

Abstract
Chronic nitric oxide synthase inhibition (NOSI) causes chronic kidney disease (CKD) in the Sprague Dawley (SD) rat. We previously showed that the Wistar-Furth (WF) rats are resistant to several models of CKD and maintain renal nitric oxide (NO) production compared with SD rats, whereas low-dose NOSI caused progression of CKD in WF rats. Here, we evaluate the impact of high-dose chronic NOSI in WF and SD rats, as well as intrarenal responses to an acute pressor dose of NOSI in the normal WF. Rats were given NG-nitro-l-arginine methyl ester (l-NAME) (150 and 300 mg/l for 6–10 wk) in the drinking water after an initial bolus tail vein injection. Both strains showed significant reductions in total NO production with chronic l-NAME. SD given 150 mg/l l-NAME for 6 wk developed proteinuria and renal injury, whereas WF rats receiving 150 mg/l l-NAME for 6–10 wk or 300 mg/l for 6 wk developed no proteinuria and minimal renal injury. Blood pressure was significantly elevated with chronic NOSI in both strains but was higher in the SD rat. There was little impact on renal nitric oxide synthase expression with l-NAME, except that cortical endothelial nitric oxide synthase abundance increased in WF after 6 wk (150 mg/l). Micropuncture experiments with acute pressor NOSI resulted in similar increases in systemic blood pressure in SD and WF rats, whereas WF rats showed a much smaller increment in glomerular blood pressure compared with SD rats. In conclusion, WF rats do not develop renal injury after chronic NOSI at, or above, a dose that causes significant injury in the SD rat. This protection may be associated with protection from glomerular hypertension.
Keywords: proteinuria, glomerular sclerosis, creatinine clearance, Sprague-Dawley
There is clear evidence from animal studies that chronic nitric oxide synthase (NOS) inhibition (NOSI) causes dose-dependent hypertension and progressive kidney damage. Incomplete NOSI leads to moderate systemic and glomerular hypertension, mild proteinuria, and moderate focal and segmental glomerular sclerosis. Higher doses create an accelerated model of hypertension and kidney damage, and the systemic and glomerular hypertension contribute to the increased severity of kidney damage (37). In addition, the potent vasoconstrictor effect of NO deficiency causes declines in renal function and the non-hemodynamic actions of NO deficiency include permissive mesangial growth and matrix overproduction, promoting glomerular sclerosis (21).
These findings have clinical relevance, given the evidence that NO deficiency occurs in humans as a result of chronic kidney disease (CKD) (4, 6, 26–28, 34). One site of systemic NO deficiency is undoubtedly associated with the widespread endothelial dysfunction seen in renal disease, at least in part due to elevations in the endogenous NOS inhibitor asymmetric dimethylarginine (7). In a variety of animal models of CKD, a decrease in total NO production has been reported as well as a reduced renal NOS abundance and activity (1, 3, 11, 12, 14, 25, 30, 32). Concomitant NOSI accelerates the course of experimentally induced CKD due to ablation/infarction of 5/6 of the renal mass (17) and very low-dose chronic NOSI renders the otherwise resistant Wistar-Furth rat extremely vulnerable to ablation/infarction of 5/6 of the renal mass (15). Overall, these findings suggest that NO deficiency, secondary to CKD, may play a role in CKD progression and that NO deficiency may therefore be both a cause and consequence of CKD.
The Wistar-Furth (WF) rat is interesting in that it is not only resistant to renal mass reduction models of CKD (15, 16) but also to chronic puromycin-induced CKD (12), as well as mineralocorticoid-induced hypertension and renal injury (13, 20). In the present study we investigated the response to chronic, high-dose NOSI in the WF, and because of the remarkable resistance exhibited by this rat strain, we also investigated the glomerular hemodynamic response to an acute pressor dose of NOSI in the normal WF (i.e., without preexisting CKD).
Methods
For chronic NOSI studies
Studies were conducted on two strains of rats, Sprague-Dawley (SD) and Wistar-Furth (WF) purchased from Harlan (Indianapolis, IN) and age-matched at 10 wk. Rats were divided into groups as shown in Table 1 and consisted either of sham rats or those treated with chronic NOSI using the nonselective NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME). A bolus dose was given intravenously (via tail vein) followed by l-NAME ad libitum in the drinking water for the duration of the experiment, as described in Table 1. All experiments were reviewed and approved by the West Virginia University Animal Care and Use Committee (approval #01-0701).
Table 1. Protocol for chronic nitric oxide synthase inhibition studies.
| Strain | Group ID | n | Treatment | Duration, wk |
|---|---|---|---|---|
| SD | SD CT | 5 | Control | 6 |
| WF | WF CT | 7 | Control | 6 |
| SD | SD 150 mg/l | 6 | 1.5 mg/100 g BW bolus; 150 mg/l ad lib | 6 |
| WF | WF 150 mg/l | 4 | 1.5 mg/100 g BW bolus; 150 mg/l ad lib | 6 |
| WF | WF 300 mg/l | 7 | 3 mg/100 g BW bolus; 300 mg/l ad lib | 6 |
| WF | WF-10 CT | 5 | Control | 10 |
| WF | WF-10 150 mg/l | 8 | 1.5 mg/100 g BW bolus; 150 mg/l ad lib | 10 |
SD, Sprague-Dawley; WF, Wistar-Furth; CT, control; BW, body weight; ad lib, ad libitum.
