

Abstract
Backbone cyclization (BC) and N-methylation have been shown to enhance the activity and/or selectivity of biologically active peptides and improve metabolic stability and intestinal permeability. In this study, we describe the synthesis, structure-activity relationship (SAR) and intestinal metabolic stability of a backbone cyclic peptide library, BL3020, based on the linear α-Melanocyte stimulating hormone analog Phe-D-Phe-Arg-Trp-Gly. The drug lead, BL3020−1, selected from the BL3020 library (compound 1) has been shown to inhibit weight gain in mice following oral administration. Another member of the BL3020 library, BL3020−17, showed improved biological activity towards the mMC4R, in comparison to BL3020−1, although neither were selective for MC4R or MC5R. N-methylation, which restrains conformational freedom while increasing metabolic stability beyond that which is imparted by BC, was used to find analogs with increased selectivity. N-methylated backbone cyclic libraries were synthesized based on the BL3020 library. SAR studies showed that all the N-methylated backbone cyclic peptides demonstrated reduced biological activity and selectivity for all the analyzed receptors. N-methylation of active backbone cyclic peptides destabilized the active conformation or stabilized an inactive conformation, rendering the peptides biologically inactive. N-methylation of backbone cyclic peptides maintained stability to degradation by intestinal enzymes.
Keywords: backbone cyclization, N-methylation, Fmoc chemistry, structure activity relationship
INTRODUCTION
It is well recognized that peptides generally lack single receptor selectivity1 and have low bioavailability profiles due to their fast rate of metabolism. Many approaches have been developed to overcome these limitations, among which are backbone cyclization (BC)2 and N-methylation.3-8 These approaches are general methods for conformationally constraining peptides.9,10 BC and cycloscan are general methods by which conformational constraints can be imposed on peptides. BC is formed by covalently connecting atoms in the backbone of a target linear peptide via a linker to form a ring. BC has been shown to dramatically enhance the metabolic stability of peptides in serum and intestinal enzymes.11 A library of backbone cyclic peptides has a basic parent peptide sequence with the same side chain pharmacophores and hence the same potential bioactivity,2,10 but they differ in their ring size and chemistry, conferring conformational diversity. Discrete conformational perturbations, introduced by changing the linker properties, create cyclic peptides that sample the conformational space of the parent peptide. The advantage of BC over other methods of peptide cyclization is that cyclization is performed mainly through backbone atoms without using side chains that are essential for biological activity. This method has been shown to improve the pharmacological selectivity of a given peptide, as demonstrated for substance P8 and somatostatin12 analogs.
N-methylated peptides were shown to have improved pharmacological properties such as lipophilicity,13 proteolytic stability,13,14 bioavailability,15-17 and conformational rigidity.18 They have also shown enhanced potency19-22 and new receptor subtype selectivity.23-25 For example, N-methylated cyclic RGD peptides22 and dermorphin analogs26 have been shown to be more potent following mono N-methylation. Multiple N-Methylation (MNM) is an approach where a library of all possible N-methylated peptide analogs, based on the sequence of a linear or cyclic bioactive peptide, is synthesized and screened to select an active N-methylated analog.17 This approach has led to the discovery of a new selective, metabolically stable, intestinally permeable somatostatin analog,17 and RGD analogs with new receptor selectivity.6 N-methylation and the MNM approach are recognized as useful methods for enhancing selectivity and improving pharmacological properties of peptides.
α-Melanocyte stimulating hormone (αMSH) is part of the melanocortin pathway, which controls the metabolic balance in the body and regulates it.27 This 13-amino acid hormone can bind to five different melanocortin receptors (MC1R-MC5R) which belong to the super family of seven transmembrane-spanning G-protein-coupled receptors. The active site of αMSH possess the same common structural motif, His-Phe-Arg-Trp, for all the five receptors.28-31 It is important to note that αMSH binding to each receptor stimulates different physiological pathways in the body. The MC1R is involved in pigmentation and animal coat coloration.32,33 The MC2R regulates lipogenesis, glucocorticoid, and aldosterone production.34 The MC3R and MC4R have been identified in knockout mice to be involved in feeding behavior, obesity, metabolism, and energy homeostasis.35 MC5R knockout mice showed the role of this receptor in exocrine gland function.36
Backbone cyclic peptides based on the sequence Phe6-D-Phe-Arg-Trp−Gly10 have been subjected to extensive structure-activity relationship (SAR) studies.11 This sequence is an analog of the minimal αMSH sequence that activates the MC4 receptor.31 BL3020−1 (compound 1, Figure 1A) from this library was found to have high intestinal permeability, high stability to intestinal enzymes, and found to have bound and activated the MC4R. Oral administration of compound 1 in mice showed reduced food intake (∼40% in 24 h) and inhibition in body weight gain (over 12 days) in comparison to the control group.11,37 EC50 analysis showed that compound 1 binds and activates the MC4 and MC5 receptors with the same selectivity (EC50 4.0 ± 0.6 nM and 7.0 ± 0.4 nM, respectively).11,37
FIGURE 1.

Structures of compound 1 and general structures of the “N-methylation” sub-libraries (A). The structure of compound 1 (B). General structures of N-Me-1 (C). General structures of N-Me-2 (D). General structures of pre-cyclic N-Me-3 libraries. The arrows indicate the N-methylation position.
Two libraries were synthesized, comprising 37 backbone cyclic (BL3020) and N-methylated backbone cyclic (N-Me) peptides with the same sequence as compound 1 (Phe6-D-Phe-Arg-Trp−Gly10-NH2) and a lactam bridge between the N terminus and the backbone nitrogen of the C-terminal Gly (see Figure 1). The peptides in the BL3020 library differed in ring size and ring chemistry, whereas the peptides in the N-Me libraries differed also in the number and position of the N-Me group. These libraries were screened to select peptide(s) with improved selectivity and activity towards MC4R, while retaining resistance against intestinal enzymes. Such peptide(s) would have the potential to serve as improved antiobesity drug leads, while avoiding adverse effects caused by activation of other MCR subtypes. Nuclear magnetic resonance (NMR)-derived structures of compound 1 were compared with the homology model of αMSH in MC4R,38 which showed a similar orientation of Arg8-Trp9 but not D-Phe6 and Phe7 (Safrai et al., unpublished results), suggesting that the bridge size and N-methylation of D-Phe6 and Phe7 may effect the activity and selectivity. SAR studies were performed by activating mouse melanocortin receptors mMC1R, mMC3R, mMC4R, and mMC5R, and screening for metabolic stability was done using the brush border membrane vesicles (BBMV)36 in vitro model. Peptide BL3020−17 from the BL library showed increased activity towards MC4R, but N-methylation decreased biological activity and selectivity for all the analyzed receptors while maintaining resistance towards enzymatic degradation.
