wuest U, Mier W, Haberkorn U, Eisenhut M. J Nucl Med. 2005;46:1066–1074. [PubMed] [Google Scholar]
- 32.Tait JF, Smith C, Levashova Z, Patel B, Blankenberg FG, Vanderheyden JL. J Nucl Med. 2006;47:1546–1553. [PubMed] [Google Scholar]
- 33.Rottey S, Slegers G, Van Belle S, Goethals I, Van de Wiele C. J Nucl Med. 2006;47:1813–1818. [PubMed] [Google Scholar]
- 34.Harrison LB, Chadha M, Hill RJ, Hu K, Shasha D. Oncologist. 2002;7:492–508. doi: 10.1634/theoncologist.7-6-492. [DOI] [PubMed] [Google Scholar]
- 35.Chapman JD. N Engl J Med. 1979;301:1429–1432. doi: 10.1056/NEJM197912273012606. [DOI] [PubMed] [Google Scholar]
- 36.Denny WA. Eur J Med Chem. 2001;36:577–595. doi: 10.1016/s0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- 37.Rajendran JG, Mankoff DA. J Nucl Med. 2007;48:855–856. doi: 10.2967/jnumed.107.039768. [DOI] [PubMed] [Google Scholar]
- 38.Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, Peters LJ. J Clin Oncol. 2006;24:2098–2104. doi: 10.1200/JCO.2005.05.2878. [DOI] [PubMed] [Google Scholar]
- 39.Rajendran JG, Schwartz DL, O’Sullivan J, Peterson LM, Ng P, Scharnhorst J, Grierson JR, Krohn KA. Clin Cancer Res. 2006;12:5435–5441. doi: 10.1158/1078-0432.CCR-05-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piert M, Machulla HJ, Picchio M, Reischl G, Ziegler S, Kumar P, Wester HJ, Beck R, McEwan AJ, Wiebe LI, Schwaiger M. J Nucl Med. 2005;46:106–113. [PubMed] [Google Scholar]
- 41.Beck R, Roper B, Carlsen JM, Huisman MC, Lebschi JA, Andratschke N, Picchio M, Souvatzoglou M, Machulla HJ, Piert M. J Nucl Med. 2007;48:973–980. doi: 10.2967/jnumed.106.038570. [DOI] [PubMed] [Google Scholar]
- 42.Yuan H, Schroeder T, Bowsher JE, Hedlund LW, Wong T, Dewhirst MW. J Nucl Med. 2006;47:989–998. [PubMed] [Google Scholar]
- 43.Hynes NE, Lane HA. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 44.Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 45.Baselga J, Hammond LA. Oncology. 2002;63 Suppl 1:6–16. doi: 10.1159/000066198. [DOI] [PubMed] [Google Scholar]
- 46.Nahta R, Hortobagyi GN, Esteva FJ. Oncologist. 2003;8:5–17. doi: 10.1634/theoncologist.8-1-5. [DOI] [PubMed] [Google Scholar]
- 47.Selvaggi G, Novello S, Torri V, Leonardo E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P, Scagliotti GV. Ann Oncol. 2004;15:28–32. doi: 10.1093/annonc/mdh011. [DOI] [PubMed] [Google Scholar]
- 48.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. J Pathol. 2003;200:290–297. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 49.Memon AA, Sorensen BS, Meldgaard P, Fokdal L, Thykjaer T, Nexo E. Br J Cancer. 2006;94:1703–1709. doi: 10.1038/sj.bjc.6603154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velikyan I, Sundberg AL, Lindhe O, Hoglund AU, Eriksson O, Werner E, Carlsson J, Bergstrom M, Langstrom B, Tolmachev V. J Nucl Med. 2005;46:1881–1888. [PubMed] [Google Scholar]
- 51.Reilly RM, Chen P, Wang J, Scollard D, Cameron R, Vallis KA. J Nucl Med. 2006;47:1023–1031. [PubMed] [Google Scholar]