Metabolic cage collections of 24 h urines were made at baseline and at weekly intervals of chronic NOSI to measure urinary total protein, the stable oxidation products of NO, NO2 + NO3 = NOx, and creatinine. Rats were placed on a low-NOx diet (AIN 76C semipurified diet, ICN Pharmaceuticals, Costa Mesa, CA) 2 days before placement in metabolic cages. Total protein was determined using the Bradford assay, and NOx was measured via the Greiss reaction (24, 25). Creatinine was measured using HPLC, as described earlier (12). Blood pressure (BP) was measured under anesthesia via aorta just before death; then blood was collected (3–5 ml), and tissues were perfused with cold phosphate-buffered saline and harvested onto dry ice, then snap frozen in liquid nitrogen. Plasma NOx and creatinine were measured, as above and blood urea nitrogen was measured using a Sigma kit (#640-A).
Western blot procedures were performed as explained previously (36). Briefly, neuronal nitric oxide synthase (nNOS) was detected with a rabbit monoclonal antibody [gift of Dr. Kim Lau, 1:10,000 dilution, 1 h incubation; secondary antibody, goat, anti-rabbit IgG-HRP, Bio-Rad, (Hercules, CA); 1:3,000 dilution, 1 h]. Membranes were stripped and reprobed for endothelial nitric oxide synthase (eNOS) [mouse, monoclonal antibody, Transduction Laboratories (San Diego, CA), 1:250 dilution, 1 h; secondary antibody goat, anti-mouse IgG-HRP conjugate, Transduction Labs, 1:2,000 dilution, 1 h]. All steps were performed at room temperature. Bands were visualized using ECL reagent and quantitated by densitometry, as integrated optical density (IOD) after subtraction of background. The IOD was factored for Ponceau red staining to correct for any variations in total protein loading and for an internal standard (eNOS = 10 μg bovine aortic endothelial cell lysate; nNOS = 1 μg of cerebellar homogenate) to allow comparison between different membranes.
For pathology, the formalin-fixed left kidney was dehydrated in alcohol and blocked in paraffin wax; 5-μm sections were cut and stained with periodic acid Schiff + hematoxylin eosin counterstain. The level of injury was assessed by histology, performed on a blinded basis, by assessing the sclerotic damage to glomeruli (n = 100) using the 0 to 4+ scale, as described previously (5). Pathology was conducted on kidneys from all rats studied (Table 1).
For acute NOSI studies
Studies were conducted on WF (n = 5) purchased from Harlan and studied at age 3–5 mo. On the day of micropuncture, rats were anesthetized with an intraperitoneal injection of the thiobarbiturate Inactin (120 mg/kg; Research Biochemicals International, Natick, MA), and further supplemental doses (5–10 mg/kg) were given intraperitoneally as required during the experiment. General anesthesia in WF rats turned out to be very difficult to control, requiring many small administrations of booster doses. Also, BP tended to become unstable after ∼3 h of general anesthesia, and the experimental design was therefore modified and shortened to allow key measurements only. Rats were placed on a temperature-controlled micropuncture table, and core temperature was maintained at 36–38°C. The rat was surgically prepared for glomerular micropuncture studies using the euvolemic preparation (5). Surgery included a tracheotomy, placement of intravenous lines in the left femoral and a jugular vein for infusion of synthetic plasma (2.5% bovine serum albumin and 2.5% bovine globulin in Ringer solution), and 0.9% NaCl at 1.2 ml/h and a femoral arterial line to monitor BP and to collect blood samples. The left kidney was exposed through a ventral midline and left subcostal incision and was immobilized and prepared for micropuncture. The surface of the kidney was illuminated and bathed in warm, 0.9% NaCl solution (34–36°C). For estimation of glomerular blood pressure (PGC) by the indirect, stop-flow pressure method, tubule fluid flow was stopped by insertion of paraffin wax blocks into 3–7 randomly selected, midproximal surface nephron segments.
After equilibration, control measurements were made as follows: measurements were made of the stop-flow pressure (Psf) in the superficial cortex of hydrostatic pressures in proximal segments of obstructed tubules (n = 3–7) with simultaneous measurement of systemic BP. Free-flow proximal tubule (PT) pressures were also measured in 3–5 tubules. At the end of the control, rats were given an intravenous bolus of the NO synthesis inhibitor, NG-monomethyl-l-arginine (l-NMMA; 30 mg/kg), followed by a continual intravenous infusion of l-NMMA (2 mg·kg−1·min−1). After equilibration of the new BP (within 1–3 min), repeat measurements were made of Psf, PT, and BP. Arterial blood samples were taken during control and experimental periods for measurement of systemic protein concentration (refractometer) and calculation of the afferent arteriolar oncotic pressure to allow calculation of glomerular blood pressure (PGC), as described previously (5). At the end of the study, a second intravenous bolus of l-NMMA (30 mg/kg) was given to assess the degree of NOSI. The BP response was 1 ± 1 mmHg, demonstrating complete NOSI.
Statistical analyses were performed by paired and unpaired t-test, one- and two-way ANOVA, and by Wilcoxon rank sum analysis for the injury data. Statistical significance was found at P < 0.05. Data were expressed as means ± SE.