MATERIALS AND METHODS
Chemistry General
Protected amino acids, 9-fluorenylmethyloxycarbonyl-N-hydroxysuccinimide (Fmoc-OSu), bromo-tris-pyrrolidone-phosphonium hexafluorophosphate (PyBrop), Rink amide methylbenzhydrylamine (MBHA) polystyrene resins, and organic reagents and supports for solid phase peptide synthesis (SPPS) were purchased from Nova Biochemicals (Laufelfingen, Switzerland). Bis(trichloromethyl)carbonate (BTC) was purchased from Lancaster (Lancashire, England), trifluoroacetic acid (TFA) and solvents for high performance liquid chromatography (HPLC) were purchased from Bio-Lab (Jerusalem, Israel). Glyoxylic acid, 1,2- diaminoethane, 1,3-diaminopentan, and 1,4-diaminobutane were purchased from Merck (Darmstadt, Germany); tetrakis (triphenylphosphine)palladium (0) was purchased from ACROS (Geel, Belgium). Organic solvents were purchased from Frutarom (Haifa, Israel). NMR spectra were recorded on a Bruker AMX-300 MHz spectrometer. Mass spectra were performed on a Finnigan LCQ DUO ion trap mass spectrometer. Thin layer chromatography (TLC) was performed on Merck F245 60 silica gel plates (Darmstadt, Germany). HPLC analysis was performed using a Vydac analytical RP column (C18, 4.6 × 250 mm, catalog number 201TP54) and were carried out on a Merck-Hitachi L-7100 pump and a Merck-Hitachi L-7400 variable wavelength detector operating at 215 nm. The mobile phase consisted of a gradient system, with solvent A corresponding to water with 0.1% TFA and solvent B corresponding to acetonitrile (ACN) with 0.1% TFA. The mobile phase started with 95% A from 0 to 5 min followed by a linear gradient from 5% B to 95% B from 5 to 55 min. The gradient remained at 95% B for an additional 5 min, and then was dropped to 95% A and 5% B from 60 to 65 min. The gradient remained at 95% A for additional 5 min to achieve column equilibration. The flow rate of the mobile phase was 1 mL/min. Peptide purification was performed by reversed phase HPLC (RP-HPLC) (L-6200A pump, Merck-Hitachi, Japan), using a Vydac preparative RP column (C8, 22 × 250 mm, catalog number 218TP1022). All preparative HPLC runs were carried out using a gradient system with solvent A corresponding to water with 0.1% TFA and solvent B corresponding to ACN with 0.1% TFA.
Solid Phase Peptide Synthesis of N-Methylation Libraries
The synthesis was performed3,11 in a reaction vessel equipped with a sintered glass bottom, following general Fmoc chemistry protocols: Rink amide methylbenzhydrilamine (MBHA) resin (1 g, 0.66 mmol/g) was preswollen in N-methylpyrrolidone (NMP) for 2 h. Fmoc deprotection step was carried out with 20% piperidine in NMP (2 × 15 min), followed by washing with NMP (5 × 2 min) and DCM (2 × 2 min). Couplings of the building unit to the resin and of Fmoc-amino-acid-OH (Fmoc-AA-OH) to the building unit were carried out according to a modified procedure published by Falb et al.39 (3 equiv., 1.98 mmol) and bis-(trichloromethyl) carbonate (BTC, triphosgene) (1 equiv., 0.66 mmol) were suspended in DCM. 2,4,6-trimethylcollidine (10 equiv., 6.6 mmol) was added to the precooled suspension in an ice bath. After all the solids were dissolved (about 1 min), the solution was poured onto the resin and shaken for 3 h at room temperature. This coupling cycle was repeated once more. At the end of the second coupling cycle, the peptidyl-resin was washed with DCM (5 × 2 min). Capping was carried out after anchoring the building unit to the resin and was repeated twice by reacting the peptidyl-resin with a mixture of acetic anhydride (1.1 mL, 0.5M), diisopropylethyl amine (DIEA) (0.5 mL, 0.125M), and N-hydroxybenzotriazole (HOBT) (0.05 g, 0.015M)in dimethyl formamide (DMF) (25 mL). Capping was followed with a resin wash with DMF (5 × 2 min), DCM (2 × 2 min), and NMP (2 × 2 min). Coupling of Fmoc-Trp(BOC)-OH to the building unit was carried out as follows: the Fmoc protecting group was removed from the peptidyl-resin with piperidine in NMP (20%, 2 × 30 min) and the resin was washed (NMP, 5 × 2 min; DCM 5 × 2 min). Fmoc-Trp(BOC)-OH (3 equiv., 1.98 mmol) and BTC (1 equiv., 0.66 mmol) were suspended in dichloromethane and cooled in an ice bath for 30 min. 2,4,6-collidine (10 equiv., 6.6 mmol) was added, and after all the solids were dissolved (1 min), the solution was added to the peptidyl-resin and the vessel was shaken for 3 h at room temperature. Washing steps were carried out with DCM (5 × 2 min). The cycle of Fmoc removal and coupling was followed with Fmoc-DPhe-OH and Fmoc-Phe-OH. All the coupling steps were carried out using BTC as the coupling agent as described earlier. The last amino acid on the peptidyl-resin (Phe) was acylated with 10 equiv. of succinic anhydride in NMP, for 2 h at room temperature, in the presence of 1 equiv. DMAP. The resin was washed with NMP (2 × 5 min) and DCM (2 × 5 min), dried overnight in a desiccator, and removal of the Alloc protecting group from the building unit was performed with tetrakis(triphenylphosphine)Pd(0) (0.1 equiv., 0.066 mmol) in DCM containing acetic acid (5%) and N-methyl morpholin (2.5%) under argon. This step was carried out for 4 h with vigorous shaking in the dark. Washing steps were carried out with chloroform (8 × 2 min) and NMP with 0.5% DIEA (3 × 2 min). After Alloc deprotection, the peptide was cyclized by adding 6 equiv. PyBoP and 12 equiv. DIEA in NMP for 24 h (repeated twice) and then with 3 equiv. PyBoP and 6 equiv. DIEA in NMP. Washing steps were carried out with NMP (5 × 2 min) and DCM (5 × 2 min). The peptidyl-resin was dried under vacuum over night. Cleavage from the resin and removal of side chain protecting groups were carried out simultaneously using a precooled mixture of 95% TFA, 2.5% TDW, and 2.5% triisopropylsilane (TIS). After the resin was added, the mixture was agitated for 30 min in an ice bath, and then was shaken for 2.5 h at room temperature. The combined TFA filtrates were evaporated to dryness by a stream of nitrogen. The oily residue was triturated three times with cold ether to remove the scavengers, and the ether was removed by centrifugation. The dry crude peptide was dissolved in ACN/H2O (1:1) and lyophilized.