- 52.Cai W, Chen K, He L, Cao Q, Koong A, Chen X. Eur J Nucl Med Mol Imaging. 2007;34:850–858. doi: 10.1007/s00259-006-0361-6. [DOI] [PubMed] [Google Scholar]
- 53.Steffen AC, Wikman M, Tolmachev V, Adams GP, Nilsson FY, Stahl S, Carlsson J. Cancer Biother Radiopharm. 2005;20:239–248. doi: 10.1089/cbr.2005.20.239. [DOI] [PubMed] [Google Scholar]
- 54.Orlova A, Nilsson FY, Wikman M, Widstrom C, Stahl S, Carlsson J, Tolmachev V. J Nucl Med. 2006;47:512–519. [PubMed] [Google Scholar]
- 55.Engfeldt T, Orlova A, Tran T, Bruskin A, Widstrom C, Karlstrom AE, Tolmachev V. Eur J Nucl Med Mol Imaging. 2007;34:722–733. doi: 10.1007/s00259-006-0266-4. [DOI] [PubMed] [Google Scholar]
- 56.Smith-Jones PM, Solit D, Afroze F, Rosen N, Larson SM. J Nucl Med. 2006;47:793–796. [PMC free article] [PubMed] [Google Scholar]
- 57.Mariani G, Erba PA, Signore A. J Nucl Med. 2006;47:1904–1907. [PubMed] [Google Scholar]
- 58.Schillaci O. J Nucl Med. 2007;48:498–500. doi: 10.2967/jnumed.106.038653. [DOI] [PubMed] [Google Scholar]
- 59.Bodei L, Paganelli G, Mariani G. J Nucl Med. 2006;47:375–377. [PubMed] [Google Scholar]
- 60.Reubi JC, Waser B. Eur J Nucl Med Mol Imaging. 2003;30:781–793. doi: 10.1007/s00259-003-1184-3. [DOI] [PubMed] [Google Scholar]
- 61.Ginj M, Chen J, Walter MA, Eltschinger V, Reubi JC, Maecke HR. Clin Cancer Res. 2005;11:1136–1145. [PubMed] [Google Scholar]
- 62.Cescato R, Schulz S, Waser B, Eltschinger V, Rivier JE, Wester HJ, Culler M, Ginj M, Liu Q, Schonbrunn A, Reubi JC. J Nucl Med. 2006;47:502–511. [PubMed] [Google Scholar]
- 63.Scarpignato C, Pelosini I. Chemotherapy. 2001;47 Suppl 2:1–29. doi: 10.1159/000049157. [DOI] [PubMed] [Google Scholar]
- 64.Lamberts SW, Bakker WH, Reubi JC, Krenning EP. N Engl J Med. 1990;323:1246–1249. doi: 10.1056/NEJM199011013231805. [DOI] [PubMed] [Google Scholar]
- 65.Forrer F, Valkema R, Kwekkeboom DJ, de Jong M, Krenning EP. Best Pract Res Clin Endocrinol Metab. 2007;21:111–129. doi: 10.1016/j.beem.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 66.Storch D, Behe M, Walter MA, Chen J, Powell P, Mikolajczak R, Macke HR. J Nucl Med. 2005;46:1561–1569. [PubMed] [Google Scholar]
- 67.Guggenberg EV, Mikolajczak R, Janota B, Riccabona G, Decristoforo C. J Pharm Sci. 2004;93:2497–2506. doi: 10.1002/jps.20148. [DOI] [PubMed] [Google Scholar]
- 68.Gabriel M, Decristoforo C, Kendler D, Dobrozemsky G, Heute D, Uprimny C, Kovacs P, Von Guggenberg E, Bale R, Virgolini IJ. J Nucl Med. 2007;48:508–518. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]
- 69.Krenning EP, Kwekkeboom DJ, Bakker WH, Breeman WA, Kooij PP, Oei HY, van Hagen M, Postema PT, de Jong M, Reubi JC, et al. Eur J Nucl Med. 1993;20:716–731. doi: 10.1007/BF00181765. [DOI] [PubMed] [Google Scholar]
- 70.Henze M, Dimitrakopoulou-Strauss A, Milker-Zabel S, Schuhmacher J, Strauss LG, Doll J, Macke HR, Eisenhut M, Debus J, Haberkorn U. J Nucl Med. 2005;46:763–769. [PubMed] [Google Scholar]
- 71.Meisetschlager G, Poethko T, Stahl A, Wolf I, Scheidhauer K, Schottelius M, Herz M, Wester HJ, Schwaiger M. J Nucl Med. 2006;47:566–573. [PubMed] [Google Scholar]