Results
Rats treated with the chronic NOS inhibitor l-NAME (150 mg/l) showed decreased levels of total NO production consistently maintained throughout the 6 wk (Fig. 1). In both strains, NOSI with 150 mg/l l-NAME elicited reductions in urinary NOx excretion of ∼0.60 μmol·24 h−1·100 gBW−1. Because of the higher mean values of the WF vs. SD at baseline (P < 0.05), the percent reduction was less in the WF (∼45%) compared with the SD (∼55%). Also shown in Fig. 1, doubling the dose of l-NAME to 300 mg/l in the WF further reduced total NO production (P < 0.01 vs. lower dose) by 0.9 μmol·24 h−1·100 gBW−1 (∼70% reduction vs. control) to an absolute level similar to that of the SD receiving 150 mg/l. Despite the clear evidence of decreased total NO generation in all rats, there was a marked strain difference in the functional responses. In the sham rats, BP was lower in the WF compared with SD rats (Fig. 2). With chronic NOSI, BP increased in both strains with a rise of 26 ± 2 mmHg in WF and a greater increment (P < 0.05) of 41 ± 8 mmHg in SD. The %change in BP was greater in SD vs. WF (47 ± 9 vs. 35 ± 2%, P = 0.05). The rise in BP seen in the WF with 300 mg/l l-NAME was similar to that obtained with the lower dose in WF (Fig. 2; equivalent to 33 ± 6% increase). As shown in Fig. 3, the SD developed significant proteinuria after 6 wk of chronic NOSI, whereas the WF developed no proteinuria at either dose (Fig. 3). As with the BP, the baseline value of urinary protein excretion (Fig. 3) and percent and severity of damaged glomeruli (Fig. 4) were lower in WF vs. SD (both P < 0.05). Representative sections showing renal cortical pathology are given in Fig. 5. The SD developed significant glomerular sclerosis after 6 wk of chronic NOSI, whereas the increase in the WF was minimal (and similar to the SD sham value) on 150 mg/l l-NAME (Fig. 4) and on the 300 mg/l dose (with 4.6 ± 0.7 damaged glomeruli, of which 3.4 ± 0.8 were +1 severity and 1.2 ± 0.4 were +2). In addition to glomerular sclerosis, there was significant focal tubulointerstitial injury in the SD with many casts, whereas the tubulointerstitium was normal in WF and no casts were present (Fig. 4). There was no difference in baseline glomerular filtration rate, estimated from 24 h creatinine clearance (CCr) between WF and SD (7.8 ± 0.2 and 8.1 ± 0.6 ml·min−1·kgBW−1, respectively) and no significant change with chronic l-NAME at 150 ml/l for 6 wk (7.4 ± 0.2 and 7.4 ± 0.6 ml·min−1·kgBW−1, respectively).
Fig. 1.

Twenty-four-hour urinary excretion of NO2 + NO3 (UNOxV) by Wistar-Furth (WF) and Sprague-Dawley (SD) rats for 6 wk with and without NG-nitro-l-arginine methyl ester (l-NAME).
Fig. 2.

Blood pressure (BP) at 6 wk of sham treatment or administration of 150 or 300 mg/l l-NAME for SD (solid bars) and WF (gray bars). #Significant difference between strain; *significant difference within each strain vs. the sham value.
Fig. 3.

Twenty-four-hour urine protein excretion (UpV) after 6 wk of sham treatment in the controls, or oral administration of 150 or 300 mg/l of l-NAME in the SD (solid bars) and WF (gray bars). #Significant difference between strains; *significant difference within each strain vs. the sham value.
Fig. 4.

The total number of damaged (sclerotic) glomeruli in SD-treated with 150 mg/l l-NAME for 6 wk [chronic nitric oxide synthase inhibition (CNOSI); solid bars] and SD shams (open bars) and WF CNOSI (gray), and WF shams (dark gray) rats are given in the first columns. Next, the severity of glomerular injury is given, with +1 representing mild damage, up to 25% of glomerulus involved; +2 representing moderate damage with 26–50% injury; +3 and +4 representing severe damage of 51–75% and 76%, global sclerosis, respectively. #Significant difference between strain; *significant difference within each strain vs. the sham value.
Fig. 5.
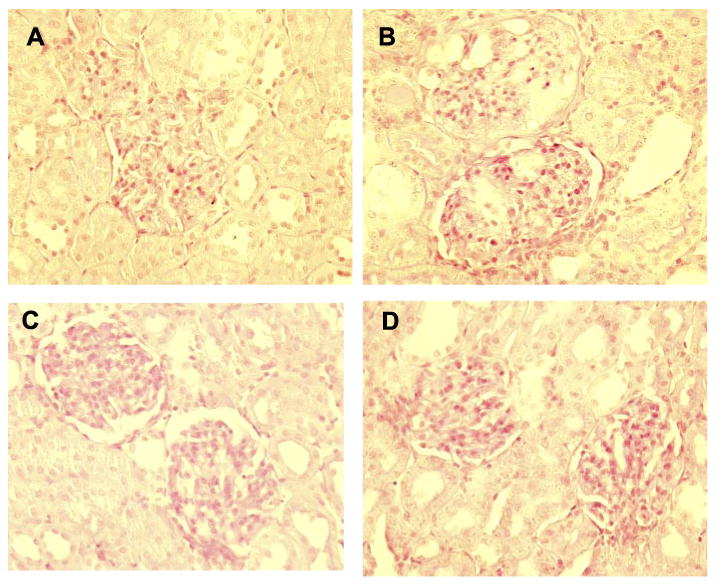
A: two normal glomeruli from a sham control SD rat. B: two damaged glomeruli with ∼50% injury in the upper glomerulus and a cast to the left (just below part label B), with mild <25% injury to the lower glomerulus. C: two normal glomeruli from a sham WF rat. D: two normal glomeruli from a WF CNOSI rat.