Functional Bioassay
HEK-293 cells stably expressing the melanocortin receptors were transfected with 4 μg of CRE/β-galactosidase reporter gene, as previously described.29 Briefly, 5000−15,000 post-transfection cells were plated into 96-well Primera plates (Falcon) and incubated overnight. At 48 h post-transfection, the cells were stimulated with 100 μL of peptide (10−4 to 10−12M) or forskolin (10−4M) control in assay medium (DMEM containing 0.1 mg/mL BSA and 0.1 mM isobutylmethylxanthine) for 6 h. The assay media was aspirated and 50 μL of lysis buffer (250 mM Tris-HCl, pH 8.0, and 0.1% Triton X-100) was added. The plates were stored at −80°C overnight. The plates containing the cell lysates were thawed the following day. Aliquots of 10 μL were taken from each well and transferred to another 96-well plate for relative protein determination. To the cell lysate plates, 40 μL of phosphate-buffered saline with 0.5% BSA was added to each well. Subsequently, 150 μL substrate buffer (60 mM sodium phosphate, 1 mM MgCl2, 10 mM KCl, 5 mM β-mercaptoethanol, and 200 mg ONPG) was added to each well, and the plates were incubated at 37°C. The sample absorbance, OD 405 nm, was measured using a 96-well plate reader (Molecular Devices). The relative protein was determined by adding 200 μL of 1:5 dilution BioRad G250 protein dye/water to the 10 μL cell lysate sample taken previously, and the OD 595 nm was measured on a 96-well plate reader (Molecular Devices). Data points were normalized both to the relative protein content and to the nonreceptor-dependent forskolin stimulation.
Preparation of Brush Border Membrane
Vesicles
BBMVs were prepared from combined duodenum, jejunum, and upper ileum by a Ca2+ precipitation method.40-42 The intestines of five male Wistar rats, 200−250 g, were rinsed with ice cold 0.9% NaCl solution and freed of mucus; the mucosa was scraped off the luminal surface with glass slides and put immediately into buffer containing 50 nM KCl and 10 mM Tris-HCl (pH 7.5, 4°C) and then homogenated (Polytron PT 1200, Kinematica AG, Switzerland). CaCl2 was added to a final concentration of 10 mM. The homogenate was left shaking for 30 min at 4°C and then centrifuged at 10,000g for 10 min. The supernatant was then centrifuged at 48,000g for 30 min, and an additional two purification steps were undertaken by suspending the pellet in 300 mM mannitol and 10 mM Hepes/Tris (pH 7.5) and centrifuging at 24,000g/h. Purification of brush border membranes was assayed using the brush border membrane enzyme markers γ-glutamyl transpeptidase (GGT), leucine amino peptidase (LAP), and alkaline phosphatase. During the course of these studies, enrichment in brush border membrane enzymes varied between 13- and 18-fold. The tested molecule was mixed with purified BBMVs and incubated in 37°C for 90 min. Duplicate samples were taken at time 0 and after 15, 30, 45, 60, and 90 min. The samples were diluted 1:1 with ice-cold ACN, centrifuged (7500g, 10 min, 4°C), and sent to HPLC analysis.
RESULTS
Backbone cyclic and N-methylated backbone cyclic pentapeptides were synthesized and their biological activities towards mouse MC1R, MC3R, MC4R, and MC5R were determined.
Chemical Synthesis and Characterization
Two libraries of backbone cyclic and N-methylated backbone cyclic pentapeptides were synthesized, designated BL3020 and N-Me. The latter was further divided into three sub-libraries, N-Me-1, N-Me-2, and N-Me-3 according to the position of N-methylation. All BL3020 peptides contain the parent sequence Phe6-D-Phe-Arg-Trp-Gly10-NH2 in which a lactam bridge connects the N-terminus to the Nα of the C-terminal Gly, resembling the ring position of the model compound 111,37 (Figure 1A and Table I). All the peptides differ in the length of the alkyl chains, the position of the lactam bridge in the ring and ring chemistry. The position of the N-methylation in the N-Me-1 sub-library was N-methyl Phe6 (Figure 1B and Table II); All the peptides in the N-Me-2 sub-library, had both N-methyl Phe6 and N-methyl D-Phe7 (Figure 1C and Table III); and the N-Me-3 sub-library was precyclic analogs of peptides from the N-Me-2 sub-library (Figure 1D and Table IV). All the peptides were synthesized on Rink amide MBHA resin by the Fmoc chemistry as described by Hess et al.11 N-methylation was performed on resin by the method described by Biron et al.3 The cyclization was performed on-resin after removing the Alloc protecting group from the Nα (ω-aminoalkyl)Gly building unit. The peptides of the N-Me-3 sub-library were the same peptides as those of the N-Me-2 sub-library, except that the cyclization step was not performed in the N-Me-3 library, and the peptides were removed from the resin after Alloc deprotection (see Figure 1). All peptides were purified to homogeneity using preparative RP-HPLC. The purity and characterization of these peptides was assessed by both mass spectrometry and analytical RP-HPLC.
Table I.
Structure of the BL3020 Library
| Peptide Name | Ring Size | m | n | Sequence |
|---|---|---|---|---|
| Compound 1a | 20 | 2 | 2 | c(Phe(C2)-D-Phe-Arg-Trp-Gly(N2)NH2) |
| BL3020−4a | 21 | 3 | 2 | c(Phe(C3)-D-Phe-Arg-Trp-Gly(N2)NH2) |
| BL3020−6 | 22 | 3 | 3 | c(Phe(C3)-D-Phe-Arg-Trp-Gly(N3)NH2) |
| BL3020−7 | 24 | 5 | 3 | c(Phe(C5)-D-Phe-Arg-Trp-Gly(N3)NH2) |
| BL3020−8a | 21 | 2 | 3 | c(Phe(C2)-D-Phe-Arg-Trp-Gly(N3)NH2) |
| BL3020−9 | 23 | 5 | 2 | c(Phe(C5)-D-Phe-Arg-Trp-Gly(N2)NH2) |
| BL3020−10 | 22 | 4 | 2 | c(Phe(C4)-D-Phe-Arg-Trp-Gly(N2)NH2) |
| BL3020−11 | 22 | 2 | 4 | c(Phe(C2)-D-Phe-Arg-Trp-Gly(N4)NH2) |
| BL3020−12 | 23 | 3 | 4 | c(Phe(C3)-D-Phe-Arg-Trp-Gly(N4)NH2) |
| BL3020−13 | 24 | 4 | 4 | c(Phe(C4)-D-Phe-Arg-Trp-Gly(N4)NH2) |
| BL3020−14 | 25 | 5 | 4 | c(Phe(C5)-D-Phe-Arg-Trp-Gly(N4)NH2) |
| BL3020−15a | 23 | 4 | 3 | c(Phe(C4)-D-Phe-Arg-Trp-Gly(N3)NH2) |
| BL3020−16a | 21 | a | 2 | A |
| BL3020−17 | 21 | b | 2 | B |
The dicarboxylic acyl spacer (m) was replaced by o-phthaloyl spacer. Hess et al.11
The dicarboxylic acyl spacer (m) was replaced by dimethylglutaric acid spacer. An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.0 mL/min. The peptide purity (>95%) was determined by HPLC at a wavelength of 220 nm. Gly(Nx) represents Nα (ω–amino(alkyl)x)Gly.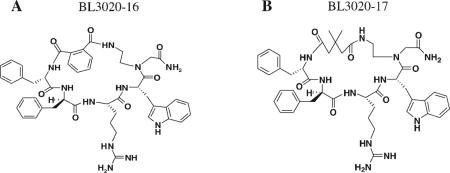
Table II.