- 72.Anderson CJ, Dehdashti F, Cutler PD, Schwarz SW, Laforest R, Bass LA, Lewis JS, McCarthy DW. J Nucl Med. 2001;42:213–221. [PubMed] [Google Scholar]
- 73.Mottaghy FM, Reske SN. Pituitary. 2006;9:237–242. doi: 10.1007/s11102-006-0269-y. [DOI] [PubMed] [Google Scholar]
- 74.Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS, Macke HR. Eur J Nucl Med. 2000;27:273–282. doi: 10.1007/s002590050034. [DOI] [PubMed] [Google Scholar]
- 75.Heppeler A, Froidevaux S, Eberle AN, Maecke HR. Curr Med Chem. 2000;7:971–994. doi: 10.2174/0929867003374516. [DOI] [PubMed] [Google Scholar]
- 76.Rufini V, Calcagni ML, Baum RP. Semin Nucl Med. 2006;36:228–247. doi: 10.1053/j.semnuclmed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 77.Forrer F, Uusijarvi H, Storch D, Maecke HR, Mueller-Brand J. J Nucl Med. 2005;46:1310–1316. [PubMed] [Google Scholar]
- 78.van Essen M, Krenning EP, Kooij PP, Bakker WH, Feelders RA, de Herder WW, Wolbers JG, Kwekkeboom DJ. J Nucl Med. 2006;47:1599–1606. [PubMed] [Google Scholar]
- 79.de Jong M, Breeman WA, Valkema R, Bernard BF, Krenning EP. J Nucl Med. 2005;46 Suppl 1:13S–17S. [PubMed] [Google Scholar]
- 80.De Jong M, Valkema R, Jamar F, Kvols LK, Kwekkeboom DJ, Breeman WA, Bakker WH, Smith C, Pauwels S, Krenning EP. Semin Nucl Med. 2002;32:133–140. doi: 10.1053/snuc.2002.31027. [DOI] [PubMed] [Google Scholar]
- 81.Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, Obradovich JE, Muzik O, Mangner TJ. Nat Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- 82.Mankoff DA, Shields AF, Krohn KA. Radiol Clin North Am. 2005;43:153–167. doi: 10.1016/j.rcl.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 83.Leyton J, Alao JP, Da Costa M, Stavropoulou AV, Latigo JR, Perumal M, Pillai R, He Q, Atadja P, Lam EW, Workman P, Vigushin DM, Aboagye EO. Cancer Res. 2006;66:7621–7629. doi: 10.1158/0008-5472.CAN-05-3962. [DOI] [PubMed] [Google Scholar]
- 84.McLaughlin F, La Thangue NB. Biochem Pharmacol. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 85.Leyton J, Latigo JR, Perumal M, Dhaliwal H, He Q, Aboagye EO. Cancer Res. 2005;65:4202–4210. doi: 10.1158/0008-5472.CAN-04-4008. [DOI] [PubMed] [Google Scholar]
- 86.Boulikas T, Vougiouka M. Oncol Rep. 2004;11:559–595. [PubMed] [Google Scholar]
- 87.Leyton J, Lockley M, Aerts JL, Baird SK, Aboagye EO, Lemoine NR, McNeish IA. Cancer Res. 2006;66:9178–9185. doi: 10.1158/0008-5472.CAN-06-1539. [DOI] [PubMed] [Google Scholar]
- 88.Waldherr C, Mellinghoff IK, Tran C, Halpern BS, Rozengurt N, Safaei A, Weber WA, Stout D, Satyamurthy N, Barrio J, Phelps ME, Silverman DH, Sawyers CL, Czernin J. J Nucl Med. 2005;46:114–120. [PubMed] [Google Scholar]
- 89.Yang YJ, Ryu JS, Kim SY, Oh SJ, Im KC, Lee H, Lee SW, Cho KJ, Cheon GJ, Moon DH. Eur J Nucl Med Mol Imaging. 2006;33:412–419. doi: 10.1007/s00259-005-0011-4. [DOI] [PubMed] [Google Scholar]
- 90.Yap CS, Czernin J, Fishbein MC, Cameron RB, Schiepers C, Phelps ME, Weber WA. Chest. 2006;129:393–401. doi: 10.1378/chest.129.2.393. [DOI] [PubMed] [Google Scholar]