We also studied a group of WF given 150 mg/l l-NAME for 10 wk. Total NO production (from urinary NOx excretion) was decreased with chronic NOSI similarly to the 6-wk protocol (Table 2). There was no proteinuria, glomerular sclerosis (%GS), or change in CCr in rats treated with chronic NOSI for 10 wk vs. shams (Table 2). The BP was elevated compared with shams studied over the 10-wk period (114 ± 4 and 80 ± 4 mmHg; P < 0.001; equivalent to an increase of 47 ± 6%) and was also greater than that seen in WF at 6-wk chronic NOSI at the same dose (P < 0.05; Fig. 2).
Table 2. Measured variables in the WF rats given chronic NOSI (150 mg/l) for 10 wk.
| UNOxV | PNOx | UpV | PCr | BUN | CCr | |
|---|---|---|---|---|---|---|
| WF sham | 1.21±0.01 | 12.7±1.0 | 15±2 | 0.31±0.02 | 17±1 | 8.3±1.0 |
| WF NOSI | 0.55±0.02 | 9.2±0.9 | 15±2 | 0.30±0.02 | 17±1 | 8.1±1.1 |
NOSI, nitric oxide synthase inhibition studies; UNOx, urinary NOx excretion; PNOx, plasma NOx concentration; UpV, urine protein excretion; BUN, blood urea nitrogen; CCr, creatine clearance.
We determined the abundance of eNOS protein in both the cortex and medulla and as indicated in Fig. 6; the only significant change was an increase in cortical eNOS protein abundance in the WF given chronic NOSI (150 mg/l l-NAME). There was no significant change in nNOS protein abundance due to chronic NOSI, although the nonsignificant tendency of the SD renal cortex abundance to decline created a significant difference between strains (Fig. 7).
Fig. 6.
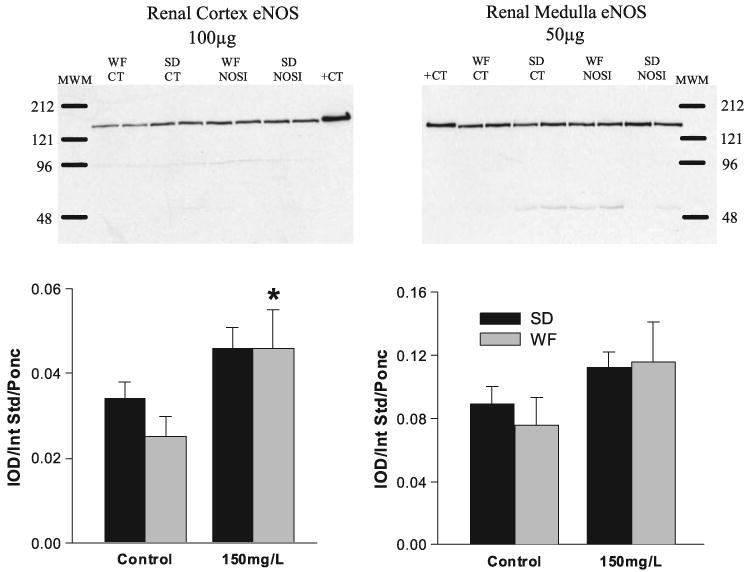
Relative abundance of endothelial nitric oxide synthase (eNOS) in the cortex and medulla of SD (solid bars) and WF (gray bars) 6 wk after 150 mg/l l-NAME. Representative gels are given for cortex (left) and medulla (right) with average densitometric values (Integrated optical density/Internal standard/Ponceau red) given below. MWM, molecular weight marker; +CT, positive control. *Significant change from control.
Fig. 7.
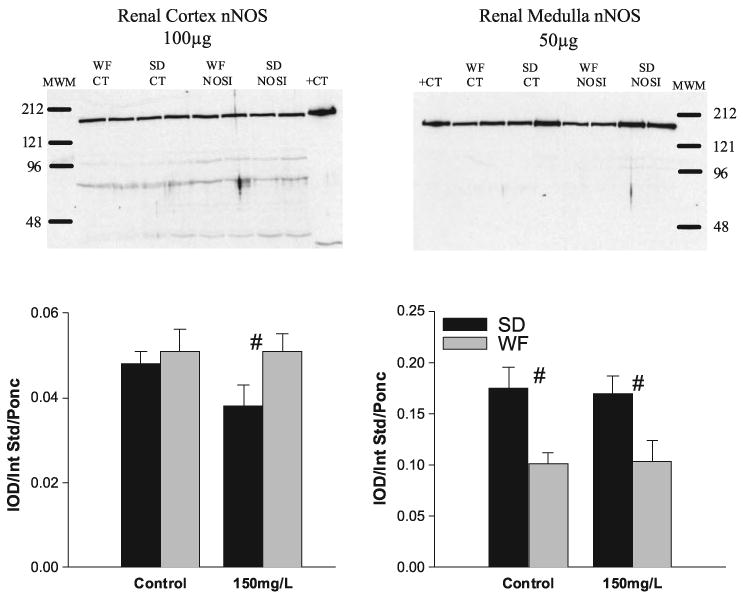
Relative abundance of neuronal nitric oxide synthase (nNOS) in the cortex and medulla of SD (solid bars) and WF (gray bars) 6 wk after 150 mg/l l-NAME. Representative gels are given for cortex (left) and medulla (right) with average densitometric value (integrated optical density/internal standard/Ponceau red) given below. MWM, molecular weight marker; +CT, positive control. #Significant difference between strains.