Structure of the N-Me-1 Sub-Library
| Peptide Name | Ring Size | m | n | Origin |
|---|---|---|---|---|
| N-Me-1−1 | 20 | 2 | 2 | BL3020−1 |
| N-Me-1−2 | 21 | 3 | 2 | BL3020−4 |
| N-Me-1−3 | 21 | a | 2 | BL3020−16 |
| N-Me-1−4 | 22 | 4 | 2 | BL3020−10 |
| N-Me-1−6 | 21 | b | 2 | BL3020−17 |
The dicarboxylic acyl spacer (m) was replaced by o-phthaloyl spacer.
The dicarboxylic acyl spacer (m) was replaced by dimethylglutaric acid spacer. An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.0 mL/min. The peptide purity (>95%) was determined by HPLC at a wavelength of 220 nm.
Table III.
Structure of the N-Me-2 Sub-Library
| Peptide Name | Ring Size | m | n | Origin |
|---|---|---|---|---|
| N-Me-2−1 | 20 | 2 | 2 | BL3020−1 |
| N-Me-2−2 | 21 | 3 | 2 | BL3020−4 |
| N-Me-2−4 | 22 | 4 | 2 | BL3020−10 |
| N-Me-2−5 | 23 | 5 | 2 | BL3020−9 |
| N-Me-2−6 | 21 | a | 2 | BL3020−17 |
| N-Me-2−7 | 21 | 2 | 3 | BL3020−8 |
| N-Me-2−8 | 22 | 3 | 3 | BL3020−6 |
| N-Me-2−9 | 22 | b | 3 | BL3020−16c |
| N-Me-2−10 | 22 | 4 | 3 | BL3020−15 |
| N-Me-2−11 | 23 | 5 | 3 | BL3020−7 |
| N-Me-2−12 | 23 | b | 3 | BL3020−17c |
| N-Me-2−13 | 22 | 2 | 4 | BL3020−11 |
| N-Me-2−14 | 23 | 3 | 4 | BL3020−12 |
| N-Me-2−15 | 23 | b | 4 | BL3020−16c |
| N-Me-2−16 | 24 | 4 | 4 | BL3020−13 |
| N-Me-2−17 | 25 | 5 | 4 | BL3020−14 |
| N-Me-2−18 | 26 | b | 4 | BL3020−17c |
The dicarboxylic acyl spacer (m) was replaced by dimethylglutaric acid spacer. An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.0 mL/min. The peptide purity (>95%) was determined by HPLC at a wavelength of 220 nm.
The dicarboxylic acyl spacer (m) was replaced by o-phthaloyl spacer.
There are differences from the structures at Table I on the ring size and N-methylation position.
Table IV.
Structure of the Pre-Cyclic N-Me-3 Sub-Library
| Peptide Name | Ring Size | m | n | Origin |
|---|---|---|---|---|
| N-Me-3−1 | – | 2 | 2 | BL3020−1 |
| N-Me-3−3 | – | a | 2 | BL3020−16 |
| N-Me-3−4 | – | 4 | 2 | BL3020−10 |
| N-Me-3−10 | – | 4 | 3 | BL3020−15 |
| N-Me-3−11 | – | 5 | 3 | BL3020−7 |
| N-Me-3−16 | – | 4 | 4 | BL3020−13 |
The dicarboxylic acyl spacer (m) was replaced by o-phthaloyl spacer. An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.0 mL/min. The peptide purity (>95%) was determined by HPLC at a wavelength of 220 nm.
Biological Evaluation
Tables VI-VIII summarize the biological activity at the mouse melanocortin receptors, mMC1R, mMC3R, mMC4R, and mMC5R of the 37 peptides. Previously, we have demonstrated that compound 1 selectively activates the MC4R and the MC5R in the low nanomolar range (EC50 = 4.0 and 7.0 nM, respectively) which indicates agonist¼ activity, whereas the control peptides NDP-MSH showed very high affinity agonist activity towards all the analyzed receptors with no selectivity.11 Figure 2 summarizes the EC50 values of a sample of BL3020 peptides towards mMC4R. Figures 3A-3C summarizes the EC50 values of a sample of peptides from each of the three N-methylation sub-libraries towards mMC4R. Figure 3D demonstrates the EC50 values of all N-Me-1 peptides towards mMC3R.
Table VI.
Functional Activity of N-Me-1 Library at the Mouse Melanocortin Receptors
| Ref | mMC1R (nM) | mMC3R (nM) | mMC4R (nM) | mMC5R (nM) |
|---|---|---|---|---|
| NDP-MSH | 0.0081 ± 0.001 | 0.063 ± 0.004 | 0.097 ± 0.016 | 0.078 ± 0.0084 |
| Compound 1 | 64 ± 14 | 770 ± 175 | 4.0 ± 0.6 | 7.0 ± 0.40 |
| N-Me-1−1 | 720 ± 397 | >100,000 | 100 ± 47 | 200 ± 35 |
| N-Me-1−2 | 743 ± 210 | >100,000 | 175 ± 96 | 392 ± 89 |
| N-Me-1−3 | 930 ± 156 | >100,000 | 96 ± 46 | 69 ± 13 |
| N-Me-1−4 | 12,680 ± 9150 | >100,000 | 1913 ± 878 | 820 ± 132 |
| N-Me-1−6 | 1490 ± 367 | >100,000 | 291 ± 127 | 1610 ± 727 |
Table VIII.
Functional Activity of N-Me-3 Library at the Mouse Melanocortin Receptors
| Ref | mMC1R (nM) | mMC3R (nM) | mMC4R (nM) | mMC5R (nM) |
|---|---|---|---|---|
| NDP-MSH | 0.0081 ± 0.001 | 0.063 ± 0.004 | 0.097 ± 0.016 | 0.078 ± 0.0084 |
| Compound 1 | 64 ± 14 | 770 ± 175 | 4.0 ± 0.6 | 7.0 ± 0.40 |
| N-Me-3−1 | 1350 ± 240 | >100,000 | 613 ± 98 | 560 ± 75 |
| N-Me-3−3 | 5090 ± 680 | >100,000 | 1350 ± 270 | 810 ± 164 |
| N-Me-3−4 | 6790 ± 1030 | >100,000 | 1790 ± 880 | 782 ± 99 |
| N-Me-3−10 | 19,700 ± 3670 | >100,000 | 1498 ± 370 | 849 ± 320 |
| N-Me-3−11 | 36,700 ± 22,900 | >100,000 | 1320 ± 450 | 440 ± 69 |
| N-Me-3−16 | 6030 ± 2080 | >100,000 | 336 ± 59 | 290 ± 52 |
FIGURE 2.

Illustration of the chosen BL3020 peptides possessing partial agonist pharmacology at the mouse MC4R (A). BL3020−6, 7, 10, 11, and 13 agonist pharmacology at the mouse MC4R (B). BL3020−4, 8, 14, 15, and 17 agonist pharmacology at the mouse MC4R. The peptides NDP and compound 1 are included as controls to illustrate the maximal response observed for full agonists using this assay. HEK-293 cell in 96-well plate stably expressing the melanocortin receptors were incubated with the tested molecules overnight. The sample absorbance, OD 405 nm, was measured using a 96-well plate reader (Molecular Devices).
FIGURE 3.

Illustration of the chosen N-methylation peptides possessing partial agonist pharmacology at the mouse MC3R and agonist pharmacology at the mouse MC4R. (A) N-Me-1 library agonist pharmacology at the mouse MC4R. (B) N-Me-2 library agonist pharmacology at the mouse MC4R. (C) N-Me-3 library agonist pharmacology at the mouse MC4R. (D). N-Me-2 library agonist pharmacology at the mouse MC3R. The peptides NDP and compound 1 are included as controls to illustrate the maximal response observed for full agonists using this assay. HEK-293 cell in 96-well plate stably expressing the melanocortin receptors were incubated with the tested molecules overnight. The sample absorbance, OD 405 nm, was measured using a 96-well plate reader (Molecular Devices).
Mouse Melanocortin-1 Receptor
The peripheral skin melanocortin receptor, MC1R, is involved in human skin pigmentation32,43 and animal coat coloration.33 Compound 1 has been shown to possess low stimulatory activity at the mMC1R (EC50 = 64 nM).11 The results of the current study showed that the stimulatory activity at the mMC1R for BL3020 peptides was between 28 nM for BL3020−17 to 3.3 μM for BL3020−11. Thus, the biological activity at the mMC1R of BL3020−17 was increased by 2.2 fold, while that of BL3020−11 was decreased by 51 fold, in comparison to compound 1 (Table V). The biological activity of the N-Me-1 peptides was between 720 nM for N-Me-1−1 to 12.68 μM for N-Me-1−4, which is 11- to 198-fold decreased potency, respectively, in comparison to compound 1 (Table VI). The N-Me-2 peptides showed stimulatory activity at the mMC1R between 1.05 μM for N-Me-2−5 to 8.67 μM for N-Me-2−8, which is 15.6- to 135-fold decreased potency, respectively, in comparison to compound 1 (Table VII). N-Me-3 peptides stimulated the mMC1R at 1.35 μM for N-Me-3−1 to 36.7 μM for N-Me-3−11, which is a 21- to 573-fold decrease in potency, respectively, in comparison to compound 1 (Table VIII). These results showed that the stimulatory activity at the mMC1R for all analyzed peptides was reduced in comparison to that found for compound 1, except for two peptides, BL3020−8 and BL3020−17 (Table V). Specifically, BL3020−17 showed the highest biological activity at the mMC1R, which was 2.2-fold more than that of compound 1.
Table V.
Functional Activity of BL3020 Library at the Mouse Melanocortin Receptors
| Refa | mMC1R (nM) | mMC3R (nM) | mMC4R (nM) | mMC5R (nM) |
|---|---|---|---|---|
| NDP-MSH | 0.0081 ± 0.001 | 0.063 ± 0.004 | 0.097 ± 0.016 | 0.078 ± 0.0084 |
| Compound 1a | 64 ± 14 | 770 ± 175 | 4.0 ± 0.6 | 7.0 ± 0.4 |
| BL3020−4a | 120 ± 28 | 1460 ± 145 | 18 ± 4 | 20 ± 4 |
| BL3020−6 | 590 ± 170 | >100,000 | 29 ± 3 | 51 ± 17 |
| BL3020−7 | 1600 ± 700 | 12,000 ± 2600 | 6.4 ± 1.0 | 73 ± 13 |
| BL3020−8a | 28 ± 12 | 1260 ± 385 | 4 ± 2 | 2 ± 1 |
| BL3020−10 | 740 ± 120 | 11,500 ± 5200 | 3706 ± 4 | 92 ± 6 |
| BL3020−11 | 3300 ± 2200 | >100,000 | 35 ± 6 | 113 ± 15 |
| BL3020−12 | 270 ± 84 | 5530 ± 300 | 17 ± 8 | 25 ± 1 |
| BL3020−13 | 590 ± 77 | 7900 ± 1940 | 9.5 ± 2.0 | 50 ± 14 |
| BL3020−14 | 340 ± 137 | 33 ± 17 | 9.0 ± 0.4 | 16 ± 4 |
| BL3020−15a | 240 ± 41 | 7750 ± 1960 | 5.0 ± 0.1 | 26 ± 17 |
| BL3020−16a | 550 ± 97 | 20,500 ± 2100 | 150 ± 22 | 390 ± 120 |
| BL3020−17 | 28 ± 5 | 840 ± 150 | 1.8 ± 0.1 | 4 ± 1 |
Hess et al.11
Table VII.