In the micropuncture study, BP in the Inactin-anesthetized WF was nonsignificantly lower than that found in our previously published data forthe SD (23) strain, using the same preparation (92 ± 6 and 99 ± 1 mmHg), although the PGC was slightly lower (53 ± 1 vs. 57 ± 1, P < 0.05). In response to acute NOSI with intravenous l-NMMA, the BP increased to 118 ± 7 mmHg, which was a lower absolute value than we previously observed in the SD (23) = 132 ± 2 mmHg, although the percent change from control was similar in the two strains (Fig. 8). Of particular note, however, the magnitude of the rise in PGC was very much attenuated (Fig. 8), rising to only 56 mmHg compared with the value of 79 ± 3 mmHg that we previously reported in the SD (23). Because the PT pressure was unchanged by acute NOSI in the WF (11 ± 1 mmHg in both control and + l-NMMA), the transglomerular hydrostatic pressure gradient (PGC-PT) was only slightly increased in the WF with acute NOSI (45 ± 2 vs. 41 ± 2 mmHg; P < 0.05).
Fig. 8.

The %change from control for BP and glomerular blood pressure (PGC) in response to an acute intravenous infusion of the NOS inhibitor NG-monomethyl-l-arginine in the normal SD (solid bars; data taken from Ref. 23) and WF (gray bars). #Significant difference between strains.
Discussion
The novel findings of this study are that as with other models of CKD, the WF are protected from chronic NOSI-induced renal injury. No proteinuria developed, and glomerular injury was minimal in WF on any dose of l-NAME compared with SD rats following chronic NOSI. Both strains developed systemic hypertension, but the WF showed a reduced absolute level compared with SD. With acute systemic NOSI, the WF exhibited a substantial pressor response but with a minimal increase in PGC, which contrasted with the large increase in PGC seen previously in the SD (23).
The primary characteristics of chronic NOSI are hypertension, proteinuria, and renal injury (37), and all three measurements were different between the two strains. Although SD rats developed proteinuria and significant increases in total glomerular injury and severity, the WF developed minimal glomerular injury and no proteinuria. In our extended protocol, with 10 wk of chronic NOSI, the WF showed no evidence of progression, whereas the similarly treated SD had 100% mortality before week 10, and thus a comparison group was not studied. Even when the dose of l-NAME was doubled in WF, reducing total NO production to the absolute level seen in SD with chronic NOSI, there was no evidence of progressive renal injury. This indicates that the absolute level of total NO production is not always a good predictor of renal injury, and it should also be noted that the UNOXV does not give specific information on regional NO production such as intrarenal NO. Indeed, in the diabetic Zucker rat with severe renal damage, total NO production actually increases, while renal NOS activity falls (11).
Our previous data have shown the central importance of renal NO in determining the rate of progression of CKD. WF rats have maintained renal NO production compared with the SD in two models of CKD, both 5/6 ablation/infarction of renal mass A/I and chronic PAN (12, 15). However, low-dose NOSI (25 mg/l l-NAME from week 4 to 6 and 12.5 mg/l from week 6 to 7.5) in the WF following 5/6 renal ablation/infarction caused severe systemic hypertension, rapidly progressing glomerular injury, and decreased renal function such that the study was terminated early (15). The importance of NO in protecting the WF kidney resembles the findings in spontaneously hypertensive rats, which despite severe hypertension, do not develop renal injury until about 1 yr of age. Chronic low-dose NOSI greatly accelerated the appearance of proteinuria and decreased survival compared with Wistar-Kyoto controls (33). These findings suggest that whereas an individual stimulus was not sufficient to cause renal injury in WF, a combination of CKD and NOSI led to accelerated damage. The WF are not entirely resistant to development of CKD, but progression is extremely slow (12, 15, 16). In support of delayed susceptibility in the WF, a constant dose of NOSI (150 mg/l) caused BP to rise progressively up to 10 wk. It has previously been shown that constant levels of NOSI cause a time dependent rise in blood pressure (24).