Functional Activity of N-Me-2 Library at the Mouse Melanocortin Receptors
| Ref | mMC1R (nM) | mMC3R (nM) | mMC4R (nM) | mMC5R (nM) |
|---|---|---|---|---|
| NDP-MSH | 0.0081 ± 0.001 | 0.063 ± 0.004 | 0.097 ± 0.016 | 0.078 ± 0.0084 |
| Compound 1 | 64 ± 14 | 770 ± 175 | 4.0 ± 0.6 | 7.0 ± 0.40 |
| N-Me-2−1 | 2510 ± 610 | >100,000 | 4900 ± 1800 | 4540 ± 1400 |
| N-Me-2−2 | 4100 ± 190 | >100,000 | 2500 ± 330 | 1430 ± 565 |
| N-Me-2−4 | 1196 ± 670 | >100,000 | 2877 ± 659 | 2960 ± 995 |
| N-Me-2−5 | 1049 ± 312 | >100,000 | 2416 ± 498 | 7990 ± 460 |
| N-Me-2−6 | 4900 ± 1030 | >100,000 | 4350 ± 385 | 7010 ± 2900 |
| N-Me-2−7 | 8220 ± 7500 | >100,000 | 2710 ± 520 | 3500 ± 1125 |
| N-Me-2−8 | 8670 ± 3340 | >100,000 | 860 ± 110 | 900 ± 280 |
| N-Me-2−9 | 6167 ± 2687 | >100,000 | 872 ± 379 | 189 ± 28 |
| N-Me-2−10 | 8175 ± 1970 | >100,000 | 2493 ± 479 | 2115 ± 392 |
| N-Me-2−11 | 6000 ± 4200 | 10,380 ± 2270 | 1050 ± 33 | 1160 ± 365 |
| N-Me-2−12 | 8070 ± 2900 | >100,000 | 4900 ± 3200 | 2420 ± 470 |
| N-Me-2−13 | 4920 ± 1900 | >100,000 | 1030 ± 109 | 2020 ± 350 |
| N-Me-2−14 | 3440 ± 2890 | >100,000 | 498 ± 67 | 740 ± 110 |
| N-Me-2−15 | 2478 ± 1566 | >100,000 | 1416 ± 922 | 605 ± 125 |
| N-Me-2−16 | 5410 ± 2092 | >100,000 | 1457 ± 731 | 1034 ± 129 |
| N-Me-2−17 | 2340 ± 559 | >100,000 | 1552 ± 845 | 1297 ± 100 |
| N-Me-2−18 | 8440 ± 4495 | >100,000 | 1850 ± 97 | 4590 ± 930 |
Mouse Melanocortin-3 Receptor
The MC3R is expressed both peripherally and centrally and appears to be involved in metabolism and energy homeostasis.43,44 Compound 1 has been shown to have an EC50 value of 770 nM at the mMC3R.11 The results of the current study showed that the stimulatory activity at the mMC3R for BL3020 peptides was between 33.0 nM for BL3020−14 to over 100 μM for BL3020−6 and BL3020−11. Thus, the biological activity at the mMC3R of BL3020−14 was increased by 23.3-fold, whereas those of BL3020−6 and BL3020−11 were fully depressed in comparison to compound 1 (Table V). N-methylation libraries results showed that all the peptides were devoid of any potency at the mMC3R (Tables VII-9 and Figure 3D), except for N-Me-2−11. This peptide stimulated the mMC3R at 10.38 μM, which is 13.5-fold decreased potency in comparison to compound 1 (Table VII). The N-Me-2 library differed from the other two libraries in N-methylamino acids on Phe6 and D-Phe7. All the peptides showed less potency to MC3R, in comparison to compound 1, except for BL3020−14. It is interesting to note that all the peptides that had EC50 values above 100 μM (Tables VII-9 and Figure 3D) had no agonist activity, which indicates a potential role as antagonists at the mMC3R.
Mouse Melanocortin-4 Receptor
The central MC4R has been identified as physiologically participating in food consumption45 and obesity in mice46 with several polymorphisms of the MC4R observed in obese humans.47-49 Compound 1 was shown to have an EC50 value of 4.0 nM at the mMC4R.11 BL3020−17 (Table I) stimulated the mMC4R with an EC50 of 1.8 nM, a 2.2-fold increase in potency compared to that found for compound 1, whereas BL3020−10 stimulated the mMC4R with an EC50 of 3.7 μM, a 925-fold decrease potency in comparison to compound 1 (Figure 2 and Table V). Among the N-Me-1 peptides, N-Me-1−3 stimulated the mMC4R with an EC50 of 96.0 nM, which is 24-fold decreased potency when compared with that found for compound 1 (Figure 3A and Table VI). N-Me-1−4 stimulated the mMC4R with an EC50 of 1.91 μM, which is 478-fold decreased potency when compared with compound 1 (Figure 3A and Table VI). The other peptides of this library showed EC50 values within this range (Figure 3A and Table VI). N-Me-2 peptides (N-Me-2−1 to N-Me-2−18) showed stimulatory activity at the mMC4R between 498 μM for N-Me-2−14 to 4.9 μM for N-Me-2−1 and N-Me-2−12, which is 124- to 1225-fold decreased potency, respectively, in comparison to compound 1 (Figure 3B, Table VII). N-Me-3 peptides stimulated the mMC4R at 336 nM for N-Me-3−16 to 1.79 μM for N-Me-3−4, which is 84- to 447-fold decreased potency, respectively, in comparison to compound 1 (Figure 3C, Table VIII). The stimulatory activity at the mMC4R for all analyzed peptides was reduced in comparison to that found for compound 1, except for BL3020−17. However, although this peptide showed higher biological activity at the mMC4R when compared with compound 1, it was not selective for this receptor.
Mouse Melanocortin-5 Receptor
The peripheral MC5R is expressed in a variety of tissues and has been implicated as physiologically participating in the role of exocrine gland function.34,36,50 The agonist activity of compound 1 at the mMC5R was shown to have an EC50 of 7.0 nM.11 The results of the current study showed that the stimulatory activity at the mMC5R for BL3020 peptides was between 2 nM for BL3020−8 to 390 nM for BL3020−16 (Table I). The biological activity at the mMC5R of BL3020−8 was increased by 3.5-fold, while that of BL3020−16 was decreased 56-fold, in comparison to compound 1 (Table V). The N-Me-1 peptides were between 69.0 nM for N-Me-1−3 to 1.61 μM for N-Me-1−6, which is 9.8- to 230-fold decreased potency, respectively, in comparison to compound 1 (Table VI). N-Me-2 peptides showed stimulatory activity at the mMC5R between 189 μM for N-Me-2−9 to 8.0 μM for N-Me-2−5, which is 27- to 1141-fold decreased potency, respectively, in comparison to compound 1 (Table VII). N-Me-3 peptides stimulated the mMC5R at 290 nM for N-Me-3−16 to 849 nM for N-Me-3−10, which is 21- to 121-fold decreased potency, respectively, in comparison to compound 1 (Table VIII). The stimulatory activity at the mMC5R for all analyzed peptides was reduced in comparison to that found for compound 1, except for BL3020−8 and BL3020−17.
Metabolic Stability
Metabolic stability was studied with BBMV.36 The stability of the N-methylated analogs N-Me-1−3 and N-Me-2−8 was compared to that of compound 1 and the precyclic analog N-Me-3−4. The cyclic N-methylated analogs had a 100% recovery after 90 min, and the precyclic N-Me-3−4 was found to be less stable to intestinal enzymes, with a 30% reduced concentration after 90 min (see Figure 4).
FIGURE 4.

Metabolic stability in brush border membrane vesicles (BBMV), (■) N-Me-3−4 (pre-cyclic), (▼) N-Me-1−3 (•) N-Me-2−8, and (◆) Compound 1.
DISCUSSION
Backbone cyclic peptide library (BL3020), and N-methylated (N-Me) libraries, were synthesized and their activity determined. This is the first case in which BC and the MNM approaches were combined. Previously, the MNM approach has been applied to end-to-end cyclic peptides.6,17 In such MNM library, the diversity is only in the position and number of the N-Me groups. In the BC-MNM approach, there are two types of libraries: Type I stems from a previously screened BC library where an active peptide is selected and used for the MNM library. In this case, the diversity is the same as in the end-to-end MNM cyclic peptide library. In the second library, type II, the diversities of both MNM and BC are combined, such that each N-methylated peptide is the basis of a library that has ring size and chemistry diversity. Obviously, the size of the library of type II is much bigger than the size of the library of type I. Here we describe the synthesis and SAR of the peptides in the BC-MNM library, based on the αMSH stem peptide, with type II diversity.
All the peptides in both the BC and the BC-MNM libraries had the same primary sequence: Phe-D-Phe-Arg-Trp-Gly-NH2. They also had the same ring position. The N-Me library was further divided into three sub-libraries. Although peptides in the BL3020 library differed in ring size and chemistry, the peptides in the N-Me library differed in these two diversity parameters and also in their N-methylation. The N-Me-1 sub-library contains N-Me-Phe6; and the N-Me-2 sub-library contain both N-Me-Phe6 and N-Me-D-Phe7; and the N-Me-3 sub-library comprised precyclic peptides with the same modifications as the N-Me-2 peptides but not cyclic (see Figure 1).
The SAR studies demonstrated significant difference between the two main libraries, where several of the BL3020 peptides had enhanced biological activity towards to the different mMCRs when compared with compound 1, whereas all the peptides in the N-Me libraries had significantly reduced biological activity to relative to compound 1. BL3020−17 (with a ring size of 21) and BL3020−14 (with a ring size of 25) stimulated the mMCRs at lower concentrations than compound 1. These results indicate that the ring size has a significant effect on their biological activity on the mMCRs. The results of the peptides from the N-Me libraries showed that none of them stimulated mMCRs at concentrations below those of compound 1. N-Me-1−6, based on BL3020−17, and N-Me-2−17, based on BL3020−14, stimulated the mMC4R at significantly higher concentrations than compound 1. BL3020−17 and N-Me-1−6 have the same ring size and ring chemistry: The only difference between them is the N-methylated amino acid on Phe6, showing that N-methylation impaired the biological activity towards mMC4R and that the active conformation is probably destabilized by N-methylation at this position.
Ring size, ring chemistry, and N-methylation also dramatically effected receptor selectivity. It is important to note that the BL3020 peptides showed both mMCR4 and mMC5R activity, indicating that similar bioactive conformations activate these receptors.
The lack of biological activity due to N-methylation is in contrast to findings of Biron et al.17 and Chatterjee and co-worker.4 Where N-methylation was shown to increase bioavailability and in several cases also the interaction of the drug with its targets, and hence its overall activity. Here, N-methylation reduced biological activity, probably by changing the active conformation and/or by increasing the rigidity of the molecule. These changes may prevent hydrogen bonding that stabilizes the peptide in the active conformation (Safrai et al., unpublished results) and thus can prevent the interaction with the receptor.
The backbone cyclic peptides (with or without N-methylation) maintain their metabolic stability: Our results demonstrate that N-methylation does not affect the intestinal stability of the backbone peptides. BC stabilization of the peptides was demonstrated as the N-methylation precyclic analog was found to be more susceptible to degradation by the intestinal enzymes than the cyclic analog.
CONCLUSIONS
The sequence Phe6-D-Phe-Arg-Trp-Gly10-NH2 derived from αMSH was the basis of two libraries: (a) An amino end to N-backbone cyclic ring size and ring chemistry library (the BL3020 library), and (b) An extensive, N-methylated BC-MNM library based on the BC library. A previously discovered BC peptide (compound 1) that had MC4R and MC5R selectivity and oral in vivo activity was used as a lead. Screening these libraries for activation of various melanocortin receptors revealed that some peptides in the BL3020 library have improved activity and selectivity, when compared with compound 1, whereas the peptides in the BC-MNM library had reduced activity. N-methylation at position 6 and 7 render the BC peptide inactive. Peptides of the BC-MNM maintained their intestinal metabolic stability when compared with their precyclic analogs.
Acknowledgments
This work is part of the Ph.D. dissertations of Yaniv Linde and Oded Ovadia.
Contract grant sponsor: US-Israeli Binational Science Foundation (BSF)
Contract grant number: 2005-193
Contract grant sponsor: NIH
Contract grant number: DK57080
Footnotes
Publisher's Disclaimer: This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
REFERENCES
- 1.Okumu FW, Pauletti GM, Vander Velde DG, Siahaan TJ, Borchardt RT. Pharm Res. 1997;14:169–175. doi: 10.1023/a:1012092409216. [DOI] [PubMed] [Google Scholar]
- 2.Gilon C, Halle D, Chorev M, Selinger Z, Byk G. Biopolymers. 1991;31:745–750. doi: 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]
- 3.Biron E, Kessler H. J Org Chem. 2005;70:5183–5189. doi: 10.1021/jo050477z. [DOI] [PubMed] [Google Scholar]
- 4.Biron E, Chatterjee J, Kessler H. J Pept Sci. 2006;12:213–219. doi: 10.1002/psc.711. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee J, Mierke D, Kessler H. J Am Chem Soc. 2006;128:15164–15172. doi: 10.1021/ja063123d. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee J, Ovadia O, Zahn G, Marinelli L, Hoffman A, Gilon C, Kessler H. J Med Chem. 2007;50:5878–5881. doi: 10.1021/jm701044r. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee J, Mierke DF, Kessler H. Chemistry. 2008;14:1508–1517. doi: 10.1002/chem.200701029. [DOI] [PubMed] [Google Scholar]