The protection of the WF to a single stimulus (i.e., induced CKD or chronic NOSI) could be associated with their relative resistance to development of hypertension. They have a lower basal BP than SD (12, 15) or Wistar (16), and the increase after renal mass reduction (15) or chronic NOSI, in the present study, was also blunted. However, the primary risk factor for renal injury is glomerular, rather than systemic, hypertension (2). There was little renal benefit with antihypertensive treatment (triple therapy) that reduced systemic but not glomerular pressure after renal mass reduction in the “vulnerable” Munich-Wistar strain (2). A similar result was reported in the “vulnerable” Wistar rats with 5/6 renal ablation/infarction for which reducing systemic BP with triple therapy to the level of “protected” WF was still associated with significant albuminuria (16). One marked difference between the WF and SD that might contribute to the difference in susceptibility to CKD was observed in the present study at the level of the glomerular microcirculation. It is clear that the renal circulation is acutely sensitive to NOSI as low-dose and intrarenal NOSI cause intrarenal vasoconstriction with no changes in systemic BP (10, 18, 35). As shown previously, acute NOSI in the SD resulted in a marked glomerular hypertension (23), and this is maintained in (female) SD during chronic NOSI (9), as also seen in the Munich-Wistar rat (5); unfortunately, no data are available on chronic NOSI in the WF strain. On the other hand, although the WF exhibited a significant pressor response to acute NOSI (+26 mmHg), the glomerular capillary pressure only increased by 4 mmHg, in sharp contrast to the 22 mmHg rise in PGC seen in the SD (23). We do not know what the effect of chronic NOSI is on PGC in the WF, but on the basis of the similarity between the acute and chronic response in the Munich-Wistar (5, 10) rats, we anticipate that PGC is normal in the WF during chronic NOSI.
Because of the difficulty in maintaining the WF under barbiturate-induced anesthesia, we were forced to make an abbreviated set of micropuncture measurements that do not allow assessment of the relative roles of the afferent and efferent arteriolar resistances in the response to acute NOSI. In the SD and Munich Wistar rat strains, acute NOSI leads to significant efferent arteriolar constriction, which causes the marked rise in PGC (10, 23, 38). The efferent arteriolar constriction is dependent on a NOSI-induced rise in systemic BP and does not occur with intrarenal NOSI, where PGC falls due to selective afferent arteriolar constriction (10). Even transient exposure of the SD rat kidney to increased renal perfusion pressure during acute NOSI leads to an amplified increase in total renal vascular resistance that is preventable by endothelin blockade (39). Furthermore, the increased efferent resistance can be prevented by blockade of angiotensin type 1 and endothelin receptors (23). Thus it is likely that the efferent arteriolar constriction does not occur in the WF.
Renal injury following chronic NOSI is known to be due to activation of the renin-angiotensin, endothelin, and sympathetic nervous systems rather than simply a lack of vasodilatory NO (37). Substrate supplementation with l-arginine, simultaneous with NOSI, prevents hypertension, but l-arginine started a week or longer after NOSI only has mild attenuating effects, implicating the role of other systems in established chronic NOSI (37). At present, there is no information on the relative activities of the renin-angiotensin and endothelin systems in the WF strain, which may underlie the protection that these rats enjoy from development of CKD. There may also be vasodilatory systems active in the WF that help offset the damaging effects of chronic NOSI. In fact, differences in other vasoactive systems may also underlie the differences in susceptibility to chronic NOSI-induced CKD reported for other strains, and even within the SD strain from different vendors (22, 31). Future experiments should investigate the interaction of NO and other vasoactive systems in modulating glomerular protection in the WF.
Earlier studies by us using different models of CKD have shown a relationship between reduced renal NO production and the level of renal injury (11, 12, 14, 15, 30). The loss of renal cortical nNOS predicts the severity of the disease in 5/6 renal mass ablation/infarction, chronic glomerulonephritis, chronic puromycin-induced CKD, a model of type 2 diabetic nephropathy and in normal aging (11, 12, 14, 15, 25, 30). However, in the present study, nNOS levels in the kidney were not changed as a result of NOSI in either vulnerable SD or protected WF. We assume that this reflects removal of the feedback inhibitory pathways by which NO controls NOS activity (19), which offsets the nNOS-reducing signals of the CKD in the chronic NOSI model. Indeed, both cortical and medullary eNOS abundance, although not always significant, certainly trended upward in both strains. Of course, with chronic NOSI, the function of all the NOS enzymes is inhibited.
In summary, WF rats did not develop any progressive symptoms of renal injury up to 10 wk of chronic NOSI. In contrast to SD, WF rats show protection of the glomerular microcirculation to pressor doses of NOSI. Certainly long-term maintenance of glomerular pressure, likely to be the case in the WF, confers renal protection. Future studies will need to be done to elucidate the mechanism(s) of protection in the WF.
Acknowledgments
The excellent technical assistance of Lennie Samsell and Kevin Engels is gratefully acknowledged.
Grants: These studies were supported by NIH Grants # R01 DK-56843 and DK-45517.
References
- 1.Aiello S, Noris M, Todeschini M, Zappella S, Foglieni C, Benigni A, Corna D, Zoja C, Cavallotti D, Remuzzi G. Renal and systemic nitric oxide synthesis in rats with renal mass reduction. Kidney Int. 1977;52:171–181. doi: 10.1038/ki.1997.317. [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Brenner BM. The role of intraglomerular pressure in the initiation and progression of renal disease. J Hypertens Suppl. 1986;4:S236–S238. [PubMed] [Google Scholar]
- 3.Ashab I, Peer G, Blum M, Wollman Y, Chernihovsky T, Hassner A, Schwartz D, Cabili S, Silverberg D, Iaina A. Oral administration of l-arginine and captopril in rats prevents chronic renal failure by nitric oxide production. Kidney Int. 1995;47:1515–1521. doi: 10.1038/ki.1995.214. [DOI] [PubMed] [Google Scholar]
- 4.Baylis C, Vallance P. Measurement of nitrite and nitrate (NOx) levels in plasma and urine; what does this measure tell us about the activity of the endogenous nitric oxide? Curr Opin Nephrol Hypertens. 1998;7:59–62. doi: 10.1097/00041552-199801000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Baylis C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest. 1992;90:278–281. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum M, Yachnin T, Wollman Y, Chernihovsky T, Peer G, Grosskopf I, Kaplan E, Silverberg D, Cabili S, Iaina A. Low nitric oxide production in patients with chronic renal failure. Nephron. 1998;79:265–268. doi: 10.1159/000045047. [DOI] [PubMed] [Google Scholar]
- 7.Böger RH, Zoccali C. ADMA: a novel risk factor that explains excess cardiovascular event rate in patients with end-stage renal disease. Atheroscler Suppl. 2003;4:23–28. doi: 10.1016/s1567-5688(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 8.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 9.Deng A, Engels K, Baylis C. Increased nitric oxide production plays a critical role in the maternal blood pressure and glomerular hemodynamic adaptations to pregnancy in the rat. Kidney Int. 1996;50:1132–1138. doi: 10.1038/ki.1996.420. [DOI] [PubMed] [Google Scholar]
- 10.Deng A, Baylis C. Locally produced EDRF controls preglomerular resistance and ultrafiltration coefficient. Am J Physiol Renal Fluid Electrolyte Physiol. 1993;264:F212–F215. doi: 10.1152/ajprenal.1993.264.2.F212. [DOI] [PubMed] [Google Scholar]
- 11.Erdely A, Freshour G, Maddox D, Olson J, Samsell L, Baylis C. Renal disease in rats with type 2 diabetes is associated with decreased renal nitric oxide production. Diabetologia. 2004;47:1672–1676. doi: 10.1007/s00125-004-1509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erdely A, Freshour G, Smith C, Engels K, Olson J, Baylis C. Protection against puromycin aminonucleoside-induced chronic renal disease in the Wistar-Furth rat. Am J Physiol Renal Physiol. 2004;287:F81–F89. doi: 10.1152/ajprenal.00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erdely A, Freshour G, Tain YL, Engels K, Baylis C. Maintained renal nitric oxide production in Wistar-Furth (WF) vs Sprague-Dawley (SD) rats following DOCA salt induced renal disease (Abstract) J Am Soc Nephrol. 2004;15:483A. [Google Scholar]
- 14.Erdely A, Greenfeld Z, Wagner L, Baylis C. Sexual dimorphism in the aging kidney; inverse relationship between injury and nitric oxide system. Kidney Int. 2003;63:1021–1026. doi: 10.1046/j.1523-1755.2003.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erdely A, Wagner L, Muller V, Szabo A, Baylis C. Protection of Wistar-Furth rat from chronic renal disease is associated with maintained renal nitric oxide synthase. JASN. 2003;14:2526–2533. doi: 10.1097/01.asn.0000086476.48686.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitzgibbon WR, Greene EL, Grewal JS, Hutchison FN, Self SE, Latten SY, Ullian ME. Resistance to remnant nephropathy in the Wistar-Furth rat. J Am Soc Nephrol. 1999;10:814–821. doi: 10.1681/ASN.V104814. [DOI] [PubMed] [Google Scholar]
- 17.Fujihara CK, De Nucci G, Zatz R. Chronic nitric oxide synthase inhibition aggravates the glomerular injury in rats with subtotal nepnrectomy. J Am Soc Nephrol. 1995;5:1498–1507. doi: 10.1681/ASN.V571498. [DOI] [PubMed] [Google Scholar]
- 18.Granger JP, Alberola AM, Salazar FJ, Nakamura T. Control of renal hemodynamics during intrarenal and systemic blockade of nitric oxide synthesis in conscious dogs. J Cardiovasc Pharmacol. 1992;20:S160–S162. doi: 10.1097/00005344-199204002-00045. [DOI] [PubMed] [Google Scholar]
- 19.Griscavage JM, Hobbs AJ, Ignarro LJ. Negative modulation of nitric oxide synthase by nitric oxide and nitroso compounds. Adv Pharmacol. 1995;34:215–234. doi: 10.1016/s1054-3589(08)61088-1. [DOI] [PubMed] [Google Scholar]
- 20.Kayes K, Ziegler L, Yu CP, Brownie AC, Gallant S. The resistance of the Wistar/Furth rat strain to steroid hypertension. Endocr Res. 1996;22:681–689. doi: 10.1080/07435809609043763. [DOI] [PubMed] [Google Scholar]