- 8.Byk G, Halle D, Zeltser I, Bitan G, Selinger Z, Gilon C. J Med Chem. 1996;39:3174–3178. doi: 10.1021/jm960154i. [DOI] [PubMed] [Google Scholar]
- 9.Friedler A, Zakai N, Karni O, Broder YC, Baraz L, Kotler M, Loyter A, Gilon C. Biochemistry. 1998;37:5616–5622. doi: 10.1021/bi972878h. [DOI] [PubMed] [Google Scholar]
- 10.Hess S, Ovadia O, Shalev DE, Senderovich H, Qadri B, Yehezkel T, Salitra Y, Sheynis T, Jelinek R, Gilon C, Hoffman A. J Med Chem. 2007;50:6201–6211. doi: 10.1021/jm070836d. [DOI] [PubMed] [Google Scholar]
- 11.Hess S, Linde Y, Ovadia O, Safrai E, Shalev DE, Swed A, Halbfinger E, Lapidot T, Winkler I, Gabinet Y, Faier A, Yarden D, Xiang Z, Portillo FP, Haskell-Luevano C, Gilon C, Hoffman A. J Med Chem. 2008;51:1026–1034. doi: 10.1021/jm701093y. [DOI] [PubMed] [Google Scholar]
- 12.Afargan M, Janson ET, Gelerman G, Rosenfeld R, Ziv O, Karpov O, Wolf A, Bracha M, Shohat D, Liapakis G, Gilon C, Hoffman A, Stephensky D, Oberg K. Endocrinology. 2001;142:477–486. doi: 10.1210/endo.142.1.7880. [DOI] [PubMed] [Google Scholar]
- 13.Cody WL, He JX, Reily MD, Haleen SJ, Walker DM, Reyner EL, Stewart BH, Doherty AM. J Med Chem. 1997;40:2228–2240. doi: 10.1021/jm970161m. [DOI] [PubMed] [Google Scholar]
- 14.Haviv F, Fitzpatrick TD, Swenson RE, Nichols CJ, Mort NA, Bush EN, Diaz G, Bammert G, Nguyen A, Rhutasel NS. J Med Chem. 1993;36:363–369. doi: 10.1021/jm00055a007. [DOI] [PubMed] [Google Scholar]
- 15.Ali FE, Bennett DB, Calvo RR, Elliott JD, Hwang SM, Ku TW, Lago MA, Nichols AJ, Romoff TT, Shah DH. J Med Chem. 1994;37:769–780. doi: 10.1021/jm00032a009. [DOI] [PubMed] [Google Scholar]
- 16.Mazur RH, James PA, Tyner DA, Hallinan EA, Sanner JH, Schulze R. J Med Chem. 1980;23:758–763. doi: 10.1021/jm00181a012. [DOI] [PubMed] [Google Scholar]
- 17.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Angew Chem Int Ed Engl. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 18.Vitoux B, Aubry A, Cung MT, Boussard G, Marraud M. Int J Pept Protein Res. 1981;17:469–479. doi: 10.1111/j.1399-3011.1981.tb02016.x. [DOI] [PubMed] [Google Scholar]
- 19.Tonelli A. Biopolymers. 1976;15:1615–1622. doi: 10.1002/bip.1976.360150814. [DOI] [PubMed] [Google Scholar]
- 20.Manavalan P, Momany FA. Biopolymers. 1980;19:1943–1973. doi: 10.1002/bip.1980.360191103. [DOI] [PubMed] [Google Scholar]
- 21.Ron D, Gilon C, Hanani M, Vromen A, Selinger Z, Chorev M. J Med Chem. 1992;35:2806–2811. doi: 10.1021/jm00093a013. [DOI] [PubMed] [Google Scholar]
- 22.Dechantsreiter MA, Planker E, Matha B, Lohof E, Holzemann G, Jonczyk A, Goodman SL, Kessler H. J Med Chem. 1999;42:3033–3040. doi: 10.1021/jm970832g. [DOI] [PubMed] [Google Scholar]
- 23.Laufer R, Wormser U, Friedman ZY, Gilon C, Chorev M, Selinger Z. Proc Natl Acad Sci USA. 1985;82:7444–7448. doi: 10.1073/pnas.82.21.7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laufer R, Gilon C, Chorev M, Selinger Z. J Biol Chem. 1986;261:10257–10263. [PubMed] [Google Scholar]
- 25.Rajeswaran WG, Hocart SJ, Murphy WA, Taylor JE, Coy DH. J Med Chem. 2001;44:1416–1421. doi: 10.1021/jm000361p. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt R, Kalman A, Chung NN, Lemieux C, Horvath C, Schiller PW. Int J Pept Protein Res. 1995;46:47–55. doi: 10.1111/j.1399-3011.1995.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 27.Cummings DE, Schwartz MW. Nat Genet. 2000;26:8–9. doi: 10.1038/79223. [DOI] [PubMed] [Google Scholar]
- 28.Haskell-Luevano C, Sawyer TK, Hendrata S, North C, Panahinia L, Stum M, Staples DJ, Castrucci AM, Hadley MF, Hruby VJ. Peptides. 1996;17:995–1002. doi: 10.1016/0196-9781(96)00141-6. [DOI] [PubMed] [Google Scholar]
- 29.Haskell-Luevano C, Lim S, Yuan W, Cone RD, Hruby VJ. Peptides. 2000;21:49–57. doi: 10.1016/s0196-9781(99)00167-9. [DOI] [PubMed] [Google Scholar]
- 30.Haskell-Luevano C, Holder JR, Monck EK, Bauzo RM. J Med Chem. 2001;44:2247–2252. doi: 10.1021/jm010061n. [DOI] [PubMed] [Google Scholar]
- 31.Holder JR, Haskell-Luevano C. Ann NY Acad Sci. 2003;994:36–48. doi: 10.1111/j.1749-6632.2003.tb03160.x. [DOI] [PubMed] [Google Scholar]
- 32.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 33.Lu D, Vage DI, Cone RD. Mol Endocrinol. 1998;12:592–604. doi: 10.1210/mend.12.4.0091. [DOI] [PubMed] [Google Scholar]
- 34.Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C, Kesterson RA. Recent Prog Horm Res. 1996;51:287–317. discussion 318. [PubMed] [Google Scholar]
- 35.Fehm HL, Smolnik R, Kern W, McGregor GP, Bickel U, Born J. J Clin Endocrinol Metab. 2001;86:1144–1148. doi: 10.1210/jcem.86.3.7298. [DOI] [PubMed] [Google Scholar]
- 36.Chen W, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Cell. 1997;91:789–798. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 37.Gilon C, Hoffman A, Linde Y, Hess S. PCT/IL. 2006:00064. [Google Scholar]
- 38.Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP, Mulholland MW, Gantz I, Mosberg HI. Biochemistry. 2005;44:11329–11341. doi: 10.1021/bi0501840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falb E, Yechezkel T, Salitra Y, Gilon C. J Pept Res. 1999;53:507–517. doi: 10.1034/j.1399-3011.1999.00049.x. [DOI] [PubMed] [Google Scholar]
- 40.Kessler M, Acuto O, Storelli C, Murer H, Muller M, Semenza G. Biochim Biophys Acta. 1978;506:136–154. doi: 10.1016/0005-2736(78)90440-6. [DOI] [PubMed] [Google Scholar]
- 41.Peerce BE, Fleming RY, Clarke RD. Biochem Biophys Res Commun. 2003;301:8–12. doi: 10.1016/s0006-291x(02)02974-1. [DOI] [PubMed] [Google Scholar]
- 42.Peerce BE. Biochim Biophys Acta. 1997;1323:45–56. doi: 10.1016/s0005-2736(96)00174-5. [DOI] [PubMed] [Google Scholar]
- 43.Chhajlani V, Wikberg JE. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 44.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 45.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 46.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 47.Mergen M, Mergen H, Ozata M, Oner R, Oner C. J Clin Endocrinol Metab. 2001;86:3448–3451. doi: 10.1210/jcem.86.7.7809. [DOI] [PubMed] [Google Scholar]
- 48.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. J Clin Invest. 2000;106:253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaisse C, Clement K, Guy-Grand B, Froguel P. Nat Genet. 1998;20:113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 50.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]