- 21.Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the kidney. Am J Physiol Renal Physiol. 1997;272:F561–F578. doi: 10.1152/ajprenal.1997.272.5.F561. [DOI] [PubMed] [Google Scholar]
- 22.Pollock DM, Rekito A. Hypertensive response to chronic NO synthase inhibition is different in Sprague-Dawley rats from two suppliers. Am J Physiol Regul Integr Comp Physiol. 1998;275:R1719–R1723. doi: 10.1152/ajpregu.1998.275.5.R1719. [DOI] [PubMed] [Google Scholar]
- 23.Qiu C, Baylis C. Endothelin and angiotensin mediate most glomerular responses to nitric oxide inhibition. Kidney Int. 1999;55:2390–2396. doi: 10.1046/j.1523-1755.1999.00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qui C, Muchant D, Beierwaltes WH, Racusen L, Baylis C. Evolution of chronic nitric oxide hypertension: relationship to renal function. Hypertension. 1998;31:21–26. doi: 10.1161/01.hyp.31.1.21. [DOI] [PubMed] [Google Scholar]
- 25.Roczniak A, Fryer JN, Levine DZ, Burns KD. Downregulation of neuronal nitric oxide synthase in the rat remnant kidney. J Am Soc Nephrol. 1999;10:704–713. doi: 10.1681/ASN.V104704. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt RJ, Baylis C. Total nitric oxide production is low in patients with chronic renal disease. Kidney Int. 2000;58:1261–1266. doi: 10.1046/j.1523-1755.2000.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt RJ, Domico J, Samsell L, Yokota S, Tracy T, Sorkin M, Engels K, Baylis C. Indices of activity of the nitric oxide system in patients on hemodialysis. Am J Kidney Dis. 1999;34:228–234. doi: 10.1053/AJKD03400228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt RJ, Yokota S, Tracy TS, Sorkin MI, Baylis C. Nitric oxide production is low in end stage renal disease patients on peritoneal dialysis. Am J Physiol Renal Physiol. 1999;276:F794–F797. doi: 10.1152/ajprenal.1999.276.5.F794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suto T, Losonczy G, Qiu C, Hill C, Samsell L, Ruby J, Charon N, Venuto R, Baylis C. Acute changes in urinary excretion of nitrite+nitrate (UNOXV) do not predict renal vascular NO production. Kidney Int. 1995;48:1272–1277. doi: 10.1038/ki.1995.411. [DOI] [PubMed] [Google Scholar]
- 30.Szabo A, Wagner L, Erdely A, Lau K, Baylis C. Renal neuronal nitric oxide synthase protein expression as a marker of renal function. Kidney Int. 2003;64:1765–1771. doi: 10.1046/j.1523-1755.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 31.Van Dokkum RP, Jacob HJ, Provoost AP. Difference in susceptibility of developing renal damage in normotensive fawn-hooded (FHL) and August × Copenhagen Irish (ACI) rats after N(omega)-nitro-l-arginine methyl ester induced hypertension. Am J Hypertens. 1997;10:1109–1116. doi: 10.1016/s0895-7061(97)00163-5. [DOI] [PubMed] [Google Scholar]
- 32.Vaziri ND, Ni Z, Wang XQ, Oveisi F, Zhou XJ. Downregulation of nitric oxide synthase in chronic renal insufficiency: role of excess PTH. Am J Physiol Renal Physiol. 1998;274:F642–F649. doi: 10.1152/ajprenal.1998.274.4.F642. [DOI] [PubMed] [Google Scholar]
- 33.Verhagen AM, Koomans HA, Joles JA. Predisposition of spontaneously hypertensive rats to develop renal injury during nitric oxide synthase inhibition. Eur J Pharmacol. 2001;411:175–180. doi: 10.1016/s0014-2999(00)00900-6. [DOI] [PubMed] [Google Scholar]
- 34.Wever R, Boer P, Hijmering M, Stroes E, Verhaar M, Kastelein J, Versluis K, Lagerwerf F, van Rijn H, Koomans H, Rabelink T. Nitric oxide production is reduced in patients with chronic renal failure. Arterioscler Thromb Vasc Biol. 1999;19:1168–1172. doi: 10.1161/01.atv.19.5.1168. [DOI] [PubMed] [Google Scholar]
- 35.Woltz M, Schmetterer L, Ferber W, Artner E, Mensik C, Eishler HG, Krejcy K. Effect of nitric oxide synthase inhibition on renal hemodynamics in man: reversal by l-arginine. Am J Physiol Renal Physiol. 1997;272:F178–F182. doi: 10.1152/ajprenal.1997.272.2.F178. [DOI] [PubMed] [Google Scholar]
- 36.Xiao S, Erdely A, Wagner L, Baylis C. Uremic levels of BUN do not cause nitric oxide deficiency in rats with normal renal function. Am J Physiol Renal Physiol. 2001;280:F996–F1000. doi: 10.1152/ajprenal.2001.280.6.F996. [DOI] [PubMed] [Google Scholar]
- 37.Zatz R, Baylis C. Chronic nitric oxide inhibition model six years on. Hypertension. 1988;32:958–964. doi: 10.1161/01.hyp.32.6.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zatz R, De Nucci G. Effects of acute nitric oxide inhibition on rat glomerular microcirculation. Am J Physiol Renal Fluid Electrolyte Physiol. 1991;261:F360–F363. doi: 10.1152/ajprenal.1991.261.2.F360. [DOI] [PubMed] [Google Scholar]
- 39.Zhang XZ, Baylis C. Endothelin mediates The renal vascular “memory” of a transient rise in perfusion pressure due to acute systemic NOS inhibition. Am J Physiol Renal Physiol. 1999;276:F629–F634. doi: 10.1152/ajprenal.1999.276.4.F629. [DOI] [PMC free article] [PubMed] [Google Scholar]